

Abstract
Liver fibrosis is a wound-healing response that results from various chronic damages. If the causes of damage are not removed or effective treatments are not given in a timely manner, it will progress to cirrhosis, even liver cancer. Currently, there are no specific medical therapies for liver fibrosis. Adeno-associated virus (AAV)-mediated gene therapy, one of the frontiers of modern medicine, has gained more attention in many fields due to its high safety profile, low immunogenicity, long-term efficacy in mediating gene expression, and increasingly known tropism. Notably, increasing evidence suggests a promising therapeutic potential for AAV-mediated gene therapy in different liver fibrosis models, which helps to correct abnormally changed target genes in the process of fibrosis and improve liver fibrosis at the molecular level. Moreover, the addition of cell-specific promoters to the genome of recombinant AAV helps to limit gene expression in specific cells, thereby producing better therapeutic efficacy in liver fibrosis. However, animal models are considered to be powerless predictive of tissue tropism, immunogenicity, and genotoxic risks in humans. Thus, AAV-mediated gene therapy will face many challenges. This review systemically summarizes the recent advances of AAV-mediated gene therapy in liver fibrosis, especially focusing on cellular and molecular mechanisms of transferred genes, and presents prospective challenges.
Keywords: liver fibrosis, AAV, gene therapy, HSCs, cellular mechanism
Graphical abstract
Bu and colleagues summarized studies of AAV-mediated gene therapy in rodent liver fibrosis caused by different etiologies, described its function and mechanism, and presented prospective challenges that affected AAV translational potential for human liver fibrosis.
Introduction
Recent estimates indicate that 844 million people worldwide suffer from chronic liver disease, with approximately 2 million deaths per year and the incidence is still rising.1 Iterative liver injury caused by various etiologies results in liver fibrosis, ultimately leading to cirrhosis, even liver cancer, if left untreated.2 During hepatic fibrogenesis, the formation of a fibrous scar that is composed of multiple extracellular matrix proteins, such as collagen type I and type III, gradually upsets normal liver architecture.3 In general, hepatotoxic injury, mostly caused by viral hepatitis, alcohol, chemical insults, or non-alcoholic steatohepatitis (NASH), and cholestatic injury, mostly caused by biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), or biliary atresia, are regarded as two common reasons for liver fibrosis initiation.4 It is worth noting that an increasing number of studies show that considerable histopathological improvement of liver fibrosis is achieved when chronic injury factors are removed or patients are properly treated.5 Moreover, studies on patients with liver fibrosis or experimental models with liver fibrosis have revealed the necessary therapeutic target genes, and satisfactory results have been obtained in rodent models by using appropriate gene carriers.6 Currently, gene-carrying vectors can be classified into two categories: non-viral vectors and viral vectors. Non-viral vectors, such as nanoparticles, naked DNA, exosomes, and liposomes, have been regarded as promising gene carriers because of their simple production methods, low cost, and safety; however, they often display relatively low transmission efficiency and mediate a transient effect, thereby necessitating repeated administrations, which could provoke an immune response.7 In the past few decades, viral gene delivery vectors, such as retrovirus, lentivirus (LV) and adenovirus (Ad), have become capable of efficiently mediating gene transduction and expression, and have been widely applied to rodent models; however, some non-negligible defects, including lack of tissue specificity, high immunorejection, possible tumorigenicity, and obscure insertional mutagenesis, limit their applications for clinical practice.8 Fortunately, adeno-associated virus (AAV), with a high safety profile, low immunogenicity, long-term efficacy in mediating gene expression, and increasingly known tissue tropism, overcomes the shortcomings of the aforementioned vectors and has been applied for clinical studies, such as hemophilia, inherited retinal diseases, acute intermittent porphyria, and spinal muscular atrophy (SMA).9, 10, 11, 12
In this review, we outline the role of AAV-mediated gene therapy in rodent liver fibrosis caused by different etiologies and put forward the prospective challenges that will affect the translational potential of AAV in human liver fibrosis.
Overview of liver fibrosis mechanism
The pathogenesis of liver fibrosis is relatively complex and may be the result of communication between various cells in the liver13 (Figure 1). Briefly, in response to chronic injury and pro-fibrogenic factors, hepatocytes produce a group of damage-associated molecular patterns (DAMPs) and vesicles containing vital microRNAs (miRNAs), which can promote the increased number of pro-inflammatory phenotypes of macrophages.14 Hepatic stellate cells (HSCs), the residents in the space of Disse, keep a quiescent phenotype under normal conditions and function as a reservoir for vitamin A lipid droplets. However, activated macrophages generate a large number of pro-inflammatory and fibrogenic cytokines, such as classic transforming growth factor β1 (TGF-β1) and tumor necrosis factor α (TNF-α), and reactive oxygen species (ROS), leading to the activation of HSCs.14 Activated HSCs (aHSCs) lose vitamin A lipid droplets, present α-smooth muscle actin (α-SMA)-positive signaling, migrate to injury sites, and produce a significant number of extracellular matrix (ECM) components, such as collagen type I and type III, matrix metalloproteinases (MMPs), and tissue inhibitors of matrix metalloproteinases (TIMPs).15 The characteristics of aHSCs are similar to those of activated myofibroblasts (MFs). They also acquire high proliferative potentials when facing a variety of cytokines stimulations, including TGF-β, platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) family, and produce multiple collagen fibers, ultimately distorting the normal liver architecture without any intervention.16 Meanwhile, DAMPs, extracellular vesicles (EVs) or profibrogenic cytokines produced by hepatocytes can directly enhance the activation of HSCs.17 Intriguingly, scientific research findings have revealed that upon the cessation of chronic injury, liver fibrosis can be regressed in patients or in experimental rodent models, which is associated with the increased ablation of fibrous scars and the reduced pro-inflammatory or pro-fibrogenic cytokines. In the reversible process, aHSCs move toward apoptosis and inactivation states, which leaves many questions worth studying, such as which gene changes drive the reversal process.13 A decreased number of aHSCs leads to the reduced production of ECMs, indicated by the activity of MMPs (e.g., MMP2, MMP12, MMP13) that degrade collagen fibers is upregulated over that of their inhibitors (TIMPs). Thus, increasing studies have focused on promoting aHSC apoptosis and inactivation. However, several mechanisms about apoptosis of aHSCs have been revealed—the activation of death receptor-mediated pathways (FAS or TRAIL), caspase 3 and caspase 8, the upregulation of pro-apoptotic signals (P53 or BAX), or the downregulation of pro-survival signals (TGF-β1 or TIMP1).13 Inactivation factors are also vital for liver fibrosis regression. It has been generally believed that peroxisome proliferator-activated receptor γ (PPAR-γ), a nuclear receptor, is essential for keeping HSCs in quiescent phenotypes, and the depletion of PPAR-γ in cultured HSCs led to the excessive production of aHSCs. Conversely, the overexpression of PPAR-γ in aHSCs enhanced their transformation into an inactivated phenotype.18 Furthermore, a recent study on the identification of lineage-specific transcription factors (TFs) demonstrated that the genetic ablation of PPAR-γ or GATA6 in HSCs in vivo accelerated the progression but impeded the regression of liver fibrosis, suggesting that PPAR-γ and GATA6 may be essential targets for inducing aHSCs to the inactivated state.19 In addition, unprecedented studies using cell fate mapping methods reported that some important cell fate decision genes, such as TF 21 (Tcf21), were identified as deactivation factors of fibrogenic HSCs and could enhance the regression of liver fibrosis. The overexpression of Tcf21 in aHSCs not only inhibited pro-fibrogenic gene expression but it also promoted the inactivation of aHSCs in vitro and in vivo.20 Therefore, it is important to use suitable vectors to deliver these important genes that promote the inactivation or apoptosis of aHSCs.
Figure 1.

Brief mechanistic concepts of liver fibrosis
In response to chronic damage factors, hepatocytes produce a number of DAMPs or EVs that contain vital miRNAs, which can lead to the activation of macrophages or HSCs. A large number of pro-inflammatory and fibrogenic cytokines that are generated by activated macrophages also lead to the activation of HSCs. Activated HSCs start to proliferate and secrete pro-fibrogenic factors, leading to ECM deposition on the liver. Upon cessation of chronic injury, activated HSCs move toward inactivation and apoptosis, resulting in the regression of liver fibrosis. On the one hand, activated HSC is transformed into an inactive state under the action of inactivating factors, such as MMP2, MMP12, MMP13, TCF21, and PPAR-γ. On the other hand, under the action of activation of death receptor-mediated pathways and pro-apoptotic factors, such as FAS, TRAIL, caspase3, caspase8, and Bax, or the decline of pro-survival genes, such as TIMP1 and TGF-β1, activated HSCs turn to an apoptotic state.
It is worth mentioning that with the assistance of AAV vectors, research on the therapeutic potential of gene therapy in liver fibrosis has been widely applied to different rodent models, and exploring AAV-mediated cell-specific gene therapy is emerging and may lead to a breakthrough in the treatment of liver fibrosis.
Biology and serotype of AAV
In the 1960s, AAV was discovered, which opened a new chapter in AAV-mediated gene therapy.21 Since then, some research groups have developed interest in it and have begun to study and understand the basic characteristics and biology of AAV. At the time, they did not realize its great potential as a gene therapy platform.22,23 AAV belongs to the parvovirus family and was regarded as a contamination of Ad preparations initially.23 The life cycle of AAV requires the assistance of helper viruses, such as Ad and herpes simplex virus (HSV); thus, AAV is also classified as a dependovirus.24 Intriguingly, AAVs are present in a variety of vertebrate species, including humans and non-human primates (NHPs), whereas they have not caused any human illness so far.25 The composition of AAV is very simple, consisting of an icosahedral capsid with a diameter of ∼26 nm and a single-stranded DNA (ssDNA) genome of ∼4.7 kb. Two indispensable coding sections, Replication (Rep) and Capsid (Cap), are included in the genome of AAV, which is flanked by 145-bp inverted terminal repeats (ITRs) on each side.26 The Rep gene encodes four proteins, Rep40, Rep52, Rep68, and Rep78, that are involved in the replication and packaging of AAV, while the Cap gene encodes three proteins, VP1, VP2, and VP3, that form the 60-mer capsid in a ratio of 1:1:10, respectively.26 The capsids determine the range of the infected host by interacting with receptors and co-receptors on the cell surface.27 Importantly, once entering the nucleus, AAV particles uncoat and release their single-stranded genome, which subsequently is converted into a double-stranded DNA (dsDNA) template; thus, the transgenic gene can be transcribed and translated from the template.27 The straightforward design of recombinant AAV (rAAV) to deliver interested transgenes is attributed to the simplicity of the AAV genome. Notably, only 145-bp ITRs that are found in the AAV genome are present in the genome of rAAV and contribute to vector production, induction of transgene expression, and cell transduction. Thus, the coding sequences of the AAV genome are removed and replaced with a therapeutic gene expression cassette. The complete removal of viral coding sequences renders rAAV incapable of gene integration during in vivo delivery, so after the transduction of host cells, rAAV transgenes remain episomal, which helps reduce immunogenicity and cytotoxicity.28 Currently, two types of rAAV are in use: single-stranded AAV (ssAAV) and self-complementary AAV (scAAV). The process of converting the single-stranded genome into a self-complementary genome is a rate-limiting process. To avoid this process, a scAAV genome was synthesized by mutating one of the ITR structures.25 These scAAV vectors can rapidly undergo transcription, thereby improving transduction efficiency.25,29 In general, the production of rAAV by transfecting HEK293T cells requires three essential plasmids, including plasmids containing the rAAV genome, the rep and cap genes, and the helper virus genes, respectively.30 It is worth noting that with the development of biotechnology, high yields of rAAV vector can be produced without contaminating cells and helper viral proteins, and it has been successfully applied in preclinical and clinical research.31,32 Moreover, current technologies allow rAAV to be packaged and purified rapidly and routinely in the laboratory. However, due to the differences in packaging and purification methods and production scale, their titers are quite different. To detect the titer of AAV more safely, simply, quickly, and conveniently and to make the titer of AAV reach a unified standard, researchers have successively developed effective detection methods, such as enzyme-linked immunosorbent assay (ELISA), quantitative real-time PCR, and the recently reported GelGreen method.33,34
Thus far, there are 12 natural AAV serotypes and 108 isolates (serovars) have been discovered and classified according to phylogenetic analyses.26 Among them, AAV2 is one of the first AAV serotypes identified and characterized, and most rAAV vectors share the same ITR from AAV2, which are often identified as AAV2/n, with “n” representing the capsid. Generally speaking, different capsids of AAV have different binding receptors, tissue tropisms, and transduction efficiency.35 Notably, several AAV serotypes have been applied to different clinical trials, including AAV1, AAV2, AAV5, AAV6, AAV8, AAV9, and AAVrh10 (isolation from rhesus monkeys).27 Moreover, the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) have approved several AAV-based in vivo gene therapy bioproducts, such as Glybera, for patients with lipoprotein lipase deficiency, Zolgensma, for patients with SMA, and Luxturna, for patients with RPE65-associated Leber’s congenital amaurosis.36, 37, 38
It is worth mentioning that clinical trials of AAV-mediated gene therapy for liver-based metabolic and genetic diseases have increasingly been performed or are ongoing at the moment, and have achieved promising results.39 For example, hemophilia B, an X-linked bleeding disorder caused by a defect in the gene-encoding coagulation factor IX, is an ideal target for gene therapy, since a minor increase in plasma factor IX above 1% of physiologic levels mitigates the bleeding phenotype.40 The clinical trial performed by Nathwani et al. reported that a single intravenous administration of AAV8 carrying factor IX in patients with severe hemophilia B contributed to a dose-dependent and long-term increase in circulating factor IX, despite side effects, such as elevated alanine aminotransferase (ALT) levels or capsid-reactive T cell response, having been observed in different dose groups during the monitoring period.9 Elevated ALT levels could be attenuated when patients were administrated by a short and tapering course of prednisolone.9 Moreover, increasing evidence suggests that AAV8 has preferential liver transduction in nonhuman primates and dog models, yielding promising therapeutic indices, although the absolute transgene expression levels by AAV8 are relatively lower than in mice.41, 42, 43 For example, scAAV8 encoding human plasma factor IX in rhesus macaques by intravenous injection showed a good tropism to liver and was sufficient for phenotypic correction in hemophilia.44 Recently, AAV8-mediated gene overexpression or silence has been widely applied to liver fibrosis models.45,46 However, other studies have recognized that AAV1-, AAV2-, AAV5-, AAV6-, and AAV9-mediated gene transfer also achieved an ideal effect in rodent liver fibrosis models (Table 1).
Table 1.
The applications of different AAV serotype-mediated gene therapy in rodent liver fibrosis or cirrhosis
| Serotypes and genes | Cell-specific promoter | OE/KD | Animal strains | Causes | Dose/route | Reference |
|---|---|---|---|---|---|---|
| AAV1 | ||||||
| IGF-I | – | OE | SD rats | CCl4 i.g. | 3.4 × 109vp/hepatic artery | Sobrevals et al.47 |
| AAV2 | ||||||
| HO-1 | – | OE | LEW rat | CCl4 i.g. | 1×1012 v.g./portal vein | Tsui et al.48,49 |
| BMP7 | – | OE | BALB/c | CCl4 i.p. | 1×1010 v.g./oral administration | Hao et al.50 |
| miR-19b | collagen alpha 1 | OE | SD rats | BDL | 1×1012 v.g./portal vein | Brandon-Warner et al.51 |
| AAV5 | ||||||
| HGF | – | OE | BALB/c | CCl4 i.g.; BDL | 1×1011 v.g./portal vein | Suzumura et al.52 |
| AAV6 | ||||||
| Tcf21 | – | OE | C57BL/6 | CCl4 i.p. | 3 × 1011 v.g./i.p. | Nakano et al.20 |
| 6TFs | OE | C57BL/6 | CCl4 i.p. | 4 ×1011 v.g./tail vein | Rezvani et al.53 | |
| Nestin | – | KD | C57BL/6 | CCl4 i.p.; DDC diet | 1.5 × 1012 v.g./tail vein | Chen et al.54 |
| AAV8 | ||||||
| GNMT | – | OE | BALB/c; C57BL/6 | CCl4 i.p. | 1 × 1011 v.g./tail vein | Fang et al.45 |
| PHP14 | – | KD | C57BL/6 | CCl4 i.p. | –/tail vein | Xu et al.46 |
| ACE2 | – | OE | C57BL/6 | CCl4 i.p.; BDL; MCD diet | 1 × 1011 v.g./i.p. | Mak et al.;55 Rajapaksha et al.56 |
| FOXA2 | TBG | OE | C57BL/6 | CCl4 i.p. | 4 × 1011 v.g./tail vein | Wang et al.57 |
| miR-29a | – | OE | C57BL/6 | CCl4 i.p. | 2 × 1011 v.g./tail vein | Knabel et al.58 |
| miR-221-3p | Ttr | KD | BALB/c | CCl4 i.p.; DDC diet | 1 × 1010 v.g./tail vein | Tsay et al.59 |
| NRF2 | TBG | OE | C57BL/6 | HFD + AGEs | 5 × 1011 gc/tail vein | Dehnad et al.60 |
| RCAN1.4 | – | OE | C57BL/6 | CCl4 i.p. | 1 × 1011 v.g./tail vein | Pan et al.61 |
| miR-21 | – | KD | BALB/c | schistosomiasis | 6 × 1011 v.g./tail vein | He et al.62 |
| miR-351 | – | KD | BALB/c | schistosomiasis | 1 × 1012 v.g./tail vein | He et al.63 |
| GDF11 | – | OE | BALB/c | CCl4 i.p; DDC diet | 1 × 1011 v.g./tail vein | Dai et al.64 |
| AAV9 | ||||||
| PSTPIP2 | – | OE | C57BL/6 | CCl4 i.p. | – | Yang et al.65 |
| SUN2 | – | OE | C57BL/6 | CCl4 i.p. | – | Chen et al.66 |
| circFBXW4 | – | OE | C57BL/6 | CCl4 i.p. | 1 × 1012 v.g./tail vein | Chen et al.67 |
| TGF-β1 | GFAP | KD | SD Rat | DEN i.p. | 8 × 1011 v.g./portal vein | Zhang et al.68 |
| LECT2 | – | KD | C57BL/6 | CCl4 i.p. | – | Xu et al.69 |
i.g., intragastrically; i.p., intraperitoneally; v.g., vector genome; vp, viral particles; gc, genome copies; SD rats, Sprague-Dawley rats; KD, knockdown; OE, overexpression.
In this review, we summarize the progress of AAV-mediated gene therapy in liver fibrosis, focusing especially on the relevant cellular and molecular mechanisms of transferred genes (Table 2), and provide a basis for the future application of AAV in the clinical treatment of liver fibrosis.
Table 2.
Target mechanisms of AAV-mediated gene therapy in rodent liver fibrosis
| Transgene | Function | Mechanism | Reference |
|---|---|---|---|
| HGF | promotes resolution of fibrosis | inhibits pro-fibrotic gene expression and upregulates MMP13 expression | Suzumura et al.52 |
| IGF-I | promotes reversal of liver cirrhosis | inhibits pro-fibrotic gene expression and upregulates HNF-4α and HGF expression | Sobrevals et al.47 |
| ACE2 | represses liver fibrogenesis | attenuates Ang-II-mediated oxidative stress, inflammation, and fibrosis | Mak et al.55 |
| ACE2 | inhibits chronic biliary fibrosis | prevents HSCs activation and EMT of cholangiocytes | Rajapaksha et al.56 |
| FOXA2 | mitigates liver fibrogenesis | suppresses ER stress and hepatocyte apoptosis | Wang et al.57 |
| GNMT | delays development of liver diseases | promotes hepatocyte proliferation | Fang et al.45 |
| miR-29a | mitigates liver fibrogenesis | not available | Knabel et al.58 |
| miR-211-3p-TuDs | suppresses hepatocyte injury and liver fibrogenesis | inhibits CCL2 production, thereby repressing HSCs activation | Tsay et al.59 |
| NRF2 | inhibits fibrogenic activity in NASH | restores AGER1 and improves liver AGEs, necroinflammation, and fibrosis | Dehnad et al.60 |
| Nedd8 shRNA | represses development of NFALD | inhibits neddylation and promotes hepatic fatty acid oxidation | Newberry et al.70 |
| PSTPIP2 | attenuates liver inflammation and fibrogenesis | represses M1 macrophages polarization by inhibiting STAT1 phosphorylation | Yang et al.65 |
| PHP14-shRNA | attenuates liver inflammation and fibrogenesis | inhibits the migration of macrophages to the injury sites | Xu et al.46 |
| HO-1 | attenuates the severity of fibrosis | inhibits HSC activation and proliferation | Tsui et al.48,49 |
| BMP7 | inhibits liver fibrosis and promotes hepatocyte regeneration | inhibits HSC activation and promotes hepatocyte proliferation | Hao et al.50 |
| miR-19b | improves liver function, injury, and fibrosis | inhibits HSC activation | Brandon-Warner et al.51 |
| miR-21-TuDs | attenuates schistosomiasis-induced liver fibrosis | inhibits HSC activation through upregulating Smad7 expression | He et al.62 |
| miR-351-sponge | attenuates schistosomiasis-induced liver fibrosis | inhibits HSC activation through upregulating VDR expression | He et al.63 |
| RCAN1.4 | mitigates liver fibrogenesis | inhibits HSC activation and proliferation, promotes HSC apoptosis through targeting calcineurin/NFAT3 signaling | Pan et al.61 |
| SUN2 | mitigates liver fibrogenesis | inhibits HSC activation and proliferation through targeting PI3K/AKT signaling | Chen et al.66 |
| circFBXW4 | mitigates liver fibrogenesis | inhibits HSC activation and proliferation through targeting miR-18b-3p/FBXW7 axis | Chen et al.67 |
| TGF-β1-shRNA | accelerates liver regeneration, mitigates liver fibrosis, and improves liver function | promotes hepatocyte proliferation | Zhang et al.68 |
| 6TFs | attenuates liver fibrosis | reprograms MFs into hepatocyte-like cells | Rezvani et al.53 |
| Tcf21 | promotes reversal of liver fibrosis | enhances aHSCs to a quiescent phenotype | Nakano et al.20 |
| Nestin shRNA | attenuates liver fibrosis | inhibits excessive activation of TGF-β/Smad signaling | Chen et al.54 |
| GDF11 | attenuates liver fibrosis | promotes expansion of liver progenitor cells | Dai et al.64 |
| LECT2 shRNA | attenuates liver fibrosis | promotes the migration and tube formations of endothelial cells | Xu et al.69 |
HNF, hepatocyte nuclear factor.
The application of AAV-mediated gene delivery system in liver fibrosis
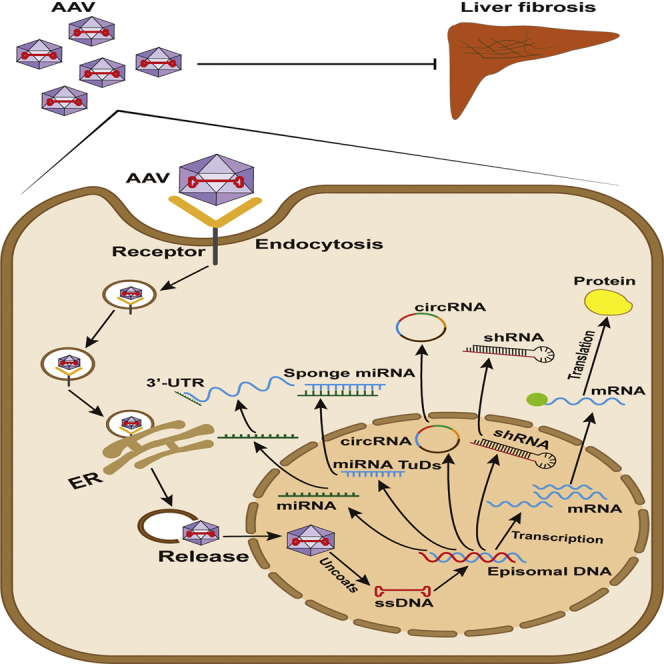
In liver fibrosis, important molecular targets have been discovered successively, which regulate the fibrotic process by regulating different mechanisms.13 AAV is a favorable vector for gene therapy due to its high safety, gene transduction efficiency, long-term efficacy in mediating gene expression, and low immunogenicity.27 It has been reported that distorted liver architecture affects the effective transduction of hepatocytes with large vectors, such as Ad, in liver cirrhosis.71 However, the study conducted by Sobrevals et al. revealed that AAV vectors transduce hepatocytes in vivo as efficiently in cirrhotic as in healthy rat livers.72 Moreover, in recent years, AAV-mediated gene delivery has been applied for the overexpression and knockdown of some important genes or non-coding RNAs in liver fibrosis, contributing to studying the vital molecular and cellular mechanism of transferred genes more conveniently (Figure 2). Here, we review the different cellular and molecular mechanisms of AAV-mediated gene therapy in liver fibrosis (Figure 3).
Figure 2.

Process of AAV-mediated gene vector transduction
In liver fibrosis, AAV-mediated genes, miRNA, miRNA TuDs, circRNA, or shRNA are taken into the endosome within liver cells by endocytosis. Once they enter the nucleus, AAVs uncoat and release their single-stranded genome (ssDNA), which subsequently is converted into a double-stranded DNA (dsDNA) template; thus, the transgenic gene can be transcribed and translated from the template.
Figure 3.

Different cellular and molecular mechanisms of AAV-mediated transgene expression in liver fibrosis
AAV-mediated gene overexpression or silence targets different cellular and molecular mechanisms, including promoting proliferation of hepatocytes, apoptosis and inactivation of HSCs, and expansion of liver progenitor cells (LPCs), inhibiting hepatocyte injury and steatosis, macrophage-mediated inflammatory response, endothelial-mediated sinusoid capillarization, activation and proliferation of HSCs, EMT of cholangiocytes, or reprogramming aHSC/MF into hepatocyte-like cells.
Promoting the production of liver protective factors
In the past 2 decades, AAV-mediated liver protective factor gene delivery has been successfully applied for treating animal models of liver fibrosis, which contributes to a more comprehensive understanding of the molecular mechanisms of these protective factors. It has been reported that AAV5 has an ability to transduce liver tissues, mediate long-term gene expression, and does not appear to elicit cellular immune response against the capsid, making AAV5 an excellent gene transfer vector.73,74 AAV5-mediated B-domain-deleted human factor VIII (AAV5-hFVIII-SQ) gene transfer in patients with severe hemophilia A has achieved a sustained and clinically relevant benefit.75,76 Recombinant hepatocyte growth factor (HGF) injection was reported to promote the recovery of liver cirrhosis, whereas the extremely short life and the high expenditure of HGF limit wide applications.77 Fortunately, mice pre-treated with AAV5-mediated HGF gene vector (AAV5-HGF) were protected from carbon tetrachloride (CCl4−) and bile duct ligation (BDL)-induced liver fibrosis.52 Moreover, a single intravenous injection of AAV5-HGF could lead to the high levels of HGF that were sustained up to 12 weeks in mice, contributing to a long-term effect in liver fibrosis.52 Mechanistically, HGF gene transduction upregulated MMP13 expression, thereby promoting the resolution of liver fibrosis. However, this study did not disclose the concrete cellular and molecular mechanism of AAV5-mediated HGF therapy, although previous studies suggested that HGF exerted antiapoptotic and cytoprotective effects on hepatocytes, and there are too few studies to assume the therapeutic potential of AAV5-mediated transgene expression in liver fibrosis, which remains a great challenge. Insulin-like growth factor I (IGF-I) produced by hepatocytes plays a protective role in cirrhotic rats by binding to IGF-I receptor on nonparenchymal cells.78 In 2009, the study by Sobrevals et al. reported that Simian virus 40 vectors encoding IGF-I (SV-IGF-I) could ameliorate rat liver cirrhosis induced by CCl4 or thioacetamide, suggested by improved liver function, increased fibrolysis, and decreased fibrogenesis in association with the upregulation of MMPs and HGF and the decreased expression of TIMP1, TIMP2, TGF-β, PDGF, connective tissue growth factor (CTGF), and VEGF.78 Notably, by comparing the effect of two different gene therapy vectors encoding IGF-I, AAV1-IGF-I, and SV-IGF-I, Sobrevals et al. found that a complete reversal of liver cirrhosis in rats was observed in the AAV1-IGF-I-treated group, although an obvious decrease in liver fibrosis and upregulation of HGF occurred in all IGF-I-treated rats.47 Moreover, uncertainties regarding the safety and technical limitations of SV40 make it unreliable for clinical applications. Excitingly, AAV-mediated gene therapy has been applied increasingly to clinical trials due to its high safety. Therefore, the considerable protection of AAV1-IGF-I in rodent cirrhosis models provided the basis for further clinical trials.
The upregulation of angiotensin II (Ang II)-dependent arm of the renin-angiotensin system (RAS) is a driver in liver fibrosis. Angiotensin-converting enzyme 2 (ACE2) modulates the activity of this arm by breaking down profibrotic Ang II to antifibrotic angiotensin-(1–7).79 Previously, a study by Osterreicher et al. reported that long-term deficiency of ACE2 in mice accelerated BDL- and CCl4-induced liver fibrosis and that the administration of recombinant human ACE2 daily, initiated at the induction of liver injury, mitigated liver fibrogenesis.80 However, their study only lasted 2 weeks and prophylactic administration is thought to be invasive in nature.80 Excitingly, the study by Mak et al. found that mice intraperitoneally injected with one dose of liver-trophic recombinant AAV genome 2 serotype 8 vector carrying murine ACE2 (rAAV2/8-ACE2) under the transcriptional control of a liver-specific human antitrypsin promoter displayed rapid and sustained high levels of ACE2 in liver than other organs, and mice with liver fibrosis, regardless of the cause, improved remarkably after treatment with rAAV8-ACE2, indicated by a profound reduction in pro-fibrotic markers. In parallel, a reduction in the levels of hepatic Ang II was observed with a concomitant increase in hepatic antifibrotic Ang-(1–7), leading to a distinct reduction in NADPH oxidase assembly, oxidative stress, ERK1/2, and p38 phosphorylation.55 Furthermore, they found that a single dose of rAAV8-ACE2 could reduce the severity of biliary fibrosis in multiple drug-resistant gene 2-knockout (Mdr2-KO) mice, an animal model of progressive familial intrahepatic cholestasis 3 (PFIC3), indicated by a marked decrease in liver injury and fibrosis at both established (3–6 months of age) and advanced (7–9 months of age) disease. Mechanistically, they revealed that rAAV8-ACE2 could promote the long-term production of ACE2 in hepatocytes, consequently leading to increased levels of Ang-(1–7). In addition, ACE2 therapy inhibited epithelial-mesenchymal transition (EMT) of cholangiocytes, and an increase in Ang-(1–7) levels further inhibited the activation of HSCs.56 These studies suggest that AAV-mediated liver protective factor gene transfer leads to long-term transgene expression and exerts anti-fibrotic effects by targeting different molecular and cellular mechanisms. Other therapeutic methods, such as administration of recombinant protein or using Simian virus 40 vectors, have the disadvantages of short efficacy and low safety relative to AAV-mediated gene therapy.
Promoting the expression of important genes or miRNAs in hepatocytes
Glycine N-methyltransferase (GNMT), a regulator in the cellular pool of methyl groups, is abundant in the periportal region of the normal liver and has been identified as a suppressor in hepatocellular carcinoma (HCC).81 Intriguingly, Gnmt−/− mice develop steatohepatitis, liver fibrosis, cirrhosis, and HCC progressively, thereby making them an available tool to investigate underlying molecular mechanisms and therapeutic targets for HCC.81 Noteworthily, Fang et al. found that AAV8-mediated Gnmt overexpression could attenuate liver injury and inflammation and delay tumor formation in Gnmt−/− mice.45 Moreover, in mice liver fibrosis induced by CCl4, the administration of AAV8-GNMT could reduce liver injury and ECM deposition and improve the proliferation of hepatocytes.45 However, they did not clarify the concrete mechanism of AAV8-mediated GNMT gene therapy.
It has been acknowledged that combining hepatocyte-specific promoters, such as transthyretin (Ttr) and thyroid-binding globulin (TBG), enables AAV-mediated transgene vectors to transduce hepatocytes more efficiently.82,83 In 2017, Wang et al. reported that the expression of liver-enriched TF Forkhead Box A2 (FOXA2) was downregulated in fibrotic liver tissues of patients and mice with CCl4 treatment, as well as hepatocytes, whereas it was significantly increased in aHSCs.57 To investigate the function of FOXA2 in hepatocytes but not HSCs during fibrogenesis, hepatocyte-specific KOs of FOXA2 mice were created by injecting adult FOXA2flox/flox mice with AAV8-TBG-Cre, an engineered vector that can assist in deleting genes specifically in hepatocytes. These mice had more severe liver fibrosis after CCl4 treatment. Notably, it was the intervention of AAV8-TBG-FOXA2 and LV-CMV-FOXA2, not LV-glial fibrillary acidic protein (GFAP)-FOXA2, a vector specifically targeting HSCs, that significantly alleviated the progression of CCl4-induced liver fibrosis. Mechanistically, the elevation of FOXA2 in hepatocytes distinctly inhibited endoplasmic reticulum (ER) stress through regulating C/EBP homologous protein (CHOP) expression, subsequently attenuating hepatocyte apoptosis during fibrogenesis, indicated by decreased expression levels of Bax and caspase 3.57 This study emphasizes the important role of AAV in combination with hepatocyte-specific promoter-mediated gene transduction in liver fibrosis, which is helpful in saving abnormally reduced genes in hepatocytes.
During liver fibrogenesis, epigenetic regulation by miRNA is involved in cell performance.84 miRNAs are a group of small, approximately 19- to 25-nt, non-coding RNA that post-transcriptionally regulate protein coding mRNA by suppressing the translation or destabilizing the target mRNA.84 In 2015, Knabel et al. reported that systemic delivery of scAAV8-encoded miR-29a could remarkably attenuate CCl4-induced mice liver fibrosis and that this anti-fibrotic response was associated with AAV8 transduction of hepatocytes but not HSCs.58 In addition, AAV-mediated miRNA tough decoys (TuDs) or antagomir delivery also showed promising anti-fibrotic effects. In 2018, Tsay et al. reported that miR-221-3p was upregulated in hepatocytes during liver fibrosis and that hepatocyte-specific knockdown of miR-221-3p by injecting mice with AAV8-Ttr-miR-211-3p-TuDs significantly inhibited HSC activation and CCl4-and DDC-induced liver fibrosis.59 Mechanistically, miR-221-3p inhibited the expression of G protein subunit alpha I2 (GNAI2) by targeting its 3′ UTR in hepatocytes, which in turn decreased the inhibitory effect of GNAI2 on C-C motif chemokine ligand 2 (CCL2), thereby liberating the expression of CCL2 and promoting HSC activation and liver fibrosis, whereas AAV8-Ttr-miR-211-3p-TuDs reversed the process.59 This study highlights the importance of crosstalk between hepatocytes and HSCs, and the use of hepatocyte-specific AAV vectors can effectively alter the expression of important genes in hepatocytes. Interestingly, the study by Malato et al. confirmed that an appropriate dosage of AAV8-Ttr-Cre could loop out the floxed sequences specifically in hepatocytes, and displayed no toxicity or function variation to liver.82 Duwaerts et al. reported that the injection of adult Xbp1flox/flox mice with AAV8-Ttr-Cre could effectively delete Xbp1 gene in hepatocytes, which helps them to investigate the function and regulatory mechanism of Xbp1 in acute and chronic mice liver diseases.85 In addition, Pradhan-Sundd et al. found that ablating β- and γ-catenin expression in mouse liver by interbreeding β-catenin-γ-catenin double-floxed mice and albumin-cre (Alb-Cre) transgenic mice (double KO [DKO]) exhibited an obvious mortality and morbidity.86 Fortunately, β-catenin-γ-catenin double-floxed mice administrated with AAV8-TBG-Cre vectors specifically and temporally inactivated both catenin in hepatocytes, and rescued deaths caused by DKO.86
These studies suggest that AAV8-Ttr-Cre or AAV8-TBG-Cre can be used for treating liver fibrosis caused by abnormal gene changes in hepatocytes by promoting the inactivation of genes in all hepatocytes of conditional gene KO mice at a selected time. Moreover, increasing studies have revealed that ALB-cre can loop out the flox sequences not only in hepatocytes but also in HSC and bile duct epithelial cells.70,87 For genes that play different or opposite roles in different cell types in the liver, conditional KO using the ALB-Cre may lead to confused results. For example, KO of liver fatty acid-binding protein in hepatocytes or HSCs has played a distinct role in fibrogenic injury.70 Thus, to study the function and mechanism of specific genes in hepatocytes, AAV8-Ttr-Cre or AAV8-TBG-Cre may be good substitutes for ALB-cre, which can be used as an editing tool for timing gene KO or knockin.
Inhibiting advanced glycation end products (AGEs) and lipid production by hepatocytes
As mentioned above, NASH is an important cause of the development of liver fibrosis.4 Of note, type 2 diabetes mellitus (T2DM) is closely associated with progressive necroinflammation and fibrosis in NASH.88 Abnormal accumulation of AGEs occurs in the presence of persistent hyperglycemia.89 The study by Dehnad et al. reported that continued exposure to AGEs in the liver led to an imbalance between AGEs clearance receptor (AGER1) and receptor for AGEs (RAGE), consequently resulting in excessive redox, inflammatory, and fibrogenic activity in NASH, which is mediated by NADPH oxidase 4 in hepatocytes.60 Functionally, RAGE KO in mice hepatocytes or inhibiting AGEs formation in vivo reversed these effects. AGER1 was downregulated in hepatocytes after prolonged exposure to AGEs, which is associated with decreased nuclear factor-E2-related factor 2 (NRF2) activity and increased degradation of NRF2, further affecting its downstream antioxidant gene expression. Excitingly, AAV8-TBG-NRF2 rescued the expression of AGER1 and inhibited the liver/serum AGEs, inflammation, and fibrosis during the AGE exposure period in mice. This study emphasizes the effects of AAV8-TBG-NRF2 therapy in improving necroinflammation and fibrosis caused by exposure to AGEs, and provides a theoretical basis for NASH treatment with AAV8-TBG-NRF2 in patients with T2DM in the future.60 Recently, Serrano-Maciá et al. reported that hepatic neddylation was induced in clinical and preclinical non-alcoholic fatty liver disease (NAFLD), suggested by increased NEDD8 expression levels.90 Notably, specific knockdown of NEDD8 in hepatocytes could be achieved by injecting AAV-DIO-shNedd8 into Alfp-Cre transgenic mice, which efficiently reduced neddylation levels and lipid accumulation in the livers of mice induced by a high-fat diet with choline deficiency, thereby inhibiting the development of NAFLD to NASH.90
The above studies are about AAV-mediated gene therapy, which changes the gene expression in hepatocytes and then causes changes in the level of important molecules in cells, thus affecting the activity of other liver cells, and subsequently suppressing the progression of fibrosis caused by various etiologies. However, whether there are side effects of AAV-mediated gene therapy has not yet been revealed.
Inhibiting inflammatory response induced by macrophages
Increasing evidence has shown that hepatic macrophages, including resident macrophages (Kupffer cells) and recruited macrophages (monocyte-derived macrophages), work through the liver fibrosis process, from initial liver injury and fibrosis formation to fibrosis regression.14 Interestingly, the heterogeneity of macrophages determines their diverse role in different stages of liver fibrosis.91,92 According to this, macrophages are classified into two categories conventionally, a classical M1 “pro-inflammatory” phenotype, and an alternative M2 “immunoregulatory” phenotype.92 In 2018, our research team found that the 5′ UTR of proline-serine-threonine-phosphatase-interacting protein2 (PSTPIP2) was hypermethylated, which led to a mixed induction of hepatic M1 and M2 biomarkers in CCl4-induced mice liver fibrosis. Noteworthily, PSTPIP2 was successfully overexpressed in mouse liver by using the rAAV9-mediated PSTPIP2 gene delivery system. Followed by 4 weeks’ CCl4 challenge (twice per week), mice with rAAV9-PSTPIP2 pretreatment were largely resistant to the toxic effects of CCl4, showing decreased liver injury indexes and ameliorated liver fibrosis scores compared with the rAAV9-empty vector group. Importantly, the expression levels of M1 macrophage markers, such as TNF-α, interleukin-1β (IL-1β), CCL2, and inducible nitric oxide synthase (iNOS), were significantly downregulated, while M2 macrophage markers, such as IL-10, Arg1, FIZZ1, and CD163, were obviously increased in liver tissues of the PSTPIP2-overexpressed mice group. Gain-of-function and loss-of-function experiments in vitro further confirmed that PSTPIP2 negatively regulated M1 macrophages polarization, while it promoted M2 macrophage polarization, thereby playing an anti-inflammatory and anti-fibrotic role in early liver fibrosis. Mechanistically, PSTPIP2 repressed signal transducer and activator of transcription 1 (STAT1) phosphorylation in M1 macrophages, but promoted STAT6 phosphorylation in M1 macrophages.65 Recently, Xu and coworkers reported that 14-kDa phosphohistidine phosphatase (PHP14), the first histidine phosphatase identified in vertebrates, promoted mouse liver fibrosis by regulating HSC migration.46 Furthermore, they found that PHP14 inhibition via AAV8-mediated small hairpin RNA (shRNA) delivery could efficiently ameliorate CCl4-induced mice liver fibrosis. Mechanistically, knockdown of PHP14 initially reduced fibrosis-associated macrophage recruitment, infiltration, and migration through the decreasing podosome formation of macrophages, subsequently affecting the activation of HSCs by modulating profibrogenic cytokine production by inflammatory cells, at least in part.46 These two studies reveal the anti-inflammatory role of AAV-mediated gene therapy in mice liver fibrosis, which contributes to investigating the molecular mechanisms underlying the pathogenesis of liver fibrosis. However, they do not rule out whether AAV itself causes phenotypic changes in macrophages. Intriguingly, a recent study by Carestia et al. reported that AAV8 administration in mice directly affected the liver macrophage population and phenotype transition, thereby reprogramming the liver microenvironment to become less inflammatory, potentially limiting liver damage and prolonging infection.93 Thus, they concluded that AAV8 is a suitable vector for gene therapy.93 However, AAV8 safety and suitability for human gene therapy is mainly supported by clinical trial results rather than preclinical studies.9,94 In addition, the mouse is considered a poor predictive model for studying AAV immunogenicity. Therefore, there is still a long way to go before AAV-mediated gene therapy can be used to treat human hepatic fibrosis.
Inhibiting activation and proliferation of HSCs
In 1999, Qing et al. found that the transduction ability of AAV2 relies on the presence on the surface of the cells of a receptor and co-receptor.95 Heparan sulfate proteoglycan (HSPG) is the receptor and human fibroblast growth factor receptor 1 (FGFR1) acts as co-receptor for the virus to successfully enter the host cell. Cells with surface expression of HSPG alone are not transduced by AAV2.95 Therefore, FGFR1 is important for the transduction efficiency of AAV2. Intriguingly, the expression of FGFR1 was upregulated in liver fibrosis, and its inhibition in vivo or in vitro was confirmed to mitigate rat liver fibrosis by suppressing the activation and proliferation of HSCs.96 In addition, in 2005, Tsui et al. found that rAAV2 showed more favorable binding activities to aHSCs compared with quiescent HSCs and that rAAV2 has preferential binding ability to the liver fibrotic areas, which is associated with the expression of its coreceptor, FGFR-1α.48 However, integrin αVβ5, another co-receptor for rAAV2 that is restricted in hepatocyte and biliary epithelial cells, did not show any changes in normal and fibrotic livers.48 Their study reconfirmed the essential role of FGFR-1 for rAAV2-mediated gene transduction. Subsequently, rAAV2 carrying expression cassette for heme oxygenase-1 (HO-1), a rate-limiting enzyme, was detected in primary rat HSCs, endothelial cells (ECs), and hepatocytes isolated from fibrotic livers, the result of which confirmed a high transduction efficacy of rAAV2 to aHSCs. Through western blot analysis, a stable increase in HO-1 protein level can be observed in HSCs isolated from the liver transduced by rAAV2/HO-1 for more than 12 weeks. Moreover, the levels of carboxyhemoglobin, a catalytic product of HO enzymatic activity, were upregulated in the peripheral blood of rAAV2/HO-1-treated rats since the first week after portal injection and were persistently sustained for more than 12 weeks.48 Excitingly, rAAV2-mediated HO-1 gene transfer by portal injection has been proven to achieve a good effect in an established rat micronodular cirrhosis model, indicated by reduced pro-inflammatory and pro-fibrogenic responses, collagen deposition, and HSCs proliferation.49 These studies suggest rAAV2-mediated gene transfer may be preferential to aHSCs and liver fibrotic areas, and mediate long-term transgene expression. Furthermore, rAAV2-mediated gene and miRNA delivery for the treatment of other established liver fibrosis models has also been reported.50,51 It is widely believed that TGF-β1 is the vital profibrogenic cytokine, while bone morphogenetic protein 7 (BMP7), a member of the TGF-β superfamily, was identified as an antagonist of TGF-β1 and displayed the antifibrotic potential in fibrotic diseases.97, 98, 99, 100 Hao et al. reported that twice oragastric administration of rAAV2-mediated BMP7 gene delivery could protect against the destruction of gastric acid and intestinal fluids and efficiently maintain a highly long-term serum level of BMP7.50 Ectopic BMP-7 expression effectively inhibited CCl4-induced liver fibrosis by suppressing HSC activation and promoting hepatocyte proliferation, highlighting safe, simple, and efficient AAV-mediated gene therapy for liver fibrosis treatment.50
AAV-mediated miRNA or miRNA inhibitor delivery also plays an important role in liver fibrosis. Previously, the expression levels of miR-19b were reported to be downregulated during HSCs transdifferentiation, and loss of miR-19b in aHSCs contributed to the activation of TGF-β1 signaling by decreasing the inhibitory effects of miR-19b on TGF-β receptor II expression (TGF-βRII) and CTGF.101 To further study the function of miR-19b in vivo, they constructed an AAV2 with a mature miR-19b transgene downstream of enhanced green fluorescent protein under the control of murine collagen 1α promoter (AAV2-miR19b-Col-EGFP) to target HSCs. By portal vein administration of AAV2-miR19b-Col-EGFP to the established BDL-induced rat liver fibrosis for 2 weeks, they unexpectedly found liver function improved and injury and fibrosis degree decreased compared with the AAV2-control-Col-EGFP-treated group. Importantly, no EGFP signal was detected in other organs, such as pancreas, heart, lung, kidney, ilium, and brain, except for liver tissues. Moreover, only HSCs were detected for positive EGFP co-staining signal, suggesting AAV2-miR19b-Col-EGFP has preferential tropism to HSCs.51 Their study revealed the therapeutic advantages of AAV-mediated miRNA delivery under the control of a cell-specific promoter in liver fibrosis, and provided new ideas for future research. Schistosomiasis is caused by several species of trematode worms, which can lead to liver fibrosis and threaten human health.102 In 2015, He et al. found that progressive hepatic schistosomiasis in mice caused a considerable elevation of miR-21 in primary aHSCs, and intravenous injection of rAAV8-miR-21-TuDs remarkably ameliorated liver fibrosis by promoting Smad7 expression, indirectly suppressing TGF-β1/Smad and IL-13/Smad pathways.62 In 2018, they reported that the elevated levels of miR-351 aggravated mice liver fibrosis induced by schistosoma infection by targeting the vitamin D receptor (VDR), an antagonist of SMAD signaling. Moreover, in vivo inhibition of miR-351 by rAAV8-miR-351-sponge significantly attenuated liver fibrosis and protected mice against lethal schistosoma infection.63
Nevertheless, the study by Pan et al. reported that AAV8-mediated RCAN1.4 gene delivery protected CCl4-induced mouse liver fibrosis by inhibiting HSC activation and proliferation, and promoting aHSC apoptosis by repressing calcineurin/NFAT3 signaling.61 In addition, Chen et al. found that rAAV9-mediated SUN2 delivery could efficiently transduce mice liver and protect mice from CCl4-induced liver fibrosis, indicated by the reduction of pro-fibrotic factors compared with the rAAV9 empty vector treated group. Mechanistically, rAAV9 mediated overexpression of SUN2 repressed the activation and proliferation of HSCs by inhibiting PI3K/AKT signaling.66 Notably, AAV9-mediated circFBXW4 effectively suppressed the progression of mice liver fibrosis by targeting the miR-18b-3p/FBXW7 axis in HSCs, suggesting that AAV9 is a good therapeutic carrier for circular RNA (circRNA).67 Although these studies have revealed that AAV-mediated RCAN1.4, SUN2, and circFBXW4 overexpression in liver play anti-fibrotic roles by inhibiting the activation and proliferation of HSCs, it is not clear whether these rAAV vectors transduce other liver cells, thereby indirectly affecting the activity of HSCs. As mentioned earlier, AAV8-mediated PHP14 shRNA delivery inhibited liver fibrosis by decreasing macrophage migration to the site of injury, subsequently affecting the activation of HSCs.46 Therefore, researchers must clarify which cells in the liver AAV vectors preferentially transduce and how to improve the transduction efficiency. Notably, the study by Zhang et al. used AAV9-GFAP-shTGF-β1 to target HSCs, since GFAP has been identified as a marker for HSCs.68,103 AAV9-GFAP-shTGF-β1 accelerated the regeneration of fibrotic functional liver remnant, mitigated fibrosis, and improved liver function in chronic diethylnitrosamine (DEN)-induced rat liver fibrosis followed by associating liver partition and portal vein ligation for staged hepatectomy (ALPPS), a novel two-stage hepatectomy. Moreover, the effect of AAV9-GFAP-shTGF-β1 on liver fibrosis is superior to that of LY2157299, a TGF-βR1 inhibitor, reflecting the advantages of AAV-mediated gene therapy under the transcriptional control of a cell-specific promoter.68 Nevertheless, an astonishing study by Mederacke et al. indicated that GFAP-Cre marks extra- and intrahepatic bile ducts but not HSCs.104 By performing essential cell identification experiments, they found that lecithin-retinol acyltransferase (Lrat) Cre efficiently labels 99% of HSCs.104 Therefore, AAV targeting HSCs under the transcriptional control of GFAP promoter is a controversial issue, and further studies should focus on the selection of cell-specific promoters.
Inducing MFs to hepatocyte-like cells or quiescent HSCs
Considering that cell lineages can be converted by altering the transcriptional network, Huang et al. established a novel strategy that induces mouse tail-tip fibroblasts (TTFs) to functional hepatocyte-like cells (iHeps) by the overexpression of Gata4, Hnf1a, and Foxa3 and the inactivation of p19Arf in vitro. Results showed that iHeps displayed a similar gene expression pattern as well as metabolic function, closely resembling hepatocytes.105 Inspired by their research, Rezvani et al. developed in vivo reprogramming of MFs into hepatocytes using AAV vector-mediated delivery of essential TFs, attempting to limit collagen deposition in liver fibrosis.53 By intravenously injecting different AAV vectors, they found that only AAV6 has a relevant MF tropism, transducing 10.2% ± 5.3% of α-SMA-positive MFs, which is contradictory to previous studies suggesting that AAV2 has preferential binding ability to aHSCs48,53. In detail, by transducing MFs generated from primary HSCs with AAV6 mediated delivery of the TF genes, including Foxa1, Foxa2, Foxa3, Gata4, Hnf1a, or Hnf4a from the cytomegalovirus (CMV) promoter (AAV6-6TFs), they observed the acquisition of hepatocyte gene expression and function in MFs whose original identity was mostly missing. Using a mouse model of MF fate tracing, identified as Lrat-Cre; R26RZsGreen mice,104 they successfully reprogramed MFs into fully functional hepatocytes (MF-iHeps) with the help of AAV6-6TFs in CCl4 or choline-deficient ethionine-supplemented (CDE) diet-induced mouse liver fibrosis. Moreover, MF-iHeps closely resembled primary hepatocytes and had normal cell function and proliferation. Functionally, they found that AAV6-6TFs reduced liver fibrosis and injury, but administration of AAV8-6TFs to hepatocytes had little effect.53 Therefore, hepatic reprogramming of MFs with AAV vectors may be a promising therapeutic strategy for liver fibrosis. Recently, the work by Nakano et al. found that Tcf21 is a novel deactivation factor of aHSCs.20 Tcf21 was decreased in HSCs that underwent culture-induced activation in vitro and also in liver fibrotic tissues of mice and humans in vivo. Interestingly, the expression of Tcf21 was recovered during the spontaneous regression of murine liver fibrosis. AAV6-mediated Tcf21 gene transfer efficiently reduced CCl4- and methionine-choline-deficient diet (MCD)-induced liver fibrosis, as evidenced by improved hepatic architecture and function. Mechanistically, AAV6-mediated Tcf21 enhanced aHSCs to a quiescent phenotype.20 In addition, the study by Chen et al. demonstrated that Nestin, a class VI intermediate filament, was positively correlated with the progression of liver fibrosis either in murine models or in human samples, and treatment with AAV6-mediated Nestin shRNA remarkably ameliorated liver fibrosis through targeting aHSCs. Mechanistically, TGF-β1 stimulated the high expression of Nestin in aHSCs; in turn, Nestin promoted the TGF-β-Smad2/3 pathway by decreasing the caveolin1 (Cav-1)-mediated degradation of TβRI, and Nestin knockdown inhibited the excessive activation of TGF-β1 signaling.54 These studies suggest that AAV6-mediated 6TFs, Tcf21, and Nestin shRNA delivery efficiently attenuates liver fibrosis by regulating different molecular mechanisms, highlighting that AAV6 has a tropism for aHSCs or MFs, but further research should be undertaken to confirm this point.
Promoting expansion of liver progenitor cells (LPCs)
Recently, the study by Dai et al. revealed that growth differentiation factor 11 (GDF11), a member of the TGF-β superfamily, was remarkably increased during liver fibrogenesis, both in mice and humans.64 They also confirmed that GDF11 is a secreted protein, mainly presenting in HSCs in normal liver, while the highest expression of GDF11 in activated HSCs was observed in fibrotic liver. By administering 1 × 1011 AAV8 particles encoding Gdf11 (AAV-GDF11) to fibrotic BALB/c mice induced by CCl4 and the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet, GDF11 was successfully overexpressed in these established models, and they surprisingly found that toxin-(CCl4) and cholestasis-(DDC)-induced mice liver fibroses were significantly attenuated. Intriguingly, the hepatic LGR5+ progenitor pool was expanded in AAV-GDF11-treated fibrotic mice compared with AAV empty vector-treated fibrotic mice. Moreover, transplanting 5 × 105 of LGR5+ cells from AAV-GDF11-treated fibrotic mice into another set of fibrotic BALB/c mice induced by CCl4 markedly improved liver fibrosis. In addition, the loss of GDF11 in hepatic MFs abrogates the expansion of LGR5+ progenitor cells in human liver organoids. Reciprocally, co-culturing MFs with LGR5+ cells or GDF11-pretreated LGR5+ cells could decrease the expression of fibrotic genes of MFs. Depletion of LGR5+ cells by injecting Gdf11-overexpressed fibrotic mice with AAV encoding diphtheria toxin A (DTA) under the transcriptional control of the Lgr5 promoter could abrogate the anti-fibrotic function of GDF11. Nevertheless, although highly expressed in MFs, GDF11 modulation itself had little, if any, role in fibrogenesis. Notably, they successfully constructed the AAV variant (AAV-NGF-GFP) that preferentially targets hepatic MFs in vivo BY performing essential experiments. The overexpression of GDF11 mediated by AAV-NGF-GDF11 promoted an increased number of LGR5+ cells, while knockdown of GDF11 mediated by AAV-NGF-GDF11-shRNA demonstrated an opposite effect.64 Overall, this study revealed that AAV-mediated GDF11 gene delivery has achieved a good effect on mice liver fibrosis, which has helped researchers to reveal the cellular and molecular mechanism of GDF11. Meanwhile, AAV variants targeting MFs were proposed, which provided new ideas for gene therapy targeting MFs in the future.
Influencing function of ECs
Previous studies often classified portal angiogenesis and sinusoidal capillarization as hepatic angiogenesis.69 In 2019, a study conducted by Xu et al. revealed the distinct roles of portal angiogenesis from sinusoids capillarization in liver fibrosis. They found that leukocyte cell-derived chemotaxin 2 (LECT2) produced by hepatocytes in response to liver damage is a functional ligand of the EC-specific orphan receptor Tie1. LV-mediated overexpression of LECT2 promoted BDL-induced, DDC-induced, and MCD-induced mice liver fibrosis by repressing portal angiogenesis and promoting sinusoidal capillarization, while these observations were reversed in Lect2-KO mice. Furthermore, AAV9-mediated LECT2 shRNA delivery (AAV9-LECT2 shRNA) significantly reduced the expression of LECT2 in mice liver and improved the liver fibrosis induced by CCl4. Prospectively, they found that mice pretreated with AAV9-LECT2 shRNA showed an increase in portal angiogenesis and a decrease in hepatic sinusoid capillarization after CCl4 challenge. Mechanistically, the binding of LECT2 to Tie1 repressed the migration and tube formations of EC, and knockdown of LECT2 by AAV9-LECT2 shRNA may reverse the above behavior of ECs.69
Challenges of AAV-mediated gene therapy in liver fibrosis
Currently, AAV-mediated gene therapy for liver fibrosis has only been applied to rodent models and no clinical trials have been reported. This is a huge challenge. First, in mice, investigators have been able to acknowledge the organ tropism of different AAV serotypes, and AAV1–3 and 5–9 were capable of transducing liver tissues.106 Moreover, increasing evidence suggests that AAV8 has higher affinity to hepatocytes than other serotypes, and AAV6 has a higher tropism to MFs in mice.53,107 However, data presenting tropism profiles in humans are far from obtaining, because examining the vector biodistribution in multiple biopsies is difficult to perform. Second, inevitable immunogenicity may have effects on the gene transfer efficiency of the AAV vector, although the above-mentioned studies merely focused attention on the function and mechanism of AAV-mediated transgene expression in rodent liver fibrosis models and did not disclose the immunogenicity. Preexisting neutralizing antibodies (NAbs) and cytotoxic T lymphocyte (CTL)-mediated immune response are two serious concerns that prevent rAAV from transducing target cells and performing long-lasting transgene expression,108 despite several approaches having been studied to eliminate these issues, such as saturating NAb binding sites through increasing rAAV dose, combining excessive empty capsids, using preventive immunosuppressive drugs, or natural shielding by EVs during rAAV production.108 Importantly, Sobrevals et al. revealed that AAV1 vectors could be diluted due to liver regeneration following hepatectomy as the frequency of AAV genomic integration is limited.72 Nevertheless, their further experiments showed that the levels of AAV transduction to liver tissue did not differ between healthy livers and fibrotic livers at any stage of liver fibrosis, suggesting that diseased livers appear to have little effect on the gene transduction ability of AAV. Intriguingly, intra-arterial injection of AAV1 led to higher transgene expression in cirrhotic than in healthy livers, while intra-portal administration showed the opposite phenomenon. Intra-hepatic injection of AAV1 leads to similar transgene expression in cirrhotic and healthy rat livers, suggesting that an appropriate procedure should be considered for future AAV-mediated gene therapy in the treatment of human liver fibrosis.72 In addition, increased genotoxic risk should be a challenge that cannot be neglected, although no major adverse events involving genotoxicity caused by rAAV vector integration in human and NHPs have been observed.109 Notably, some studies have revealed that rAAV administration is related to the development of HCC in newborn mice, despite the fact that HCC was not observed in rAAV-treated adult mice and other mammals.110,111 Therefore, long-term monitoring after rAAV-mediated gene delivery is important for evaluating potential genotoxicity.
Conclusions and prospects
In this study, we systematically summarized the therapeutic potential of AAV-mediated gene therapy in liver fibrosis models based on the molecular functions and mechanisms of transferred genes. The safety, efficacy, and sustained transgene expression of AAV-mediated gene therapy have been confirmed in different liver fibrosis models, which encourages the exploration of therapeutic strategies that could focus on AAV-mediated gene delivery systems in the future. However, the present studies only exist at the animal level, and there is no representative clinical trial of AAV-mediated gene therapy for the treatment of liver fibrosis. In practice, animal models are powerless to predict the tropism and immunogenicity of AAVs in humans; although animals are available, results from animal experiments are easily reproducible and help researchers better understand the pathological mechanisms of liver fibrosis. Thus, the paucity of human data makes AAV-mediated gene therapy a great challenge, and there is still a long way to go before it can be used for human liver fibrosis.
Acknowledgments
The study was supported by funding from the National Natural Science Foundation of China (U19A2001 and 82070628); the University Synergy Innovation Program of Anhui Province (GXXT-2020-063 and GXXT-2020-025); and the Research Fund of Anhui Institute of Translational Medicine (2021zhyx-B06).
Author contributions
F.-T.B. drafted the manuscript, figures, and tables; P.-C.J. and Y.Z. contributed to the writing and reviewing of the manuscript; Y.-R.Y., H.-W.M., and Y.-H.B. contributed to the figure design; C.H. and J.L. conceived and designed the review. All of the authors listed in this review approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Cheng Huang, Email: huangcheng@ahmu.edu.cn.
Jun Li, Email: lj@ahmu.edu.cn.
References
- 1.Marcellin P., Kutala B.K. Liver diseases: a major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018;38:2–6. doi: 10.1111/liv.13682. [DOI] [PubMed] [Google Scholar]
- 2.Tsochatzis E.A., Bosch J., Burroughs A.K. Liver cirrhosis. Lancet. 2014;383:1749–1761. doi: 10.1016/s0140-6736(14)60121-5. [DOI] [PubMed] [Google Scholar]
- 3.Böttcher K., Pinzani M. Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents. Adv. Drug Deliv. Rev. 2017;121:3–8. doi: 10.1016/j.addr.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Parola M., Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol. Aspect. Med. 2019;65:37–55. doi: 10.1016/j.mam.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Lo R.C., Kim H. Histopathological evaluation of liver fibrosis and cirrhosis regression. Clin. Mol. Hepatol. 2017;23:302–307. doi: 10.3350/cmh.2017.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahdinloo S., Kiaie S.H., Amiri A., Hemmati S., Valizadeh H., Zakeri-Milani P. Efficient drug and gene delivery to liver fibrosis: rationale, recent advances, and perspectives. Acta Pharm. Sin. B. 2020;10:1279–1293. doi: 10.1016/j.apsb.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maestro S., Weber N.D., Zabaleta N., Aldabe R., Gonzalez-Aseguinolaza G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021;3:100300. doi: 10.1016/j.jhepr.2021.100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benskey M.J., Sandoval I.M., Miller K., Sellnow R.L., Gezer A., Kuhn N.C., Vashon R., Manfredsson F.P. Basic concepts in viral vector-mediated gene therapy. Methods Mol. Biol. 2019;1937:3–26. doi: 10.1007/978-1-4939-9065-8_1. [DOI] [PubMed] [Google Scholar]
- 9.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/nejmoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/s0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Avola D., López-Franco E., Sangro B., Pañeda A., Grossios N., Gil-Farina I., Benito A., Twisk J., Paz M., Ruiz J., et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J. Hepatol. 2016;65:776–783. doi: 10.1016/j.jhep.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/nejmoa1706198. [DOI] [PubMed] [Google Scholar]
- 13.Kisseleva T., Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021;18:151–166. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 14.Cheng D., Chai J., Wang H., Fu L., Peng S., Ni X. Hepatic macrophages: key players in the development and progression of liver fibrosis. Liver Int. 2021;41:2279–2294. doi: 10.1111/liv.14940. [DOI] [PubMed] [Google Scholar]
- 15.Duarte S., Baber J., Fujii T., Coito A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015;44-46:147–156. doi: 10.1016/j.matbio.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higashi T., Friedman S.L., Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017;121:27–42. doi: 10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.An P., Wei L.L., Zhao S., Sverdlov D.Y., Vaid K.A., Miyamoto M., Kuramitsu K., Lai M., Popov Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020;11:2362. doi: 10.1038/s41467-020-16092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F., Kong D.S., Lu Y., Zheng S.Z. Peroxisome proliferator-activated receptor-gamma as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell. Mol. Life Sci. 2013;70:259–276. doi: 10.1007/s00018-012-1046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X., Xu J., Rosenthal S., Zhang L.J., McCubbin R., Meshgin N., Shang L.S., Koyama Y., Ma H.Y., Sharma S., et al. Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology. 2020;158:1728–1744.e14. doi: 10.1053/j.gastro.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakano Y., Kamiya A., Sumiyoshi H., Tsuruya K., Kagawa T., Inagaki Y. Regression of liver fibrosis induced by a novel deactivation factor of fibrogenic hepatic stellate cells. Hepatology. 2019;70:894a. doi: 10.1002/hep.30965. [DOI] [PubMed] [Google Scholar]
- 21.Atchison R.W., Casto B.C., Hammon W.M. Adenovirus-associated defective virus particles. Science. 1965;149:754–756. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 22.Berns K.I. My life with adeno-associated virus: a long time spent studying a short genome. DNA Cell Biol. 2013;32:342–347. doi: 10.1089/dna.2013.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hastie E., Samulski R.J. Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success--a personal perspective. Hum. Gene Ther. 2015;26:257–265. doi: 10.1089/hum.2015.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grieger J.C., Samulski R.J. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Adv. Biochem. Eng. Biot. 2005;99:119–145. [PubMed] [Google Scholar]
- 25.Wang D., Tai P.W.L., Gao G.P. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balakrishnan B., Jayandharan G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014;14:86–100. doi: 10.2174/1566523214666140302193709. [DOI] [PubMed] [Google Scholar]
- 27.Li C.W., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 28.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palaschak B., Herzog R.W., Markusic D.M. AAV-mediated gene delivery to the liver: overview of current technologies and methods. Methods Mol. Biol. 2019;1950:333–360. doi: 10.1007/978-1-4939-9139-6_20. [DOI] [PubMed] [Google Scholar]
- 30.Naso M.F., Tomkowicz B., Perry W.L., Strohl W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H.R., Lee K.J., Lin Y., Wolfe T., Sheng J., Plewa C., Wang S.L., Meisen W.H. Creation of a high-yield AAV vector production platform in suspension HEK293T cells using a design of experiment approach. Mol. Ther. 2020;28:196–197. doi: 10.1016/j.omtm.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buck T.M., Wijnholds J. Recombinant adeno-associated viral vectors (rAAV)-Vector elements in ocular gene therapy clinical trials and transgene expression and bioactivity assays. Int. J. Mol. Sci. 2020;21:4197. doi: 10.3390/ijms21124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suoranta T., Laham-Karam N., Ylä-Herttuala S. Optimized protocol for accurate titration of adeno-associated virus vectors. Hum. Gene Ther. 2021;19-20:1270–1279. doi: 10.1089/hum.2020.318. [DOI] [PubMed] [Google Scholar]
- 34.Xu J., DeVries S.H., Zhu Y.L. Quantification of adeno-associated virus with safe nucleic acid dyes. Human Gene Ther. 2020;31:1086–1099. doi: 10.1089/hum.2020.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asokan A., Schaffer D.V., Jude Samulski R. The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ylä-Herttuala S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012;20:1831–1832. doi: 10.1038/mt.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keeler A.M., Flotte T.R. Recombinant adeno-associated virus gene therapy in light of Luxturna (and Zolgensma and Glybera): where are we, and how did we get here? Annu. Rev. Virol. 2019;6:601–621. doi: 10.1146/annurev-virology-092818-015530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng C., Zhao P.Y., Branham K., Schlegel D., Fahim A.T., Jayasundera T.K., Khan N., Besirli C.G. Real-world outcomes of voretigene neparvovec treatment in pediatric patients with RPE65-associated Leber congenital amaurosis. Graefes Arch. Clin. Exp. Ophthalmol. 2022;260:1543–1550. doi: 10.1007/s00417-021-05508-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Junge N., Mingozzi F., Ott M., Baumann U. Adeno-associated virus vector-based gene therapy for monogenetic metabolic diseases of the liver. J. Pediatr. Gastroenterol. Nutr. 2015;60:433–440. doi: 10.1097/mpg.0000000000000703. [DOI] [PubMed] [Google Scholar]
- 40.Herzog R.W., Yang E.Y., Couto L.B., Hagstrom J.N., Elwell D., Fields P.A., Burton M., Bellinger D.A., Read M.S., Brinkhous K.M., et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat. Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Bell P., Lin J., Calcedo R., Tarantal A.F., Wilson J.M. AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta) Mol. Ther. 2011;19:2012–2020. doi: 10.1038/mt.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nathwani A.C., Gray J.T., Ng C.Y.C., Zhou J., Spence Y., Waddington S.N., Tuddenham E.G.D., Kemball-Cook G., McIntosh J., Boon-Spijker M., et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nathwani A.C., Gray J.T., McIntosh J., Ng C.Y.C., Zhou J., Spence Y., Cochrane M., Gray E., Tuddenham E.G., Davidoff A.M. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood. 2007;109:1414–1421. doi: 10.1182/blood-2006-03-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang C.C., Wu C.F., Liao Y.J., Huang S.F., Chen M., Chen Y.M.A. AAV serotype 8-mediated liver specific GNMT expression delays progression of hepatocellular carcinoma and prevents carbon tetrachloride-induced liver damage. Sci. Rep. 2018;8:13802. doi: 10.1038/s41598-018-30800-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu A.J., Zhou J.C., Li Y.M., Qiao L.Y., Jin C.C., Chen W., Sun L., Wu S.N., Li X.J., Zhou D.H., et al. 14-kDa phosphohistidine phosphatase is a potential therapeutic target for liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2021;320:G351–G365. doi: 10.1152/ajpgi.00334.2020. [DOI] [PubMed] [Google Scholar]
- 47.Sobrevals L., Enguita M., Quiroga J., Prieto J., Fortes P. Insulin-like growth factor I (IGF-I) expressed from an AAV1 vector leads to a complete reversion of liver cirrhosis in rats. PLoS One. 2016;11:e0162955. doi: 10.1371/journal.pone.0162955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsui T.Y., Lau C.K., Ma J., Wu X., Wang Y.Q., Farkas S., Xu R., Schlitt H.J., Fan S.T. rAAV-mediated stable expression of heme oxygenase-1 in stellate cells: a new approach to attenuate liver fibrosis in rats. Hepatology. 2005;42:335–342. doi: 10.1002/hep.20803. [DOI] [PubMed] [Google Scholar]
- 49.Tsui T.Y., Lau C.K., Ma J., Glockzin G., Obed A., Schlitt H.J., Fan S.T. Adeno-associated virus-mediated heme oxygenase-1 gene transfer suppresses the progression of micronodular cirrhosis in rats. World J. Gastroenterol. 2006;12:2016. doi: 10.3748/wjg.v12.i13.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao Z.M., Cai M., Lv Y.F., Huang Y.H., Li H.H. Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Mol. Ther. 2012;20:2043–2051. doi: 10.1038/mt.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandon-Warner E., Benbow J.H., Swet J.H., Feilen N.A., Culberson C.R., McKillop I.H., deLemos A.S., Russo M.W., Schrum L.W. Adeno-associated virus serotype 2 vector-mediated reintroduction of microRNA-19b attenuates hepatic fibrosis. Hum. Gene Ther. 2018;29:674–686. doi: 10.1089/hum.2017.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzumura K., Hirano T., Son G., Iimuro Y., Mizukami H., Ozawa K., Fujimoto J. Adeno-associated virus vector-mediated production of hepatocyte growth factor attenuates liver fibrosis in mice. Hepatol. Int. 2008;2:80–88. doi: 10.1007/s12072-007-9042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rezvani M., Español-Suñer R., Malato Y., Dumont L., Grimm A.A., Kienle E., Bindman J.G., Wiedtke E., Hsu B.Y., Naqvi S.J., et al. In vivo hepatic reprogramming of myofibroblasts with AAV vectors as a therapeutic strategy for liver fibrosis. Cell Stem Cell. 2016;18:809–816. doi: 10.1016/j.stem.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H., Cai J., Wang J., Qiu Y., Jiang C., Wang Y., Wang Y., Yi C., Guo L., Pan L., et al. Targeting Nestin+ hepatic stellate cells ameliorates liver fibrosis by facilitating TβRI degradation. J. Hepatol. 2021;74:1176–1187. doi: 10.1016/j.jhep.2020.11.016. [DOI] [PubMed] [Google Scholar]
- 55.Mak K.Y., Chin R., Cunningham S.C., Habib M.R., Torresi J., Sharland A.F., Alexander I.E., Angus P.W., Herath C.B. ACE2 therapy using adeno-associated viral vector inhibits liver fibrosis in mice. Mol. Ther. 2015;23:1434–1443. doi: 10.1038/mt.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajapaksha I.G., Gunarathne L.S., Asadi K., Cunningham S.C., Sharland A., Alexander I.E., Angus P.W., Herath C.B. Liver-targeted angiotensin converting enzyme 2 therapy inhibits chronic biliary fibrosis in multiple drug-resistant gene 2-knockout mice. Hepatol. Commun. 2019;3:1656–1673. doi: 10.1002/hep4.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W., Yao L.J., Shen W., Ding K., Shi P.M., Chen F., He J., Ding J., Zhang X., Xie W.F. FOXA2 alleviates CCl4-induced liver fibrosis by protecting hepatocytes in mice. Sci. Rep. 2017;7:15532. doi: 10.1038/s41598-017-15831-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knabel M.K., Ramachandran K., Karhadkar S., Hwang H.W., Creamer T.J., Chivukula R.R., Sheikh F., Clark K.R., Torbenson M., Montgomery R.A., et al. Systemic delivery of scAAV8-encoded MiR-29a ameliorates hepatic fibrosis in carbon tetrachloride-treated mice. PLoS One. 2015;10:e0124411. doi: 10.1371/journal.pone.0124411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsay H.C., Yuan Q., Balakrishnan A., Kaiser M., Möbus S., Kozdrowska E., Farid M., Tegtmeyer P.K., Borst K., Vondran F.W.R., et al. Hepatocyte-specific suppression of microRNA-221-3p mitigates liver fibrosis. J. Hepatol. 2019;70:722–734. doi: 10.1016/j.jhep.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Dehnad A., Fan W.G., Jiang J.X., Fish S.R., Li Y., Das S., Mozes G., Wong K.A., Olson K.A., Charville G.W., et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J. Clin. Invest. 2020;130:4320–4330. doi: 10.1172/jci133051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan X.Y., You H.M., Wang L., Bi Y.H., Yang Y., Meng H.W., Meng X.M., Ma T.T., Huang C., Li J. Methylation of RCAN1.4 mediated by DNMT1 and DNMT3b enhances hepatic stellate cell activation and liver fibrogenesis through Calcineurin/NFAT3 signaling. Theranostics. 2019;9:4308–4323. doi: 10.7150/thno.32710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He X., Xie J., Zhang D.M., Su Q., Sai X., Bai R.P., Chen C., Luo X.F., Gao G.P., Pan W.Q. Recombinant adeno-associated virus-mediated inhibition of microRNA-21 protects mice against the lethal schistosome infection by repressing both IL-13 and transforming growth factor beta 1 pathways. Hepatology. 2015;61:2008–2017. doi: 10.1002/hep.27671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He X., Sun Y., Lei N., Fan X., Zhang C., Wang Y., Zheng K., Zhang D., Pan W. MicroRNA-351 promotes schistosomiasis-induced hepatic fibrosis by targeting the vitamin D receptor. Proc. Natl. Acad. Sci. USA. 2018;115:180–185. doi: 10.1073/pnas.1715965115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dai Z., Song G., Balakrishnan A., Yang T., Yuan Q., Möbus S., Weiss A.C., Bentler M., Zhu J., Jiang X., et al. Growth differentiation factor 11 attenuates liver fibrosis via expansion of liver progenitor cells. Gut. 2020;69:1104–1115. doi: 10.1136/gutjnl-2019-318812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Y., Wu X.Q., Li W.X., Huang H.M., Li H.D., Pan X.Y., Li X.F., Huang C., Meng X.M., Zhang L., et al. PSTPIP2 connects DNA methylation to macrophage polarization in CCL4-induced mouse model of hepatic fibrosis. Oncogene. 2018;37:6119–6135. doi: 10.1038/s41388-018-0383-0. [DOI] [PubMed] [Google Scholar]
- 66.Chen X., Li W.X., Chen Y., Li X.F., Li H.D., Huang H.M., Bu F.T., Pan X.Y., Yang Y., Huang C., et al. Suppression of SUN2 by DNA methylation is associated with HSCs activation and hepatic fibrosis. Cell Death Dis. 2018;9:1021. doi: 10.1038/s41419-018-1032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen X., Li H.D., Bu F.T., Li X.F., Chen Y., Zhu S., Wang J.N., Chen S.Y., Sun Y.Y., Pan X.Y., et al. Circular RNA circFBXW4 suppresses hepatic fibrosis via targeting the miR-18b-3p/FBXW7 axis. Theranostics. 2020;10:4851–4870. doi: 10.7150/thno.42423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang B., Meng F., Liu Y., Yuan Y., Wang J., Wu D., Cui Y., Zhang S., Guo H., Liang S., et al. Inhibition of TGFβ1 accelerates regeneration of fibrotic rat liver elicited by a novel two-staged hepatectomy. Theranostics. 2021;11:4743–4758. doi: 10.7150/thno.52102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu M., Xu H.H., Lin Y., Sun X., Wang L.J., Fang Z.P., Su X.H., Liang X.J., Hu Y., Liu Z.M., et al. LECT2, a ligand for Tie1, plays a crucial role in liver fibrogenesis. Cell. 2019;178:1478–1492.e20. doi: 10.1016/j.cell.2019.07.021. [DOI] [PubMed] [Google Scholar]
- 70.Newberry E.P., Xie Y., Lodeiro C., Solis R., Moritz W., Kennedy S., Barron L., Onufer E., Alpini G., Zhou T.H., et al. Hepatocyte and stellate cell deletion of liver fatty acid binding protein reveals distinct roles in fibrogenic injury. FASEB J. 2019;33:4610–4625. doi: 10.1096/fj.201801976r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Bañuelos J., Siller-Lopez F., Miranda A., Aguilar L.K., Aguilar-Cordova E., Armendariz-Borunda J. Cirrhotic rat livers with extensive fibrosis can be safely transduced with clinical-grade adenoviral vectors. Evidence of cirrhosis reversion. Gene Ther. 2002;9:127–134. doi: 10.1038/sj.gt.3301647. [DOI] [PubMed] [Google Scholar]
- 72.Sobrevals L., Enguita M., Rodriguez C., Gonzalez-Rojas J., Alzaguren P., Razquin N., Prieto J., Fortes P. AAV vectors transduce hepatocytes in vivo as efficiently in cirrhotic as in healthy rat livers. Gene Ther. 2012;19:411–417. doi: 10.1038/gt.2011.119. [DOI] [PubMed] [Google Scholar]
- 73.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H., et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davidoff A.M., Gray J.T., Ng C.Y., Zhang Y., Zhou J., Spence Y., Bakar Y., Nathwani A.C. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol. Ther. 2005;11:875–888. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 75.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/nejmoa1708483. [DOI] [PubMed] [Google Scholar]
- 76.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/nejmoa1908490. [DOI] [PubMed] [Google Scholar]
- 77.Matsuda Y., Matsumoto K., Yamada A., Ichida T., Asakura H., Komoriya Y., Nishiyama E., Nakamura T., YAmAdA A. Preventive and therapeutic effects in rats of hepatocyte growth factor infusion on liver fibrosis/cirrhosis. Hepatology. 1997;26:81–89. doi: 10.1002/hep.510260111. [DOI] [PubMed] [Google Scholar]
- 78.Sobrevals L., Rodriguez C., Romero-Trevejo J.L., Gondi G., Monreal I., Paneda A., Juanarena N., Arcelus S., Razquin N., Guembe L., et al. Insulin-like growth factor I gene transfer to cirrhotic liver induces fibrolysis and reduces fibrogenesis leading to cirrhosis reversion in rats. Hepatology. 2010;51:912–921. doi: 10.1002/hep.23412. [DOI] [PubMed] [Google Scholar]
- 79.Grace J.A., Herath C.B., Mak K.Y., Burrell L.M., Angus P.W. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin. Sci. 2012;123:225–239. doi: 10.1042/cs20120030. [DOI] [PubMed] [Google Scholar]
- 80.Osterreicher C.H., Taura K., De Minicis S., Seki E., Penz-Österreicher M., Penz-Osterreicher M., Kodama Y., Kluwe J., Schuster M., Oudit G.Y., et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology. 2009;50:929–938. doi: 10.1002/hep.23104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez-Chantar M.L., Vazquez-Chantada M., Ariz U., Martinez N., Varela M., Luka Z., Capdevila A., Rodriguez J., Aransay A.M., Matthiesen R., et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malato Y., Naqvi S., Schürmann N., Ng R., Wang B., Zape J., Kay M.A., Grimm D., Willenbring H. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest. 2011;121:4850–4860. doi: 10.1172/jci59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yanger K., Zong Y., Maggs L.R., Shapira S.N., Maddipati R., Aiello N.M., Thung S.N., Wells R.G., Greenbaum L.E., Stanger B.Z. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–724. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Su Q.Z., Kumar V., Sud N., Mahato R.I. MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 2018;129:54–63. doi: 10.1016/j.addr.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 85.Duwaerts C.C., Siao K., Soon R.K., Her C., Iwawaki T., Kohno K., Mattis A.N., Maher J.J. Hepatocyte-specific deletion of XBP1 sensitizes mice to liver injury through hyperactivation of IRE1α. Cell Death Differ. 2021;28:1455–1465. doi: 10.1038/s41418-020-00671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pradhan-Sundd T., Zhou L.L., Vats R., Jiang A., Molina L., Singh S., Poddar M., Russell J., Stolz D.B., Oertel M., et al. Dual catenin loss in murine liver causes tight junctional deregulation and progressive intrahepatic cholestasis. Hepatology. 2018;67:2320–2337. doi: 10.1002/hep.29585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu X.L., Kobayashi S., Qiao W.H., Li C.L., Xiao C.Y., Radaeva S., Stiles B., Wang R.H., Ohara N., Yoshino T., et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J. Clin. Invest. 2006;116:1843–1852. doi: 10.1172/jci27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bazick J., Donithan M., Neuschwander-Tetri B.A., Kleiner D., Brunt E.M., Wilson L., Doo E., Lavine J., Tonascia J., Loomba R. Clinical model for NASH and advanced fibrosis in adult patients with diabetes and NAFLD: guidelines for referral in NAFLD. Diabetes Care. 2015;38:1347–1355. doi: 10.2337/dc14-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puddu A., Sanguineti R., Maggi D., Nicolò M., Traverso C.E., Cordera R., Viviani G.L. Advanced glycation end-products and hyperglycemia increase angiopoietin-2 production by impairing angiopoietin-1-tie-2 system. J. Diabetes Res. 2019;2019:1–7. doi: 10.1155/2019/6198495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Serrano-Maciá M., Simón J., González-Rellan M.J., Azkargorta M., Goikoetxea-Usandizaga N., Lopitz-Otsoa F., De Urturi D.S., Rodríguez-Agudo R., Lachiondo-Ortega S., Mercado-Gomez M., et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol. Metabol. 2021;53:101275. doi: 10.1016/j.molmet.2021.101275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ramachandran P., Matchett K.P., Dobie R., Wilson-Kanamori J.R., Henderson N.C. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020;17:457–472. doi: 10.1038/s41575-020-0304-x. [DOI] [PubMed] [Google Scholar]
- 92.Tacke F., Zimmermann H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 93.Carestia A., Kim S.J., Horling F., Rottensteiner H., Lubich C., Reipert B.M., Crowe B.A., Jenne C.N. Modulation of the liver immune microenvironment by the adeno-associated virus serotype 8 gene therapy vector. Mol. Ther. Methods Clin. Dev. 2021;20:95–108. doi: 10.1016/j.omtm.2020.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J., et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/nejmoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qing K., Mah C., Hansen J., Zhou S.Z., Dwarki V., Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999;5:71–77. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- 96.Lin N., Chen S., Pan W., Xu L., Hu K., Xu R. NP603, a novel and potent inhibitor of FGFR1 tyrosine kinase, inhibits hepatic stellate cell proliferation and ameliorates hepatic fibrosis in rats. Am. J. Physiol. Cell Physiol. 2011;301:C469–C477. doi: 10.1152/ajpcell.00452.2010. [DOI] [PubMed] [Google Scholar]
- 97.Liu X., Hu H., Yin J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26:8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 98.Zeisberg M., Hanai J.i., Sugimoto H., Mammoto T., Charytan D., Strutz F., Kalluri R. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 99.Izumi N., Mizuguchi S., Inagaki Y., Saika S., Kawada N., Nakajima Y., Inoue K., Suehiro S., Friedman S.L., Ikeda K. BMP-7 opposes TGF-beta 1-mediated collagen induction in mouse pulmonary myofibroblasts through Id2. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290:L120–L126. doi: 10.1152/ajplung.00171.2005. [DOI] [PubMed] [Google Scholar]
- 100.Kinoshita K., Iimuro Y., Otogawa K., Saika S., Inagaki Y., Nakajima Y., Kawada N., Fujimoto J., Friedman S.L., Ikeda K. Adenovirus-mediated expression of BMP-7 suppresses the development of liver fibrosis in rats. Gut. 2007;56:706–714. doi: 10.1136/gut.2006.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lakner A.M., Steuerwald N.M., Walling T.L., Ghosh S., Li T., McKillop I.H., Russo M.W., Bonkovsky H.L., Schrum L.W. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology. 2012;56:300–310. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368:1106–1118. doi: 10.1016/s0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 103.Yang N., Mahato R.I. GFAP promoter-driven RNA interference on TGF-β1 to treat liver fibrosis. Pharm. Res. (N Y) 2011;28:752–761. doi: 10.1007/s11095-011-0384-y. [DOI] [PubMed] [Google Scholar]
- 104.Mederacke I., Hsu C.C., Troeger J.S., Huebener P., Mu X.R., Dapito D.H., Pradere J.P., Schwabe R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang P., He Z., Ji S., Sun H., Xiang D., Liu C., Hu Y., Wang X., Hui L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386–389. doi: 10.1038/nature10116. [DOI] [PubMed] [Google Scholar]
- 106.Pipe S., Leebeek F.W.G., Ferreira V., Sawyer E.K., Pasi J. Clinical considerations for capsid choice in the development of liver-targeted AAV-based gene transfer. Mol. Ther. Methods Clin. Dev. 2019;15:170–178. doi: 10.1016/j.omtm.2019.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nam H.J., Lane M.D., Padron E., Gurda B., McKenna R., Kohlbrenner E., Aslanidi G., Byrne B., Muzyczka N., Zolotukhin S., Agbandje-McKenna M. Structure of adeno-associated virus serotype 8, a gene therapy vector. J. Virol. 2007;81:12260–12271. doi: 10.1128/jvi.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nidetz N.F., McGee M.C., Tse L.V., Li C., Cong L., Li Y., Huang W. Adeno-associated viral vector-mediated immune responses: understanding barriers to gene delivery. Pharmacol. Ther. 2020;207:107453. doi: 10.1016/j.pharmthera.2019.107453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gil-Farina I., Fronza R., Kaeppel C., Lopez-Franco E., Ferreira V., D'Avola D., Benito A., Prieto J., Petry H., Gonzalez-Aseguinolaza G., Schmidt M. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Mol. Ther. 2016;24:1100–1105. doi: 10.1038/mt.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chandler R.J., LaFave M.C., Varshney G.K., Trivedi N.S., Carrillo-Carrasco N., Senac J.S., Wu W., Hoffmann V., Elkahloun A.G., Burgess S.M., Venditti C.P. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest. 2015;125:870–880. doi: 10.1172/jci79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Donsante A., Miller D.G., Li Y., Vogler C., Brunt E.M., Russell D.W., Sands M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]