

Abstract
Purpose of Review:
Primary graft dysfunction (PGD) is the leading cause of early mortality following lung transplantation and is typically caused by lung ischemia-reperfusion injury (IRI). Current management of PGD is largely supportive and there are no approved therapies to prevent lung IRI after transplantation. The purinergic signaling network plays an important role in this sterile inflammatory process, and pharmacologic manipulation of said network is a promising therapeutic strategy. This review will summarize recent findings in this area.
Recent Findings:
In the past 18 months, our understanding of lung IRI has improved, and it is becoming clear that the purinergic signaling network plays a vital role. Recent works have identified critical components of the purinergic signaling network (Pannexin-1 channels, ectonucleotidases, purinergic P1 and P2 receptors) involved in inflammation in a number of pathologic states including lung IRI. In addition, a functionally-related calcium channel, the transient receptor potential vanilloid type 4 (TRPV4) channel, has recently been linked to purinergic signaling and has also been shown to mediate lung IRI.
Summary:
Agents targeting components of the purinergic signaling network are promising potential therapeutics to limit inflammation associated with lung IRI and thus decrease the risk of developing PGD.
Keywords: purinergic signaling, ischemia-reperfusion injury, adenosine, ATP, TRPV4
1. Introduction:
Primary graft dysfunction (PGD) increases the risk of negative outcomes following lung transplantation and is typically caused by ischemia-reperfusion injury (IRI) [1**]. Characterized by a robust inflammatory response, hallmarks and sequalae of lung IRI include increased acute innate immunity responses and oxidative stress resulting in inflammation, vascular permeability, pulmonary edema, alveolar damage and ultimate lung dysfunction. The management of PGD is primarily supportive, and there are no approved therapies to prevent the leading cause — lung IRI.
Following a period of cold ischemia and upon reperfusion, a rapid inflammatory response ensues in the allograft. Reactive oxygen species (ROS) are promptly generated causing activation of multiple cell types, with subsequent release of pro-inflammatory cytokines and damage-associated molecular patterns (DAMPs). Surrounding tissue damage occurs resulting in dysfunction of, most notably, pulmonary epithelium and endothelium. Additionally, activation of the innate immune system with stimulation of alveolar macrophages and neutrophil recruitment cumulatively contributes to surrounding tissue damage [1**, 2]. This deleterious series of rapid events culminates in lung IRI and subsequent PGD, which is the leading cause of death in the first 30 days following transplant as well as an independent risk factor for chronic allograft dysfunction [1**, 2]. Clearly, there exists a need for therapies aimed at mitigating lung IRI.
2. Purinergic Signaling:
Recent discoveries have improved our understanding of IRI, a sterile inflammatory process, and while there are a number of signaling pathways involved, it is clear that cellular release of nucleotides and nucleosides and the purinergic signaling network plays an important role [1**, 3]. Extracellular adenosine triphosphate (ATP) is a potent DAMP molecule that rapidly accumulates at sites of inflammation such as IRI [3]. When activated by various stress stimuli, cell surface pannexin 1 (Panx1) channels are known to release ATP into the extracellular space [4*]. The pro-inflammatory actions of extracellular ATP are exerted by members of the purinergic P2 receptor family such as P2X7 and P2Y2 receptors (Figure 1, left) [3, 5**]. As extracellular ATP is hydrolyzed and adenosine levels rise, the balance tilts as adenosine in the extracellular space promotes anti-inflammatory actions via stimulation of purinergic P1 receptors such as adenosine 2A receptor (A2AR) and A2BR (Figure 1, right) [3, 5**, 6]. A number of hopeful agents targeting various components of the purinergic signaling pathway are under investigation with the aim of dampening inflammation in various disease processes [3, 5**] – including lung IRI [7*].
Figure 1.
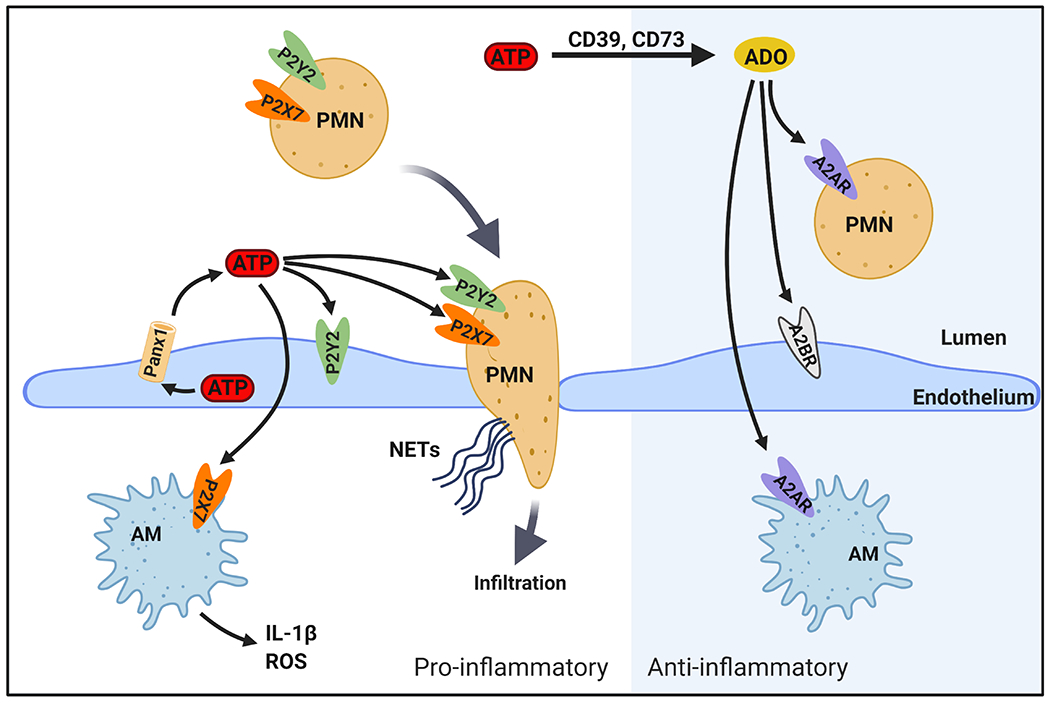
Highlighted purinergic signaling pathways important in lung IRI. Left: Pro-inflammatory pathways. After IR, endothelial pannexin-1 (Panx1) channels release ATP, which accumulates in the extracellular space [10]. ATP exerts pro-inflammatory actions primarily through activating P2Y2 and P2X7 purinergic receptors, among others not shown. P2Y2 activation leads to increased inflammatory cell chemotaxis as well as endothelial cell dysfunction and barrier disruption. P2X7 activation leads to a) activation of alveolar macrophages (AM) to produce reactive oxygen species (ROS) and pro-inflammatory cytokines such as IL-1β and b) activation of neutrophils (PMN) that infiltrate into tissue and airspaces due to endothelial barrier disruption and produce neutrophil extracellular traps (NETs). These effects culminate in pulmonary edema, inflammation, and lung dysfunction. Right: Anti-inflammatory pathways. Ectonucleotidases (CD39 and CD73) catalyze conversion of extracellular ATP to adenosine (ADO), which dampens inflammation. Anti-inflammatory effects of adenosine are largely exerted by activation of adenosine 2A receptor (A2AR) on immune cells and adenosine 2B receptor (A2BR) on endothelium.
2A. Pannexin 1 Channels
The pannexin channel family consists of three transmembrane proteins: Panx1, Panx2, and Panx3. Panx1 is ubiquitously expressed in human tissues including pulmonary endothelium and epithelium and has been the most thoroughly investigated of the group [4*]. Activation of this channel can occur from a number of different mechanisms including but not limited to TNF-α, mechanical stretch, membrane depolarization, rise in intracellular calcium, temperature, and low oxygen tension [4*, 8]. Panx1 channels on the cell membrane primarily release ATP but can also release other small anions and metabolites [9**]. Previous works have shown that the release of ATP via Panx1 channel increases inflammation in lung microvascular endothelium and airway epithelium [9**] suggesting that Panx1 may be an appropriate target to mitigate lung IRI.
Our laboratory recently demonstrated that endothelial Panx1 is implicated in the development of pulmonary edema and vascular inflammation in a murine model of lung IRI [10]. In this study, both genetic deletion (global Panx1 knockout and endothelial-specific Panx1 knockout) and pharmacologic inhibition of Panx1 significantly improved lung function and decreased endothelial permeability, pulmonary edema, and inflammation following 1 hour of lung ischemia and 2 hours of reperfusion [10]. Not only is Panx1 an important channel for releasing ATP, but it has also been directly associated with inflammasome activation, release of IL-1β, and leukocyte recruitment [11*, 12**]. Although there are a number of pharmacologic agents available that inhibit Panx1, their modest selectivity for Panx1 has been a limitation for isolating the precise inhibitory effect of this channel, and additional work in this area is needed [4*].
2B. Extracellular nucleotides and nucleosides
The accumulation of extracellular ATP at sites of inflammation (such as IRI) is well described [1**, 3, 5**, 6], and as previously mentioned, one major source is the release of intracellular ATP via activated Panx1 channels [9**]. Another mechanism by which ATP is released is through the disruption of the cellular membrane during ferroptosis, pyroptosis, and necroptosis — all forms of inflammatory programmed cell death that occur during lung IRI [1**]. A growing body of literature suggests that a high concentration of extracellular ATP acts as a pro-inflammatory danger signal (i.e. DAMP) [3, 6] — potentially exacerbating detrimental inflammatory responses following lung IRI [1**, 5**]. ATP exerts this effect largely via purinergic P2 receptors on pulmonary endothelium and epithelium, but has also been shown to activate inflammatory cells including resident macrophages [13] and neutrophils [14**].
While it was previously believed that extracellular ATP activation of macrophages inhibited release of pro-inflammatory cytokines such as TNF-α, recent work by Soni and colleagues suggests that this is not the complete story [13]. Using lipopolysaccharide as a stimulus for ATP release and macrophage activation, the authors found that while release of TNF-α via the classic secretory pathway was inhibited, extracellular ATP accumulation caused dramatic increases in TNF-α packaging into microvesicles and subsequent release via a non-classic secretory pathway [13]. We have previously demonstrated that alveolar macrophage-derived TNF-α plays an important role in the activation of alveolar epithelial cells [15], and, importantly, a recent study by Lohman et al. demonstrated that Panx1 activation and ATP release lies downstream of endothelial cell activation by TNF-α [16]. Similarly, extracellular ATP is a stimulus for neutrophil activation with a number of down-stream effects. One of these is the release of neutrophil extracellular traps (NETs), which have been shown to contribute to lung IRI and PGD [17] (see Figure 1). Though not yet demonstrated in the lung, a recent study by Sofoluwe and colleagues showed that a Panx1 channel inhibitor decreased extracellular ATP and NETosis in activated, murine bone marrow-derived neutrophils following IRI [14**]. Additional work is needed to demonstrate this effect in lung IRI.
Although extracellular ATP has been established to serve as a pro-inflammatory DAMP signal, a product of ATP degradation, adenosine, can have an opposing role by providing anti-inflammatory effect [3, 5**]. Thus, targeting ATP degradation pathways has been the subject of recent therapeutic investigation [18, 19*, 20]. Ectonucleotidases are cell surface enzymes that hydrolyze extracellular nucleotides. In physiologic states, ectonucleotidases rapidly convert ATP to adenosine monophosphate (AMP) and adenosine, thus keeping levels of ATP relatively low [3, 5**]. However, the excess extracellular ATP released during IRI and other inflammatory conditions can overwhelm this process resulting in sustained elevated ATP concentrations. These enzymes are often thought of as an “immunologic switch” as they catalyze conversion of the pro-inflammatory damage signal ATP to adenosine – a known immunosuppressant [21]. Thus, intense research efforts have focused on the use of pharmacologic adenosine P1 receptor agonists as potential therapeutic agents for IRI in various organs [22].
Two of the most important ectonucleotidases for ATP degradation are ectonucleoside triphosphate diphosphohydrolase-1 (CD39) and ecto-5’-nucleotidase (CD73). CD39 is primarily responsible for the breakdown of ATP to AMP, and CD73 catalyzes the conversion of extracellular AMP to adenosine. Both CD39 and CD73 have been investigated as anti-inflammatory agents in a number of different organ systems [18, 20, 23]. CD73 has a wide tissue distribution and is present in high quantities in endothelium [5**]. A recent study by Mierzejewski and colleagues demonstrated that CD73 deletion leads to the development of age-dependent endothelial dysfunction in mice, associated with impaired L-arginine metabolism; results suggesting that CD73 activity protects the endothelium [19*]. Similarly, it has been suggested that CD73 helps maintain the endothelial barrier during lung inflammation, and previous studies have demonstrated an adenosine-dependent protective effect of CD73 on the pulmonary endothelial barrier in models of hypoxic lung injury [20]. Thus, methods targeting ectonucleotidase agonism may be a promising strategy to mitigate lung IRI.
2C. Purinergic P2 Receptors
The proinflammatory effects of ATP and anti-inflammatory actions of adenosine are largely exerted via different families of cell surface purinergic receptors — P2 and P1 receptors, respectively. These are widely expressed receptors present on pulmonary endothelium, epithelium, and a variety of inflammatory cells [5**, 23, 24]. Eight different transmembrane G protein coupled P2Y receptors (P2Y1,2,3,6,11-14) and seven ligand gated ion P2X channels (P2X1-7) make up the P2 family [3, 23]. Of the P2Y subgroup, the P2Y2 receptor (P2Y2R) has been most commonly linked to inflammation and IRI [3, 23]. Upon binding of ATP, P2Y2R activation stimulates inflammatory cell chemotaxis including, but not limited to, the recruitment of macrophages and neutrophils [3, 23] (Figure 1). Deletion of P2Y2 in murine models has demonstrated protection in several different models of acute inflammation, and this action is believed to be mediated primarily through impaired myeloid cell chemotaxis [23]. Additionally, a recent study by Jin et al. provided evidence that activation of human endothelial cell P2Y2R increases proinflammatory cytokine production [25]. While the focus of this study was development of atherosclerosis, similar inflammatory processes may be occurring in the pulmonary microvasculature during IRI. Though a promising potential therapeutic target, additional study is needed to fully understand the role of P2Y2R in lung IRI.
In general, P2X receptors have a lower affinity for ATP than P2Y receptors [3]. However, the threshold of activation is reached when large quantities of extracellular ATP are present, such as during IRI and other inflammatory processes. One of these receptors – P2X7 receptor (P2X7R) – plays a key role in the inflammatory process and has gained attention as a potential therapeutic target [23, 26]. Though widely present, P2X7R is highly expressed in inflammatory cells including macrophages, neutrophils, and mast cells [21, 26]. These receptors are believed to contribute to a number of innate immune processes such as activation of the nucleotide-binding domain and leucine-rich repeat protein 3 (NLRP3) inflammasome — a complex effecting caspase activation and pro-inflammatory cytokine release [21, 23]. Prolonged or highly concentrated exposure of P2X7R to ATP causes cell membrane blebbing, macropore formation and ultimately results in cell death [21, 27].
P2X7R antagonists have been therapeutically employed in several preclinical inflammatory disease models [23, 28]. In a recent study by Duan and colleagues, the P2X7R inhibitor —Brilliant Blue G (BBG) — was found to ameliorate lung IRI in rats [29]. Here, the authors demonstrated increased P2X7R expression in lung tissue following IRI and that treatment with BBG resulted in less pro-inflammatory cytokine release (IL-1β, TNF-α), decreased pulmonary edema, and improved lung function. Interestingly, this effect was magnified in a rat pulmonary artery hypertension (PAH) model following lung IRI [29]. These findings suggest that P2X7R inhibitors may provide lung protection during surgery requiring cardiopulmonary bypass in patients with PAH. Similarly, we feel that this work provides evidence that inhibition of the P2X7R may be a promising strategy to limit lung IRI following transplant.
2D. Purinergic P1 Receptors
The anti-inflammatory signaling by adenosine primarily occurs through 1 of 4 purinergic G-protein coupled P1 adenosine receptors (A1R, A2AR, A2BR, and A3R) [3, 5**, 30]. Of these, activation of A2AR and A2BR have been implicated in dampening pulmonary inflammatory processes and improving tissue tolerance of ischemia [5**, 7*, 31*]. A2ARs are widely present but strongly expressed on immune cells such as neutrophils and lymphocytes, and studies have shown that A2AR activation decreases inflammatory cell activation at multiple sites [5**, 23] (Figure 1). For example, in a recent study by Xu and colleagues, an A2AR agonist (CSG21680) inhibited activation and NET production by human neutrophils isolated from peripheral venous blood [31*]. Similarly, potentiation of A2AR with the FDA-approved drug dipyridamole (known to increase extracellular adenosine) has been shown to dampen neutrophil activation, ROS production, and NET release in murine models of inflammation [32]. A2BRs have higher expression on vascular endothelial cells and aid in the maintenance of the endothelial barrier, and a number of studies have demonstrated the lung protective effect of adenosine through A2BR stimulation in models of acute pulmonary inflammation [5**, 23].
Our laboratory has established that A2AR agonists potently protect lungs from IRI after transplantation via attenuation of oxidative stress and inflammation [33–35]. These efforts have culminated in a recent phase 1 clinical trial assessing the safety of the A2AR agonist regadenoson in mitigating lung IRI [7*]. Here, lung transplant recipients were administered a 12-hour infusion of regadenoson initiated at the time of skin incision. The authors found no dose-limiting toxicities, supporting pursuit of a phase II randomized trial to determine treatment efficacy [7*]. While this clinical trial evaluated recipient treatment, advances in ex vivo lung perfusion (EVLP) allow for the possibility of targeted lung therapy prior to transplantation [36]. EVLP would theoretically allow for higher treatment doses while decreasing the risk of systemic side effects. Our laboratory has demonstrated the feasibility of this approach via use of an A2AR agonist in a porcine model [37, 38]. While additional study is needed, it is clear that manipulation of the P1 receptor signaling pathway, specifically with A2AR agonists, is a promising strategy to combat lung IRI.
3. Transient Receptor Potential Vanilloid 4 Channel
Transient receptor potential channels are a superfamily of cell surface Ca2+ permeable cation channels that play a role in a variety of physiologic and pathophysiologic processes [39]. Our previous studies showed that transient receptor potential vanilloid 4 (TRPV4) channels are a major Ca2+ influx pathway in the intact pulmonary endothelium [40, 41*]. Moreover, Ca2+ influx through TRPV4 channels activated endothelial nitric oxide synthase (eNOS) activity to induce NO release. Activation of TRPV4 channels has been linked to acute lung injury development through inflammatory cell activation, disruption of the capillary alveolar barrier, and progression of pulmonary edema [39, 42–46]. Interestingly, in addition to its role in the alveolar capillary barrier, TRPV4 has been functionally linked to integral components of the purinergic signaling network including Panx1 channels [47] and P2 receptors [48] (see Figure 2). TRPV4 activation has been shown to induce Panx1 activation in human pulmonary fibroblasts [47]. Additionally, it has been suggested that extracellular ATP can activate pulmonary endothelial TRPV4 channels via activation of P2 receptors [40, 48]. As such, targeting the TRPV4 channel may be an effective therapeutic approach, either alone or in combination with Panx1 and/or P2 inhibitors, to mitigate lung IRI.
Figure 2.
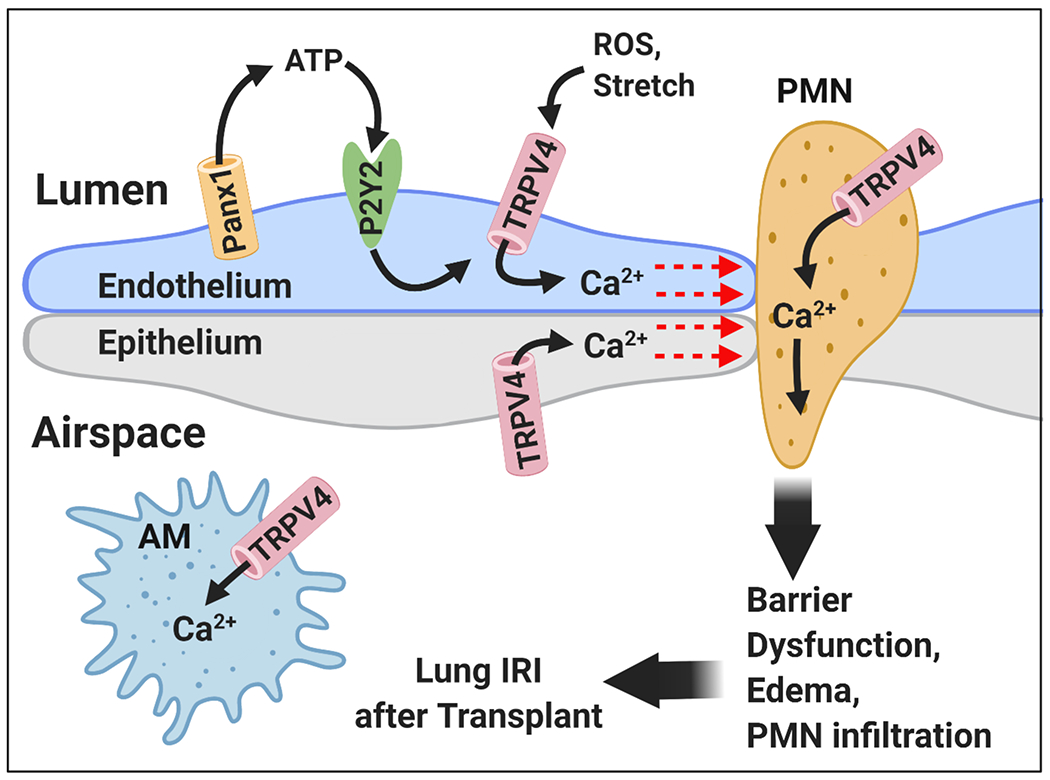
Proposed TRPV4 channel-dependent inflammatory signaling during lung IRI. IR-induced activation of Panx1 increases local ATP levels in the extracellular space. Extracellular ATP can activate TRPV4 channels through purinergic P2 receptors [40, 48] (P2Y2 is a leading candidate), thereby increasing the calcium influx in the endothelium, epithelium, neutrophils (PMN), and alveolar macrophages (AM). Elevated intracellular calcium leads to barrier disruption, AM activation, PMN infiltration, and vascular inflammation, ultimately resulting in acute lung injury following transplant.
A number of stimuli can result in pulmonary endothelial TRPV4 channel activation including, but not limited to, shear stress, pressure-induced mechanical stretch, heat, hyperosmolarity, and increased extracellular ATP [48]. Activation of TRPV4 channels allows for an influx of calcium that activates numerous intra- and inter-cellular signaling pathways [39]. In pulmonary capillaries, the increased endothelial calcium activates pathways disrupting cell-cell junctions, resulting in increased permeability and development of pulmonary edema [39]. In addition, TRPV4 channels have been shown to activate inflammatory cells including macrophages and neutrophils in lung injury, resulting in secretion of pro-inflammatory cytokines and production of ROS [42, 44–46] (see Figure 2). Pharmacologic TRPV4 inhibitors are now being investigated in a number of preclinical and clinical settings [49–51]. These inhibitors have been shown to decrease lung congestion in animal models of congestive heart failure. A recent randomized controlled pilot trial evaluated the safety and utility of a TRPV4 inhibitor (GSK2798745) in patients with chronic, compensated heart failure [52**]. The authors reported no adverse events and a nonsignificant trend toward improved lung function. Previous studies in animal models have demonstrated the ability of TRPV4 inhibitors to mitigate IRI in several organ systems including lung, heart, kidney, and brain [51, 53*, 54, 55]. Interestingly, Weber and colleagues reported that TRPV4 channel activation in alveolar epithelial cells is protective against edema development [56]. As such, the role of the TRPV4 channel in alveolar epithelial cells is less understood and may differ from that in the pulmonary endothelium. Our laboratory recently examined the use of the TRPV4 inhibitor GSK2193874 in a murine lung IRI model where we demonstrated that GSK2193874 treatment resulted in significantly less pulmonary edema and improved lung function following one hour of ischemia and 2 hours of reperfusion [57*]. Thus, it appears that the TRPV4 channel, which is functionally linked to the purinergic signaling network, is another promising potential target to mitigate lung IRI.
4. Conclusion:
Lung IRI after transplant leads to PGD, which dramatically impacts patient outcome. Therapies aimed at limiting IRI are needed to decrease the incidence of PGD. Recent discoveries have improved our understanding of this sterile inflammatory process, and the purinergic signaling network plays an important role. Targeting various components of this network (Panx1, CD39, CD73, P2Y2R, P2X7R, A2AR) and those that are functionally related (TRPV4) have shown promise in dampening lung IRI. One of these therapies — the A2AR agonist regadenoson – has been studied in a phase I clinical trial to mitigate lung IRI, but the vast majority are being investigated in pre-clinical models. Thus, while promising potential therapies exist, continued study is paramount to determine whether these agents can be translated into clinical practice.
Key Points:
PGD is the leading cause of early mortality following lung transplantation and is typically caused by lung IRI.
The purinergic signaling network plays an important role in lung IRI development.
Pharmacologic manipulation of various components of the purinergic signaling network (Panx1 channels, ectonucleotidases, purinergic P1 and P2 receptors) and close associates (TRPV4 channel) is a promising strategy to mitigate lung IRI.
Funding:
This work was supported by several research grants from the NHLBI/NIH including: T32 HL007849 (NH, ER), R01 HL133293 (VEL), R01 HL146914 (SKS), R01 HL142808 (SKS).
Footnotes
Conflicts of Interest: None
References
- 1.**.Frye CC, Bery AI, Kreisel D, Kulkarni HS. Sterile inflammation in thoracic transplantation. Cell Mol Life Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review highlights the most current state of the science regarding mechanisms of IRI-induced graft damage and provides further insight into future development of preventive and treatment strategies.
- 2.Abassi Z, Armaly Z, Heyman SN. Glycocalyx degradation in ischemia-reperfusion injury. Am J Pathol. 2020;190(4):752–67. [DOI] [PubMed] [Google Scholar]
- 3.Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic signaling, DAMPs, and inflammation. Am J Physiol Cell Physiol. 2020;318(5):C832–c5. [DOI] [PubMed] [Google Scholar]
- 4.*.Navis KE, Fan CY, Trang T, et al. Pannexin 1 Channels as a therapeutic target: structure, inhibition, and outlook. ACS Chem Neurosci. 2020;11(15):2163–72. [DOI] [PubMed] [Google Scholar]; This paper provides a thorough and recent review of Panx1 channel structure, function, physiological regulation and pharmacologic inhibition aimed toward therapeutic strategies.
- 5.**.Le TT, Berg NK, Harting MT, et al. Purinergic signaling in pulmonary inflammation. Front Immunol. 2019;10:1633. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides the most up-to-date examination of the role of purinergic signaling in acute and chronic pulmonary inflammation, with an emphasis on ATP and adenosine signaling.
- 6.Dos Anjos F, Simões JLB, Assmann CE, et al. Potential therapeutic role of purinergic receptors in cardiovascular disease mediated by SARS-CoV-2. J Immunol Res. 2020;2020:8632048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.*.Lau CL, Beller JP, Boys JA, et al. Adenosine A2A receptor agonist (regadenoson) in human lung transplantation. J Heart Lung Transplant. 2020;39(6):563–70. [DOI] [PubMed] [Google Scholar]; This is the first clinical trial to study the safety of an adenisine 2A receptor agonist in the setting of lung transplantation. It provides optimistic data for an eventual phase II randomized controlled trial.
- 8.Sanchez Arias JC, Wicki-Stordeur LE, Candlish RC, et al. PANX1 in inflammation heats up: New mechanistic insights with implications for injury and infection. Cell Calcium. 2020;90:102253. [DOI] [PubMed] [Google Scholar]
- 9.**.Kameritsch P, Pogoda K. The Role of connexin 43 and pannexin 1 during acute inflammation. Front Physiol. 2020;11:594097. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides the most up-to-date knowledge on the role connexin 43 and Panx1 in endothelial cells and leukocytes in during acute inflammation.
- 10.Sharma AK, Charles EJ, Zhao Y, et al. Pannexin-1 channels on endothelial cells mediate vascular inflammation during lung ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 2018;315(2):L301–l12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.*.Chen KW, Demarco B, Broz P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur J Immunol. 2020;50(2):170–7. [DOI] [PubMed] [Google Scholar]; This study provides genetic and pharmacological evidence that Panx1 is dispensable for canonical or noncanonical inflammasome activation. Instead, it demonstrates that Panx1 channel activaton during apoptosis promotes NLRP3 inflammasome activation.
- 12.**.Yang Y, Delalio LJ, Best AK, et al. Endothelial pannexin 1 channels control inflammation by regulating intracellular calcium. J Immunol. 2020;204(11):2995–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that endothelial Panx1 is a direct target of the TNF-α signaling pathway and demonstrates for the first time, that Panx1 channels facilitate the transport of extracellular calcium to promote a feed-forward effect on the synthesis of IL-1β.
- 13.Soni S, O’Dea KP, Tan YY, et al. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. 2019;33(5):6442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.**.Sofoluwe A, Bacchetta M, Badaoui M, et al. ATP amplifies NADPH-dependent and -independent neutrophil extracellular trap formation. Sci Rep. 2019;9(1):16556. [DOI] [PMC free article] [PubMed] [Google Scholar]; NET formation (NETosis) plays an important role in lung IRI, and this study demonstrates that ATP and Panx1 channels contribute to NETosis and may represent a therapeutic target.
- 15.Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH oxidase mediates synergistic effects of IL-17 and TNF-α on CXCL1 expression by epithelial cells after lung ischemia-reperfusion. Am J Physiol Lung Cell Mol Physiol. 2014;306(1):L69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohman AW, Leskov IL, Butcher JT, et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun. 2015;6:7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayah DM, Mallavia B, Liu F, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2015;191(4):455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granja T, Korner A, Gluck C, et al. Targeting CD39 toward activated platelets reduces systemic inflammation and improves survival in sepsis: A Preclinical Pilot Study. Crit Care Med. 2019;47(5):e420–e7. [DOI] [PubMed] [Google Scholar]
- 19.*.Mierzejewska P, Zabielska MA, Kutryb-Zajac B, et al. Impaired L-arginine metabolism marks endothelial dysfunction in CD73-deficient mice. Mol Cell Biochem. 2019;458(1-2):133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that CD73 deletion leads to the development of age-dependent endothelial dysfunction in mice. Vascular damage is related to severe distrubances in L-arginine metabolism, and increased CD73 activity could be protective in vascular pathologies.
- 20.Minor M, Alcedo KP, Battaglia RA, Snider NT. Cell type- and tissue-specific functions of ecto-5’-nucleotidase (CD73). Am J Physiol Cell Physiol. 2019;317(6):C1079–c92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rivas-Yáñez E, Barrera-Avalos C, Parra-Tello B, et al. P2X7 receptor at the crossroads of T cell fate. Int J Mol Sci. 2020;21(14):4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laubach VE, French BA, Okusa MD. Targeting of adenosine receptors in ischemia-reperfusion injury. Expert Opin Ther Targets. 2011;15(1):103–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu Rev Immunol. 2019;37:325–47. [DOI] [PubMed] [Google Scholar]
- 24.Olotu C, Kiefmann M, Ronneburg C, et al. Analysis of purine receptor expression and functionality in alveolar epithelial cells. Purinergic Signal. 2020;16(2):213–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin H, Ko YS, Park SW, Kim HJ. P2Y2R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free Radic Biol Med. 2019;136:109–17. [DOI] [PubMed] [Google Scholar]
- 26.Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A. P2X7 interactions and signaling - making head or tail of it. Front Mol Neurosci. 2019;12:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shokoples BG, Paradis P, Schiffrin EL. P2X7: An untapped target for the management of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021. Jan;41(1):186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cisneros-Mejorado AJ, Pérez-Samartín A, Domercq M, et al. P2X7 receptors as a therapeutic target in cerebrovascular diseases. Front Mol Neurosci. 2020;13:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duan L, Hu GH, Li YJ, et al. P2X7 receptor is involved in lung injuries induced by ischemia-reperfusion in pulmonary arterial hypertension rats. Mol Immunol. 2018;101:409–18. [DOI] [PubMed] [Google Scholar]
- 30.Jamwal S, Mittal A, Kumar P, et al. Therapeutic potential of agonists and antagonists of A1, A2a, A2b and A3 adenosine receptors. Curr Pharm Des. 2019;25(26):2892–905. [DOI] [PubMed] [Google Scholar]
- 31.*.Xu K, Cooney KA, Shin EY, et al. Adenosine from a biologic source regulates neutrophil extracellular traps (NETs). J Leukoc Biol. 2019;105(6):1225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study found a strong effect of adenosine (signaling via A2AR activation) to powerfully inhibit NET formation in neutrophils from a large number of healthy human donors. Mesenchymal stromal cells, which set up the anti-inflammatory environment in bone marrow, also suppress NETosis in a purinergic-dependent manner.
- 32.Ali RA, Gandhi AA, Meng H, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. 2019;10(1):1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gazoni LM, Laubach VE, Mulloy DP, et al. Additive protection against lung ischemia-reperfusion injury by adenosine A2A receptor activation before procurement and during reperfusion. J Thorac Cardiovasc Surg. 2008;135(1):156–65. [DOI] [PubMed] [Google Scholar]
- 34.LaPar DJ, Laubach VE, Emaminia A, et al. Pretreatment strategy with adenosine A2A receptor agonist attenuates reperfusion injury in a preclinical porcine lung transplantation model. J Thorac Cardiovasc Surg. 2011;142(4):887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma AK, LaPar DJ, Stone ML, et al. NOX2 activation of natural killer T cells is blocked by the adenosine A2A receptor to inhibit lung ischemia-reperfusion injury. Am J Respir Crit Care Med. 2016;193(9):988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tane S, Noda K, Shigemura N. Ex Vivo Lung Perfusion: A key tool for translational science in the lungs. Chest. 2017;151(6):1220–8. [DOI] [PubMed] [Google Scholar]
- 37.Emaminia A, Lapar DJ, Zhao Y, et al. Adenosine A2A agonist improves lung function during ex vivo lung perfusion. Ann Thorac Surg. 2011;92(5):1840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner CE, Pope NH, Charles EJ, et al. Ex vivo lung perfusion with adenosine A2A receptor agonist allows prolonged cold preservation of lungs donated after cardiac death. J Thorac Cardiovasc Surg. 2016;151(2):538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenbaum T, Benítez-Angeles M, Sánchez-Hernández R, et al. TRPV4: A physio and pathophysiologically significant ion channel. Int J Mol Sci. 2020;21(11):3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marziano C, Hong K, Cope EL, et al. Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (TRPV4) channel cooperativity and endothelial function in small pulmonary arteries. J Am Heart Assoc. 2017;6(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.*.Ottolini M, Daneva Z, Chen YL, et al. Mechanisms underlying selective coupling of endothelial Ca(2+) signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J Physiol. 2020;598(17):3577–96. [DOI] [PMC free article] [PubMed] [Google Scholar]; Results of this study reveal that differential spatial organization of signalling elements determines endothelial cell TRPV4-IK/SK vs. TRPV4-eNOS coupling in resistance arteries.
- 42.Dutta B, Arya RK, Goswami R, et al. Role of macrophage TRPV4 in inflammation. Lab Invest. 2020;100(2):178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuebler WM, Jordt SE, Liedtke WB. Urgent reconsideration of lung edema as a preventable outcome in COVID-19: inhibition of TRPV4 represents a promising and feasible approach. Am J Physiol Lung Cell Mol Physiol. 2020;318(6):L1239–l43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rayees S, Joshi JC, Tauseef M, et al. PAR2-mediated cAMP generation suppresses TRPV4-dependent Ca(2+) signaling in alveolar macrophages to resolve TLR4-induced inflammation. Cell Rep. 2019;27(3):793–805.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheraga RG, Abraham S, Grove LM, et al. TRPV4 protects the lung from bacterial pneumonia via MAPK molecular pathway switching. J Immunol. 2020;204(5):1310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheraga RG, Southern BD, Grove LM, Olman MA. The role of TRPV4 in regulating innate immune cell function in lung inflammation. Front Immunol. 2020;11:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rahman M, Sun R, Mukherjee S, et al. TRPV4 stimulation releases ATP via pannexin channels in human pulmonary fibroblasts. Am J Respir Cell Mol Biol. 2018;59(1):87–95. [DOI] [PubMed] [Google Scholar]
- 48.Chen YL, Sonkusare SK. Endothelial TRPV4 channels and vasodilator reactivity. Curr Top Membr. 2020;85:89–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brooks CA, Barton LS, Behm DJ, et al. Discovery of GSK2798745: A clinical candidate for inhibition of transient receptor potential vanilloid 4 (TRPV4). ACS Med Chem Lett. 2019;10(8):1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Wen AM, Potla R, et al. AAV-mediated gene therapy targeting TRPV4 mechanotransduction for inhibition of pulmonary vascular leakage. APL Bioeng. 2019;3(4):046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soni H, Peixoto-Neves D, Olushoga MA, Adebiyi A. Pharmacological inhibition of TRPV4 channels protects against ischemia-reperfusion-induced renal insufficiency in neonatal pigs. Clin Sci (Lond). 2019;133(9):CS20180815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.**.Stewart GM, Johnson BD, Sprecher DL, et al. Targeting pulmonary capillary permeability to reduce lung congestion in heart failure: a randomized, controlled pilot trial. Eur J Heart Fail. 2020;22(9):1641–5. [DOI] [PubMed] [Google Scholar]; This study (along with editorial comment by Bistola et al.) provides a significant step forward in the prevention and resolution of lung congestion. This pilot trial showed that a novel TRPV4 antagonist (GSK2798745) was safe and well-tolerated, with a trend toward improved gas transfer in patients with chronic, compensated heart failure.
- 53.*.Tanaka K, Matsumoto S, Yamada T, et al. Reduced post-ischemic brain injury in transient receptor potential vanilloid 4 knockout mice. Front Neurosci. 2020;14:453. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study used TRPV4 knockout mice to demonstrate that TRPV4 contributes to post-ischemic brain injury following transient focal cerebral ischemia, suggesting that TRPV4 inhibition may be an effective method to treat acute ischemic stroke.
- 54.Wang B, Wu Q, Liao J, et al. Propofol induces cardioprotection against ischemia-reperfusion injury via suppression of transient receptor potential vanilloid 4 channel. Front Pharmacol. 2019;10:1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Q, Lu K, Zhao Z, et al. Blockade of transient receptor potential vanilloid 4 enhances antioxidation after myocardial ischemia/reperfusion. Oxid Med Cell Longev. 2019;2019:7283683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber J, Rajan S, Schremmer C, et al. TRPV4 channels are essential for alveolar epithelial barrier function as protection from lung edema. JCI Insight. 2020;5(20):e134464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.*.Charles E, Zhao Y, Hong K, et al. TRPV4 Channels Mediate Pulmonary Edema and Inflammation During Ischemia-Reperfusion Injury. [abstract]. Am J Transplant. 2017;17 (SI supplement 3):399. [Google Scholar]; This is the first study to demonstrate that TRPV4 plays a pro-inflammatory role in lung ischemia-reperfusion injury and, specifically, that endothelial TRPV4 is especially critical. TRPV4 knockout mice and treatment of wild-type mice with TRPV4 inhibitor significantly attenuates lung injury and improves lung function after ischemia-reperfusion.