

ABSTRACT
Mismatch repair-deficient (dMMR) tumors show a good response toward immune checkpoint inhibitors (ICI), but developing resistance impairs patients’ outcomes. Here, we compared the therapeutic potential of an α-PD-L1 antibody with the CDK4/6 inhibitor abemaciclib in two preclinical mouse models of dMMR cancer, focusing on immune-modulatory effects of either treatment. Abemaciclib monotherapy significantly prolonged overall survival of Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1-cre) mice (Mlh1−/−: 14.5 wks vs. 9.0 wks (α-PD-L1), and 3.5 wks (control); Msh2loxP/loxP;TgTg(Vil1-cre): 11.7 wks vs. 9.6 wks (α-PD-L1), and 2.0 wks (control)). The combination was not superior to either monotherapy. PET/CT imaging revealed individual response profiles, with best clinical responses seen with abemaciclib mono- and combination therapy. Therapeutic effects were accompanied by increasing numbers of tumor-infiltrating CD4+/CD8+ T-cells and lower numbers of M2-macrophages. Levels of T cell exhaustion markers and regulatory T cell counts declined. Expression analysis identified higher numbers of dendritic cells and neutrophils within tumors together with high expression of DNA damage repair genes as part of the global stress response. In Mlh1−/− tumors, abemaciclib suppressed the PI3K/Akt pathway and led to induction of Mxd4/Myc. The immune-modulatory potential of abemaciclib renders this compound ideal for dMMR patients not eligible for ICI treatment.
GRAPHICAL ABSTRACT
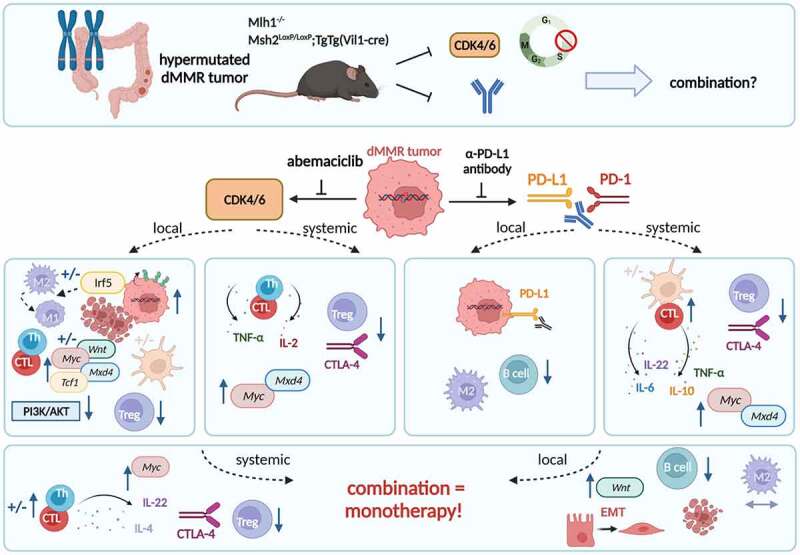
Two genetic mouse models of spontaneous dMMR-driven tumorigenesis were used in this preclinical therapy trial. Tumor-bearing Mlh1− or Msh2loxP/loxP;TgTg(Vil1−cre) mice received the CDK4/6 inhibitor abemaciclib, the α-PD-L1 monoclonal antibody or a combination of both agents, in which abemaciclib was given as lead-in therapy. Abemaciclib led to an increase of tumor-infiltrating T helper (Th) cells and cytotoxic T cells (CTL), dendritic cells, as well as IRF5-driven polarization of M2 macrophages, upregulation of Mxd4, and activation of the Wnt-signaling. Blood phenotyping revealed reduced levels of T cell exhaustion markers as well as regulatory T cell counts, attributable to Tc-mediated IL-2 secretion. While monotherapy with the α-PD-L1 monoclonal antibody had slightly different local and systemic effects, the combination approach was not superior to either monotherapy. Notably, most beneficial abemaciclib-mediated immune-modulatory effects were even repealed. Instead, the combination triggered epithelial-mesenchymal transition (EMT), raising caution for CDK-immune-checkpoint-inhibitor combination approaches. ± indicates strain-specific differences.
Introduction
Mismatch-repair-deficiency (dMMR) arises as a consequence of MLH1 gene promoter hypermethylation (= sporadic form) or secondary to a germline MMR mutation (= hereditary form). In either case, dMMR is associated with high immunogenicity. Patients harboring dMMR tumors are thus predestined to be treated with immune-checkpoint inhibitors (ICIs). ICIs reactivate exhausted T cells, prevent T cell inhibition, and force T cell-mediated tumor killing.1 Consequently, Programmed death 1 (PD-1) blockade has emerged as a highly effective treatment strategy and the positive results seen in many patients have contributed to the FDA and EMA approval for first-line therapy of unresectable or metastatic dMMR cancer.2,3
Recent data describe superior effects of α-PD-L1 antibodies compared to α-PD-1 antibodies in blocking PD-1/PD-L1 signaling.4 PD-L1 is upregulated in the tumor microenvironment and is found in a large variety of tumors. In recent clinical trials on dMMR patients, the α-PD-L1 antibody Avelumab has first proven safe5 and was then compared with standard chemotherapy for treatment of dMMR colorectal cancer patients.6 Clinical responses up to complete remissions were seen in many cases. Our preclinical data additionally support the use of α-PD-L1 antibodies either alone or in conjunction with cytostatic drugs and immunotherapy.7,8 Still, in most cases, resistance mechanisms evolve finally contributing to relapse.9
To improve outcomes, ICIs should be combined with other (targeted) agents. A very attractive option is the selective cyclin-dependent kinase 4/6 inhibitor (CDKI) abemaciclib.10 Abemaciclib stops the cell cycle at G1, induces swollen and dysfunctional lysosomes, and triggers apoptosis/necrosis in tumor cells.11–13 This CDK4/6 inhibitor is currently approved for treatment of both early and advanced/metastatic breast cancer, however, more immunogenic tumor types, such as dMMR-related cancers, should be considered as well. The rationale for this is based on the increasing body of evidence that abemaciclib stimulates antitumor immune responses by inducing an “inflamed” microenvironment finally contributing to T cell activation and improved T cell function.14–17 Actually, a recent study reported transformation of CD8+ T cells into memory cells upon CDK4/6 inhibition to expand the long-term immunity and efficacy of the cancer treatment – an effect frequently seen after ICI treatment.18 CDK blockade may thus become an interesting option for dMMR patients not eligible for ICI treatment. The notion that many patients harbor pre-formed antitumoral immune responses that can be re-activated (or boosted) by immunotherapy additionally argues in favor of using immune-modulating CDKIs for treatment of dMMR-related malignancies.19–21
Using the two preclinical Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mouse models of spontaneous tumorigenesis,22–25 we initiated a proof-of-concept study and compared the efficacy of a murine α-PD-L1 antibody (clone 6E11) with the CDK4/6 inhibitor abemaciclib either alone or in combination.
We could show that low-dose abemaciclib treatment is as effective as α-PD-L1 therapy, but its combination is not superior to the respective monotherapy. Hence, we propose abemaciclib monotherapy as a good alternative for treatment of dMMR patients that cannot be treated with ICI.
Methods
In vitro experiments
Cell culture
Two MLH1−/− tumor cell lines 328 and A7450 T1 M1 were established and characterized in our lab.25,26 Cell culture was performed in DMEM/Ham’s F12 medium, supplemented with 10% FCS (fetal calf serum), 6 mM Glutamine, and antibiotics (all from Biochrom, Berlin, Germany). Treatment was done with selected CDKIs at doses corresponding to IC30 values: (I) abemaciclib (= abema): 1 µM; (II) dinaciclib (= dina): 100 nM; (III) THZ-1: 0.83 µM. Doses were validated before via dose response curve analyses.
Apoptosis/necrosis, cell cycle analysis, and immunogenic cell death
Cells were treated with abema, dina, or THZ-1 for 72 hours, harvested and stained with 0.2 µM Yo-Pro 1 iodide (Thermo Scientific, Ex/Em 491/509 nm; blue laser 488 nm, 20 min, RT). Cells were washed and mixed with 7-AAD viability staining solution (250 ng, Biolegend, San Diego, United States) before measurements. For cell cycle analysis, cells were harvested and incubated with ice-cold 70% ethanol over night at −20°C. 1 mg/ml RNase (Carl Roth, Karlsruhe, Germany) was preheated at 37°C for 90 min. Ethanol fixed cells were washed twice with 2 ml PBS and centrifuged at 300 g for 10 min. Each sample was mixed with 500 µl of preheated RNase and incubated for 45 min in the water bath at 37°C. Cells were washed twice with 2 ml PBS and centrifuged at 300 g for 10 min. Cells were resuspended with 400 µl propidium iodide (50 µg/ml) and incubated for at least 30 min in the fridge covered from light.
For immunogenic cell death assessment, cells were treated with abemaciclib for 48 h and 72 hours. Supernatants were collected and amounts of high-mobility group protein 1 (HMGB1) were measurement by ELISA according to the manufacturers’ instructions (Abbexa, Cambridge, UK). Cells were harvested and incubated with a polyclonal rabbit CalR primary antibody (1:50; Abgent, San Diego, CA, USA, 30 min, 4°C), followed by a secondary FITC-labeled antibody (donkey anti rabbit, 1:50; Biolegend, 30 min, 4°C). Control cells were stained with the secondary FITC-labeled antibody. Measurements were performed on a FACSVerse Cytometer (BD Pharmingen, San Diego, USA). Data analysis was performed using BD FACSuite software (BD Pharmingen).
Colony formation assay
Five hundred cells/well were seeded in 6-well plates and were allowed to rest overnight. Cells were treated with abema, dina or were left untreated. After 6 days, medium was removed and cells were stained with 500 µl 0.2% crystal violet for 10 min on a rocking plate, followed by washing steps. Some cells were allowed to rest with medium for additional 6 days. Then, they were stained the same way with crystal violet. The number of colonies was evaluated using ImageJ-win64.
Co-culture assay
Tumor cells were harvested and stained with 5 µM CMFDA (15 min, 37°C, 5% CO2). Twenty-thousand cells/well were seeded in a 24-well plate. After 24 hours, 200,000 blood cells/well, which were lysed with erythrocyte lysis buffer (155 mM NH4Cl (MERCK Millipore, Darmstadt, Germany), 10 mM KHCO3 (MERCK Millipore), and 0.1 mM EDTA (Applichem, Darmstadt, Germany)) were added. Abemaciclib was used at 1 µM. After 24 hours, 10 µg/ml α-PD-L1 was added. After additional 48 hours, tumor cells were harvested. To quantify residual tumor cells, fluorescent microsphere beads (1.4 × 105 beads/ml, size: 10 µm, Polyscience, Hirschberg an der Bergstrasse, Germany) were used. Measurements were performed on a FACSVerse Cytometer (BD Pharmingen). Data analysis was performed using BD FACSuite software (BD Pharmingen).
Immunofluorescence of cytoskeleton and ROS
The cytoskeleton and formation of reactive oxygen species (ROS) was visualized. Cells were treated twice with 1 µM abemaciclib for 72 hours and stained with 7.5 µM ROS Brite 670 (AAT Bioquest, CA, USA, 30 min, 37°C) followed by 125 nM Mitolite Green (AAT Bioquest) staining (45 min, 37°C). Cells were fixed with 4% PFA (30 min, RT), washed thrice with PBS and permeabilized with 0.2% TritonX-100 (15 min, RT). Afterward, Phalloidin-iFluor 594 conjugate (AAT Bioquest) dissolved 1:1000 in 1% BSA was added (30 min, RT), followed by three washing steps and 2 min staining with 1.5 µg/ml DAPI.
In vivo experiments
Ethical Statement
The German local authority approved all animal experiments: Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg‐Vorpommern (7221.3‐1‐062/19; −025/20), under the German animal protection law and the EU Guideline 2010/63/EU. Mice were bred in the animal facility of the University Medical Center in Rostock under specific pathogen‐free conditions. Mlh1 genotyping was done according to [21] and Msh2 genotyping was done according to.22,23 During their whole life-time, all animals received enrichment in the form of mouse-igloos (ANT Tierhaltungsbedarf, Buxtehude, Germany), nesting material (shredded tissue paper, Verbandmittel GmbH, Frankenberg, Deutschland), paper roles (75 × 38 mm, H 0528–151, ssniff‐Spezialdiäten GmbH, Soest, Germany), and wooden sticks (40 × 16 × 10 mm, Abedd, Vienna, Austria). During the experiment, mice were kept in type III cages (Zoonlab GmbH, Castrop‐Rauxel, Germany) at 12‐h dark:light cycle, the temperature of 21 ± 2°C, and relative humidity of 60 ± 20% with food (pellets, 10 mm, ssniff‐Spezialdiäten GmbH, Soest, Germany) and tap water ad libitum. When mice were subjected to treatment (= time of tumor development), they were given daily-prepared soaked pellets to ensure proper food intake.
Experimental protocol
Mice with validated gastrointestinal tumor via PET/CT were taken into therapy. Mice received abemaciclib (oral gavage, 75 mg/kg body weight (bw), 1x/week, 8 times in total (q1wx8), Mlh1−/− n = 7 mice, Msh2loxP/loxP;TgTg(Vil1−cre) n = 9 mice) and α-PD-L1 antibody (i.p., 2.5 mg/kg bw, biweekly), three times in total (q2wx3), Mlh1−/− n = 7 mice, Msh2loxP/loxP;TgTg(Vil1−cre) n = 10 mice). The α-PD-L1 antibody, clone 6E11, was kindly provided by Genentech, a subsidiary of Roche, South San Francisco, USA. Mice receiving the combination were given abemaciclib first as lead-in, followed by α-PD-L1 injection (Mlh1−/− n = 7 mice, Msh2loxP/loxP;TgTg(Vil1−cre) n = 10 mice). Control mice were left untreated (Mlh1−/− n = 6 mice, Msh2loxP/loxP;TgTg(Vil1−cre) n = 10 mice) or received the isotype control antibody (i.p. 2.5 mg/kg bw, Mlh1−/− n = 7 mice, Msh2:loxP/loxP;TgTg(Vil1−cre) n = 7 mice). The health status was monitored daily using a score sheet. Reduction of suffering was ensured by applying humane endpoints (weight loss >15%, pain, changes in social behavior). All mice were sacrificed before they became moribund to prevent pain and distress. Mice were sacrificed due to human endpoints (as outlined above + progressive disease, defined by tumor volume >300 mm3), followed by removal of blood, spleen, and tumor.
PET/CT imaging
The tumor size was measured via PET/CT measurements on a small animal PET/CT scanner (Inveon PET/CT, Siemens Medical Solutions, Knoxville, TN, USA) in accordance to a standard protocol as described before.25 Mice were anesthetized using isoflurane and received FDG intravenously. Evaluation of the images was performed as described before.8 Treatment outcome determined according to clinical staging as follows: (I) progressive disease (PD) = tumor volume >25% vs. baseline (= day 0); (II) stable disease (SD): tumor volume similar to initial staging (≤25% vs. day 0); (III) partial response (PR): tumor volume 50% lower or more vs. baseline (=day 0). Follow-up was done at day 30 (= short-term) and, in some cases, at ~day 50 (= long-term follow-up).
Immune phenotyping
Blood was taken routinely from anesthetized mice (retrobulbar venous plexus). Spleen and tumor tissues were dissociated. Single cells were stained with a panel of conjugated monoclonal antibodies (mAb, 0.125 μg to 1.5 μg each). Zombie NIR™ Fixable Viability Kit by Biolegend (San Diego, United States) staining was performed following the protocol Zombie NIR™ Fixable Viability Kit by Biolegend, extracellular staining was performed following the protocol BD Horizon Briliant Stain Buffer (BD Bioscience), followed by lysis and intracellular staining using the protocol of True-Nuclear™ Transcription Factor Buffer Set by Biolegend. Measurements were performed on a spectral flow cytometer (Cytek™ Aurora). For extracellular stainings Gr1 Alexa Fluor700, CD8 FITC, CD4 APC Fire, CD11b BV570, PD-L1 BV421, NK1.1 BV605, CD19 Spark Blue (Biolegend), CD25 PerCP-eFluor710 (Thermofisher), CD83 BV750, PD-1 BV650 (BD Bioscience) and for intracellular stainings CTLA-4 PE/Cy7, CD3 PerCP, and Foxp3 Alexa Fluor 647 (Biolegend) were used. Data were analyzed using SpectroFlow™ Version 2.2.0.3. and FlowJo™ Version 10.6.1.
Multiplex cytokine assay
Cytokine levels of the plasma were measured using a multi-analyte flow assay kit following the instructions of the manufacturer (LEGENDplexTM, Biolegend). Measurements were performed on a spectral flow cytometer (Cytek™ Aurora). Data were analyzed using the manufacturer’s online Software. Absolute plasma cytokine levels are presented [ng/ml].
Fragment length analysis
Tumor gDNA was isolated using the Wizard Genomic DNA Purification Kit (Promega). An established panel of coding- and non-coding MS marker was evaluated using the fragment length analysis as described before.27
Nanostring targeted gene expression profiling
Tumor RNA was isolated from cryostat sections using the RNeasy Mini Kit (Qiagen). Then, the RNA was analyzed using Nanostring analysis as described before.8
Quantitative real-time PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen). 1 µg mRNA and 50 ng random Hexamer Primer were incubated for 10 min at 70°C. Sample-mixes were completed with 5x RT buffer complete, dNTPs and 200 units reverase. cDNA was synthesized using the PCR cycler 120 min at 45°C and for 10 min at 70°C. 25 ng cDNA was used for quantitative real-time PCR with the SensiFAST Probe Lo-ROX Kit (Bioline, Memphis, Tennessee, USA). Predesigned Taqman gene expression assays were used: 6-FAM-3ʹBHQ-1 Mxd4 (Mm00487523_m1), 6-FAM-3ʹBHQ-1 cMyc (Mm00487804_m1), 5-VIC-3ʹBHQ-1 Agr2 (Mm01291804_m1), 6-FAM-3ʹBHQ-1 Tgfb1 (Mm01178820_m1), 6-FAM-3ʹBHQ-1 Vimentin (Mm01333430_m1), 5-VIC-3ʹBHQ-1 N-Cadherin (Mm01162497_m1), 6-FAM-3ʹBHQ-1 Fpr2 (Mm00484464_s1), 5-VIC-3ʹBHQ-1 Csf1 (Mm00432686_m1), 6-FAM-3ʹBHQ-1 Csf2 (Mm01290062_m1), 5-VIC-3ʹBHQ-1 Tcf1/Pcbd2 (Mm01342270_m1), and 6-FAM-3ʹBHQ-1 Alox5 (Mm01182747_m1). Self-designed 5-VIC-3ʹBHQ-1 GAPDH was applied as housekeeping gene. Reaction was performed in the light cycler Viia7 (Applied Biosystems, Foster City, USA) with the following PCR conditions: 95°C for 10 min, 40 cycles of 15 s at 95°C, and 1 min at 60°C. All reactions were run in triplicates. The mRNA levels of target genes were normalized to GAPDH. Reactions were performed in triplicate wells. The expression level of each sample was considered by calculating 2−ΔCT (ΔCt = Cttarget – CtHousekeeping gene), followed by 2−ΔΔCT quantification, taking values of untreated controls as calibrator.
Immunofluorescence
Cryostat sections of 4 µm were fixed in cold pure methanol for 8 min, air-dried and unspecific binding site blocked (2% BSA, 2 h) followed by staining with Alexa Fluor 488, Alexa Fluor 594 and Alexa Fluor 647 labeled antibodies CD3, CD4, CD8, CD206, F4/80, CD11b, Gr1, PD-L1, PD-1, and Irf5 (Biolegend). Sections were washed and embedded in Roti Mount Fluor Care DAPI (Roth, Karlsruhe). Visualization was performed on a confocal laser scanning microscope (ZEISS Elyra 7 Confocal Laser Microscope, Zeiss, Jena, Germany). The infiltration pattern was quantified. For infiltrating CD3+CD4+ T helper cells and CD3+CD8+ cytotoxic T cells, numbers were counted in 2–3 high power fields (HPFs)/slide. For regulatory granulocytes and tumor-associated macrophages (TAM), the infiltration pattern was semi-quantitatively analyzed using a scoring system. 0 = no; 1 = mild (1–20 cells/HPF); 2 = moderate (21–40 cells/HPF); 3 = strong (>40 cells/HPF).
Statistics
GraphPad PRISM software, version 8.0.2 (GraphPad Software, San Diego, CA, USA) was used to perform statistical evaluation. All data are presented as mean + SEM. Data are depicted as scatter plots and bar charts, with individual values representing a single value of an individual mouse. Data showing baseline and follow-up are given as dots connected with a line. The value of significance was set to p < .05. The data were first tested for normality conducting Shapiro-Wilk test. Then, in case of normality, one-way ANOVA (Tukey´s multiple comparison) or unpaired T-Test was accomplished or in case of non-parametric data Kruskal-Wallis or U-Test was performed. Kaplan Meyer survival curves were analyzed using the log rank (Mantel Cox) test. In case of blood phenotyping, outliers were eliminated, when they were above or below the average plus/minus two times the standard deviation.
Dimensionality Reduction Analysis (t-SNE)
Individual fcs files were imported into FlowJo software (version 10.6.1) (FlowJo, Ashland, Oregon). Ten thousand cells per file (six files per treatment group concerning tumor data, and eight files per treatment group concerning blood and spleen data) were randomly selected and merged into one concatenated file. T-SNE algorithm provided by FlowJo software was performed only on gated live cells. The output was a t-SNE map which we show as a dot-plot. The t-SNE dimensions were used on the original gates to create the overlay t-SNE maps.
Results
In vitro effects of CDK blockade and α-PD-L1 treatment
Before performing animal experiments, the two murine Mlh1−/− tumor cell lines A7450 T1 M1 and 328 were used to evaluate the effects of different CDK inhibitors on cell cycle, proliferation, morphology, and immunogenic cell death (Figure 1a–e). In some experiments, the α-PD-L1 antibody was added and combinations were tested (Figure 1e, f).
Figure 1.
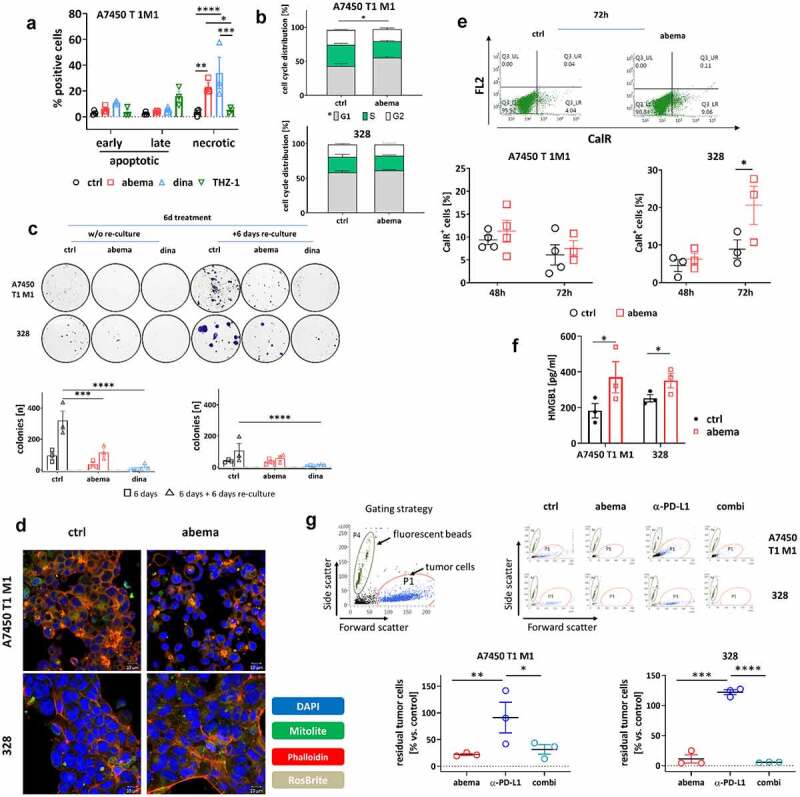
In vitro analysis on Mlh1−/− tumor cells. (a) Apoptosis/necrosis quantification. Mlh1−/− A7450 T1 M1 tumor cells were treated with CDKIs for 72 h and apoptosis/necrosis was quantified from Yo-Pro1/PI-stained cells. n = 3 independent experiments, *p < .05; ** p > .01; ***p < .001; two-way ANOVA (Tukey’s multiple comparisons test). (b) Cell cycle analysis after 48 h of treatment with abemaciclib. n = 4 independent experiments, *p < .05; t-test. (c) Colony formation assay after treatment with abemaciclib or dinaciclib. Two experimental conditions were studied: (i) 6 days treatment and direct analysis and (II) 6 days treatment + 6 days re-culture without (w/o) treatment (= reconvalescence). Thereafter, colonies were counted using ImageJ software. n = 3 independent experiments, *p < .05; ***p < .001; ****p < .0001; two-way ANOVA (Tukey’s multiple comparisons test). (d) Detection of ROS (RosBrite), cytoskeleton (Phalloidin) and mitochondria (mitolite) after 48 h of treatment with abemaciclib. Representative images are shown out of n = 3 independent experiments. Read out was done with the ZEISS Elyra 7 Confocal Laser Microscope (Zeiss). Original magnification 400 x. (e) Flow cytometric measurement of CalR-positive cells after 48 h and 72 h of treatment. n = 3–4 independent experiments. (f) HMGB1 secretion after 72 h of treatment. HMGB1 levels were determined from supernatants of MLH1−/− tumor cells. Control cells were left untreated. Experiments were repeated three times each of them performed in duplicates. *p < .05. (g) Co-culture of tumor and immune cells. Tumor and immune cells were simultaneously treated for 1 × 72 h with abemaciclib, α-PD-L1 antibody or a combination. The effector to target ratio was 1:10. Residual tumor cells were counted by adding fluorescent beads. Read out was done via flow cytometry. Representative dot plots of tumor cells treated with immune cells and drugs are shown. n = 3 independent experiments, *p < .05; **p < .01, ***p < .001; ****p < .0001, One-way ANOVA (Tukey’s multiple comparisons test). (a-g) All data are given as mean + SEM. Doses used in each experiment are as follows: abemaciclib: 1 µM, dinaciclib: 0.1 µM, THZ-1: 0.83 µM; α-PD-L1 antibody: 10 µg/ml.
The selective CDK4/6 inhibitor abemaciclib and the global CDK1/2/5/9 inhibitor dinaciclib significantly increased the number of necrotic cells. By contrast, selective CDK7 inhibition by THZ-1 did not induce significant cell death (Figure 1a). Abemaciclib led to a G1-arrest in A7450 T1 M1 cells, whereas in 328, the number of cells in the G1-phase was slightly reduced (Figure 1b). Upon dinaciclib, the number of cells in S-phase increased in both cell lines. Using a classical colony formation assay, the number of A7450 T1 M1 colonies was reduced after abemaciclib and even more pronounced after dinaciclib treatment (Figure 1c), both after 6 days of treatment and significantly after additional 6 days of rest.
Then, we focused on abemaciclib for further studies. This agent affected the cytoskeleton and reduced mitochondria of A7450 T1 M1 cells. By contrast, reactive oxygen species remained unchanged (Figure 1d). In 328, we did not observe such strong changes in colony formation, cytoskeleton, and mitochondria, but reactive oxygen species were slightly decreased. The amount of CalR+ A7450 T1 M1 cells was comparable to controls, while in 328 cells, surface-bound CalR was more abundant after 72 hours of abemaciclib treatment (Figure 1e). Likewise, HMGB1 levels significantly increased, indicative for induction of immunogenic cell death (figure 1f).
In a subsequent co-culture assay, the impact of immune cells on the tumor cell viability was investigated (Figure 1g). Abemaciclib alone and its combination with α-PD-L1 reduced tumor cell numbers significantly.
Taken together, CDKIs have individual effects on MMR-D tumor cells. The immune-stimulating potential of abemaciclib may interact with immune-checkpoint blockade. To address this further, we initiated a proof-of-concept in vivo therapy trial.
Prolonged survival and effective tumor growth control under mono- and combination therapy
We included two preclinical mouse models of spontaneous, dMMR-driven tumorigenesis. Mlh1−/− mice harbor a constitutional knock out, whereas in Msh2loxP/loxP;TgTg(Vil1−cre) mice, a conditional knock out in the gut is the driver for tumor formation.23,25,28 These mice share some features commonly related to dMMR, such as a high tumor mutational burden, and an inflamed tumor microenvironment. However, the infiltration pattern of specific immunological subtypes differs, nicely reflecting the clinical presentation of dMMR-related cancer (Table 1). Hence, these models are ideal tools for preclinical response analysis.
Table 1.
Overview on similarities and disparities between Mlh1−/− and Msh2loxP/loxP Villin Cre mice
| Characteristic | Mlh1−/− | Msh2loxP/loxP Villin Cre | ||
|---|---|---|---|---|
| Strain specificity and model establishment | Mouse | Mouse strain | C57Bl/6 | C57Bl/6 |
| Type of knock out | Constitutional | Conditional, gut | ||
| Spontaneous tumor formation | Yes | Yes | ||
| Clinicopathological characteristics | Human | Frequency of the underlying MMR defect in human | ~35 % | ~ 40% |
| Human disease counterpart | Lynch syndrome and constitutional mismatch repair deficiency | Lynch syndrome | ||
| Human lifetime risk of tumorigenesis | ♂: 27 – 74 % | ♂: 27 – 74 % | ||
| ♀: 22 – 53 % | ♀: 22 – 53 % | |||
| Mouse | Murine mean age of onset | 26 weeks (lymphoma), 35 weeks (GIT) | 35 weeks (GIT) | |
| Tumor penetrance | High, >90 % | High, >95 % | ||
| Tumor spectrum | LS-associated tumors ≥ hematological malignancies (lymphoma) > others (skin) | LS-associated tumors | ||
| Manifestation | Jejunum | Jejunum | ||
| Metastastic spread | Not in GIT, lymphomas: yes | No | ||
| MHC class I expression | 100% positive | 100% positive | ||
| MSI in mononucleotide repeats | High | High | ||
| MSI in coding mononucleotide repeats | Low | High | ||
| Tumor microenvironment | Mouse | Cytotoxic T-cells | Low | Moderate |
| T helper cells | Low | Low | ||
| MDSC | Moderate | Moderate | ||
| TAM | Moderate | High | ||
| PD-L1 | Low | Moderate | ||
| Treatment response towards | Immune checkpoint inhibition (α-PD-L1) | Poor > moderate | Moderate | |
| CDK4/6 inhibition | Good | Good | ||
| Combined ICI/CDKI | Missing | Missing | ||
| Immunological effects upon CDKI treatment | Circulation | IL-2 and TNF-α secretion; Treg, B cells ; MDSC | IL-2, TNF-α, IL-6 secretion, IL-10 secretion; B cells,Treg, CTLA-4+ cells ; MDSC | |
| Spleen | Mxd4 | Treg, CTLA-4+ cells | ||
| Tumor | Mxd4, cMyc, TGF-b; CTLA-4+ cells; DC myeloid cells ; TAM ; | Agr2 ; B cells; Treg ; TAM | ||
| Pathway alteration upon CDKI treatment | DNA Damage repair | Induced | Unaffected | |
| PI3K-Akt | Downregulated | Unaffected | ||
| Wnt signaling | Activated | Unaffected | ||
| Epigenetic regulation | Downregulated | Induced | ||
| JAK-STAT signaling | Downregulated | Induced | ||
| EMT | Activated (boosted in the combination) | Not induced | ||
TIL - tumor-infiltrating lymphocytes; GIT - gastrointestinal tumor; MDSC - myeloid-derived suppressor cells; TAM - tumor-associated macrophages; CDK - cyclin-dependent kinase; CDKI - cyclin-dependent kinase inhibitor; ICI - immune checkpoint inhibition; DC - dendritic cells; EMT - epithelial-mesenchymal transition.
Treatment with abemaciclib was given weekly at a dose of 75 mg/kg bw because of its therapeutic activity in vitro even after drug removal and its capacity to stimulate the immune system.14,16,18,29 The treatment schedule of the α-PD-L1 antibody was adopted to our previous study8 with a biweekly application route at a dose of 2.5 mg/kg bw. Tumor growth was monitored in both mouse models before and after treatment using18F-FDG PET/CT (please see Figure 2a for experimental protocol).
Figure 2.
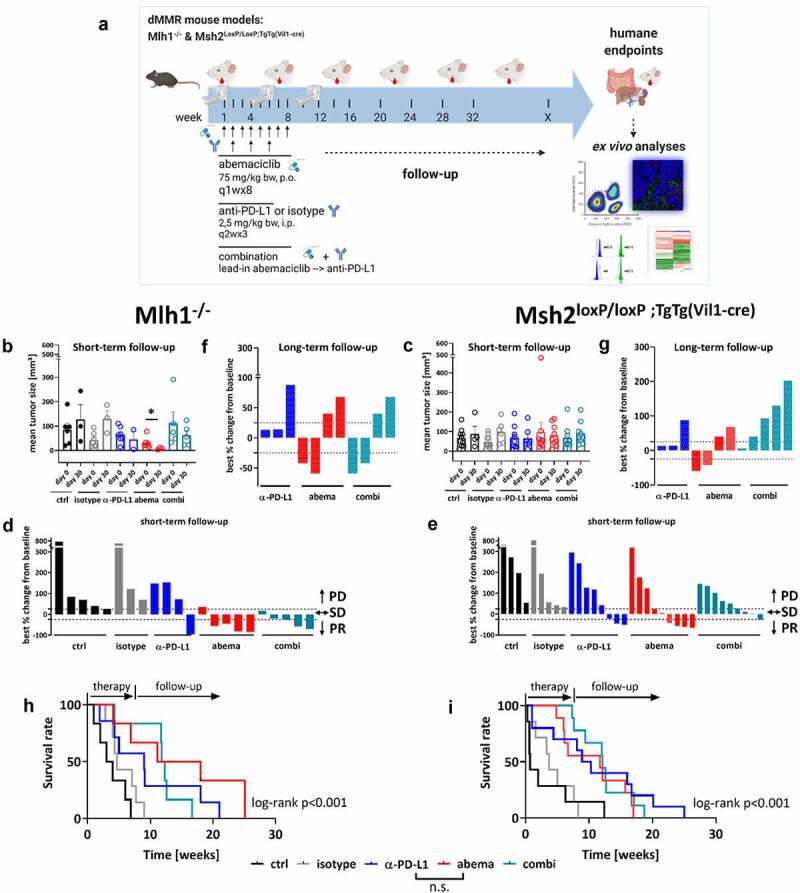
Treatment schedule, longitudinal18F-FDG PET/CT imaging in vivo and overall survival of Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. (a) Experimental protocol. Mice with gastrointestinal tumors were conducted to mono- or combination therapy. Abemaciclib: 1x/week, 8 times in total (q1wx8); α-PD-L1: biweekly, 3 times in total (q2wx3); combination: abemaciclib first (=lead-in), followed by α-PD-L1 injection. (b – g) Baseline and follow-up PET/CT imaging of individual mice. (b, c) Data are presented in column (each dot stands for an individual mouse). (D, E, F, G) data are presented as best % change from baseline according to clinical definitions and depicted for each individual mouse either after short-term (day 30) (d, e) or long-term follow-up (~day 50) (f, g); PD – progressive disease (tumor volume > 25% vs. baseline), SD – stable disease (tumor volume similar to initial staging (≤ 25% vs. day 0); PR – partial response (tumor volume 50% lower or more vs. baseline). (h, i) Kaplan-Meier survival curve. Mlh1−/−: isotype vs. α-PD-L1: p < .05; control vs. abemaciclib: p < .01; control vs. combination: p < .01; ctrl: n = 6, isotype: n = 7, α-PD-L1 n = 7, abemaciclib: n = 6, combination n = 6. Msh2loxP/loxP VillinCre: isotype vs. α-PD-L1: p < .05; control vs. abemaciclib: p < .01; control vs. combination: p < .001. ctrl: n = 10, isotype: n = 7, α-PD-L1 n = 10, abemaciclib: n = 9, combination n = 9. Log-rank analysis (Mantel Cox).
Monotherapy with α-PD-L1 and abemaciclib reduced tumor sizes in Mlh1−/− mice (Figure 2b, p < .05 abemaciclib vs. control). The combination approach, in which abemaciclib was given as lead-in, had comparable effects. Control and isotype-treated mice showed the expected increased tumor sizes. By dissecting the response rates in more detail, we identified partial response or stable disease in all mice receiving abemaciclib or the combination. By contrast, partial response was only seen in one case after α-PD-L1 monotherapy and in neither mouse of the control groups (Figure 2b, d).
The response pattern of Msh2loxP/loxP;TgTg(Vil1−cre) mice was comparable to those of Mlh1−/− mice (Figure 2c, e). Short-term follow-up was available for all mice receiving treatment, whereas, in the controls, only half of them underwent follow-up because of disease progression. 66.0% of mice had partial response or stable disease under abemaciclib and 44.4% mice under combination therapy (vs. 37.5% in the α -PD-L1 and neither in the control groups).
Long-term follow-up (> day 50) principally confirmed the good response toward abemaciclib in both mouse strains (figure 2f, g). Fifty percent of mice showed partial response. The α-PD-L1 primarily induced stable disease. The response pattern toward monotherapy was not confirmed in the combination. Here, 50% of Mlh1−/− mice had partial response, while all Msh2loxP/loxP;TgTg(Vil1−cre) mice suffered from progressive disease (figure 2f, g).
All three therapies significantly improved the outcome in both mouse strains (Mlh1−/−: p < .001, Msh2:loxP/loxP;TgTg(Vil1−cre) p < .001, Figure 2h,i). Median overall survival of Mlh1−/− mice receiving abemaciclib alone was 14.5 weeks and thus even better than under α-PD-L1 monotherapy (9.0 weeks). In Msh2loxP/loxP;TgTg(Vil1−cre) mice, both monotherapies yielded comparable outcomes (abemaciclib vs. α-PD-L1: 11.7 vs. 9.6 wks). Still, the combination was not superior to either monotherapy.
To check whether potential hepatotoxic effects of the applied regimens may account for treatment failure in the combination group, routine histology was done. This analysis revealed massive focal lymphocytic and granulocytic infiltration in livers from mice treated with abemaciclib or α-PD-L1 monotherapy (supplementary Figure S1, 63x magnification of single lymphocytes and granulocytes in the left corner, infiltrates are marked with a black arrow). While this was a likely result of the systemic immune stimulation, such strong lymphocytic infiltration was only partially preserved in the combination treatment, with single necrotic areas arising (black arrow). We conclude antagonistic instead of synergistic effects of combined CDK4/6 – immune-checkpoint blockade in these two preclinical dMMR tumor models.
Treatment-related immunological changes in the periphery and spleen
To get an idea on the mechanisms underlying this individual response pattern in vivo, plasma samples were taken from control and treatment groups at baseline (=before treatment) and after therapy (= experimental endpoint). A panel of cytokine markers was studied to cover the most relevant Th1 and Th2-specific cytokines and to track the changes under therapy (Figure 3).
Figure 3.
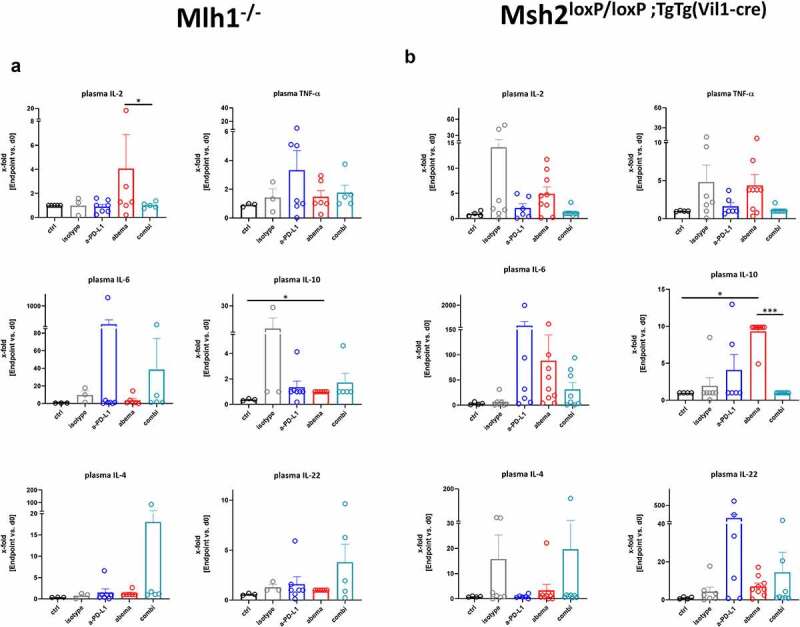
Cytokine levels of plasma from Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. (a) Mlh1−/− and (b) Msh2.loxP/loxP;TgTg(Vil1−cre) Plasma samples were collected before treatment (= baseline) and at the experimental endpoint. Cytokine levels were determined as described in material and methods. Given is the x-fold change of the indicated marker in comparison to day 0 (= baseline). Mlh1−/−: ctrl: n = 3, isotype: n = 3, α-PD-L1 n = 7, abemaciclib: n = 6, combination n = 5. Msh2loxP/loxP VillinCre: ctrl: n = 4, isotype: n = 7, α-PD-L1 n = 6, abemaciclib: n = 9, combination n = 8. *p < .05; ***p < .001, Kruskal-Wallis test (Dunn’s multiple comparisons test). (a-b) All data are given as mean + SEM.
Monotherapy with abemaciclib induced IL-2 secretion in Mlh1−/− mice, while the α-PD-L1 antibody evoked TNF-α and IL-6 release (Figure 3a). However, this did not reach statistical significance. In mice receiving the combination, we detected increased levels of IL-4 and IL-22. IL-10 was not altered under therapy. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, IL-2, TNF-α, IL-6, and IL-10 level were higher under abemaciclib treatment (Figure 3b). The α-PD-L1 monotherapy led to a strong increase of IL-6 and IL-22 (Figure 3b and supplementary Figure S2A). The combination triggered the release of IL-6 and IL-22 secretion, but not to a degree comparable to either monotherapy.
While these data already hinted toward individual effects on the immune system, we then focused on blood phenotyping to dissect the immunological changes in detail. Therefore, a panel of specific antibodies was used to quantify numbers of circulating T cell subpopulations (including exhausted and activated T cells), NK cells, B cells, and myeloid-derived suppressor cells (MDSC) via flow cytometry (Figure 4, supplementary Figure S2B, C).
Figure 4.
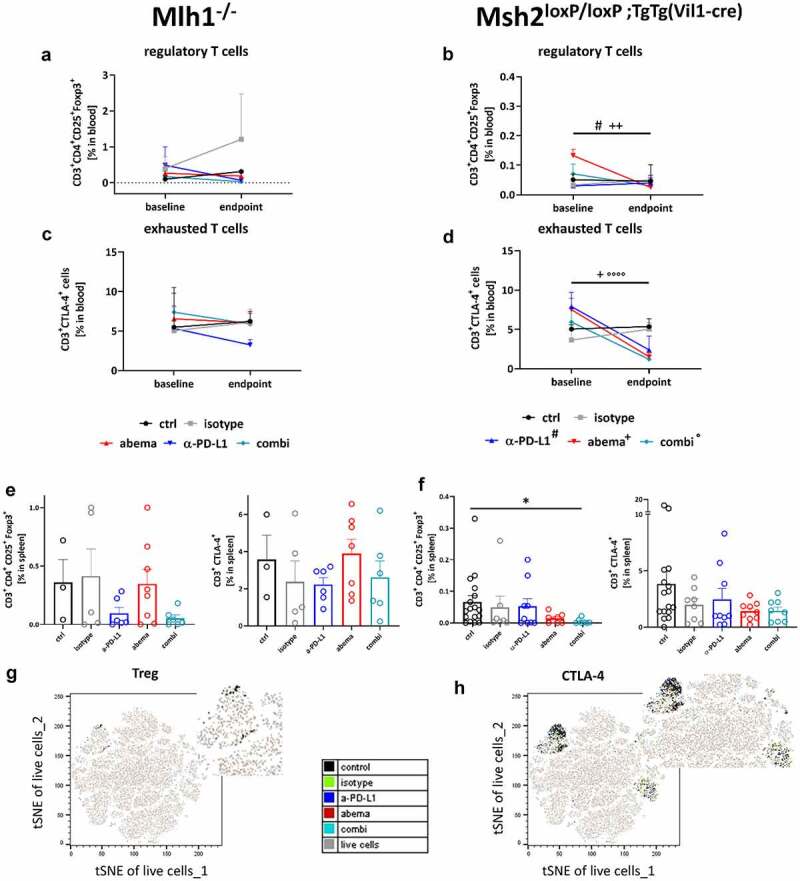
Spectral flow cytometry of peripheral blood and spleens from Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. (a – d) Blood phenotyping. Given is the number of % immune cells before treatment (= baseline) and at the experimental endpoint resulting from 100,000 events measured on a flow cytometer. Mlh1−/− n = 3–8 mice/group, Msh2loxP/loxP;TgTg(Vil1−cre) n = 7–9 mice/group. #p < .05 endpoint vs. day 0 α-PD-L1; +p < .05 endpoint vs. day 0 abemaciclib; ++p < .01 endpoint vs. day 0 abemaciclib, °°°°p < .0001 endpoint vs. day 0 combination. Kruskal-Wallis test (Dunn’s multiple comparisons test). (e – h) Spleen phenotyping. Given is the number of % immune cells at the experimental endpoint resulting from 100,000 events measured on a flow cytometer. *p < .05; Kruskal-Wallis test (Dunn’s multiple comparisons test). (g, h) tSNE plots showing single T cell subpopulations of spleens from Msh2loxP/loxP;TgTg(Vil1−cre) mice. The expression profile of the exhaustion marker CTLA-4 as well as Tregs were illustrated for the treatment and control groups, respectively.
This analysis identified decreased levels of regulatory T cells (Tregs) in Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice and in all treatment groups (Figure 4a, b). In Mlh1−/− mice, CTLA-4+ cells showed only reduced levels under α-PD-L1 mono- and combination therapy (Figure 4c). This was additionally seen for CD19+ cells (supplementary Figure S2B). In contrast, the percentage of MDSCs was highly increased in the combination. NK cells remained unchanged (supplementary Figure S2B).
Comparable effects on Tregs and exhaustion markers were evident in Msh2loxP/loxP;TgTg(Vil1−cre) mice (Figure 4b, d). Exemplarily shown for CTLA-4+ T cells, the percentage of circulating cells was reduced under all three treatments (Figure 4d). Notably, both monotherapies reduced the amounts of exhausted PD-1+ T cells (not shown). The MDSCs, B cells, and NK cells showed diverging results, with the former showing treatment-related increases (supplementary Figure S2C).
Next, we focused on splenic T cells (Figure 4e, f). In Mlh1−/− mice, Tregs were only reduced under a-PD-L1 mono – and combination treatment (Figure 4e). Abemaciclib alone did not change the amount of Tregs. Also, exhausted T cells remained unchanged under therapy. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, abemaciclib alone and in combination with α-PD-L1 reduced the numbers of Tregs and CTLA-4+ T cells (figure 4f), which is additionally illustrated as tSNE plot in Figure 4g,h Every dot represents one cell. Cells with similar surface markers are located next to each other. In the left upper part of the plot the Tregs are highlighted. As expected, they are all located very close in one cluster. Larger distances might be due to other expressed surface markers, which were not relevant for our gating of Tregs.
Treatment-related changes in the tumor microenvironment
The individual effects of either treatment on circulating immune cells were then studied in the local tumor microenvironment. Here, we quantified infiltrating T cells, regulatory CD11b+Gr1+PD-L1+ granulocytes, tumor-associated macrophages (TAMs), and IRF5+ M1 macrophages (Figure 5).
Figure 5.
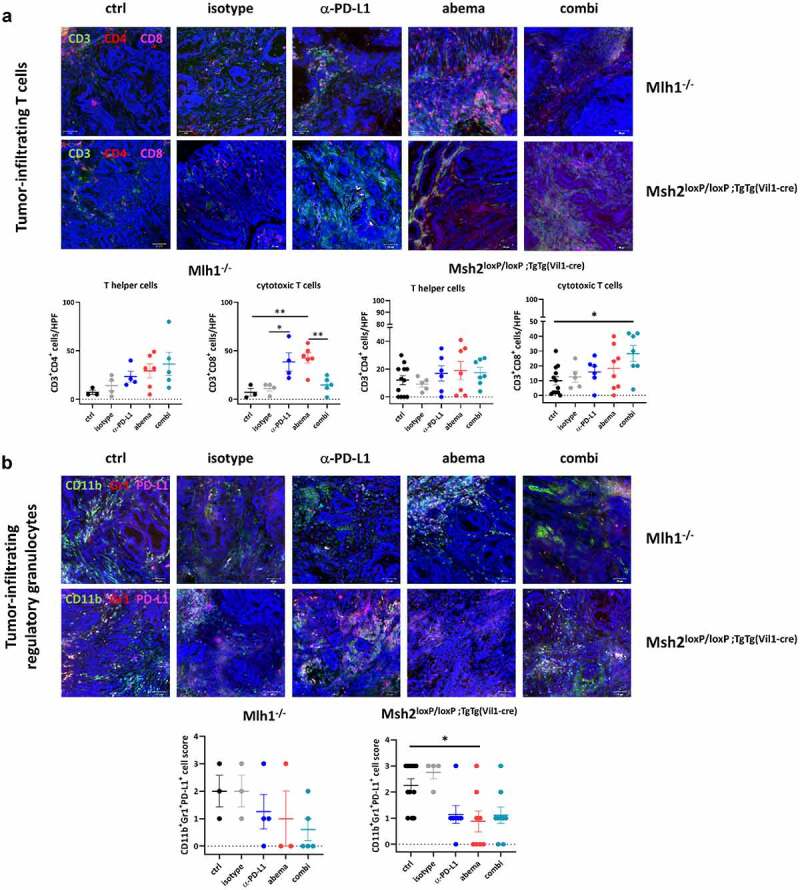
Immunofluorescence of tumor specimens from Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. Residual tumor slides were fixed, stained and embedded. Confocal laser scanning microscopy was done on a Zeiss Elyra 7 microscope. The infiltration pattern of T cells, regulatory and tumor-associated macrophages differed between individual treatment groups. In most cases, the differences reached statistical significance. Upper panel: representative images of tumor slides; lower panel: quantitative analysis of tumor-infiltrating immune cells. (a, d) Given is the number of infiltrating CD3+CD4+ T helper cells, CD3+CD8+ cytotoxic T cells and IRF5+ macrophages counted in 2–3 HPFs/slide with n = 3–10 mice/group. (b, c) The infiltration pattern was semi-quantitatively analyzed using a scoring system. 0 = no; 1 = mild; 2 = moderate; 3 = strong. Each symbol represents one case. *p < .05; **p < .01, Two-way ANOVA (Tukey’s multiple comparisons test).
Figure 5.
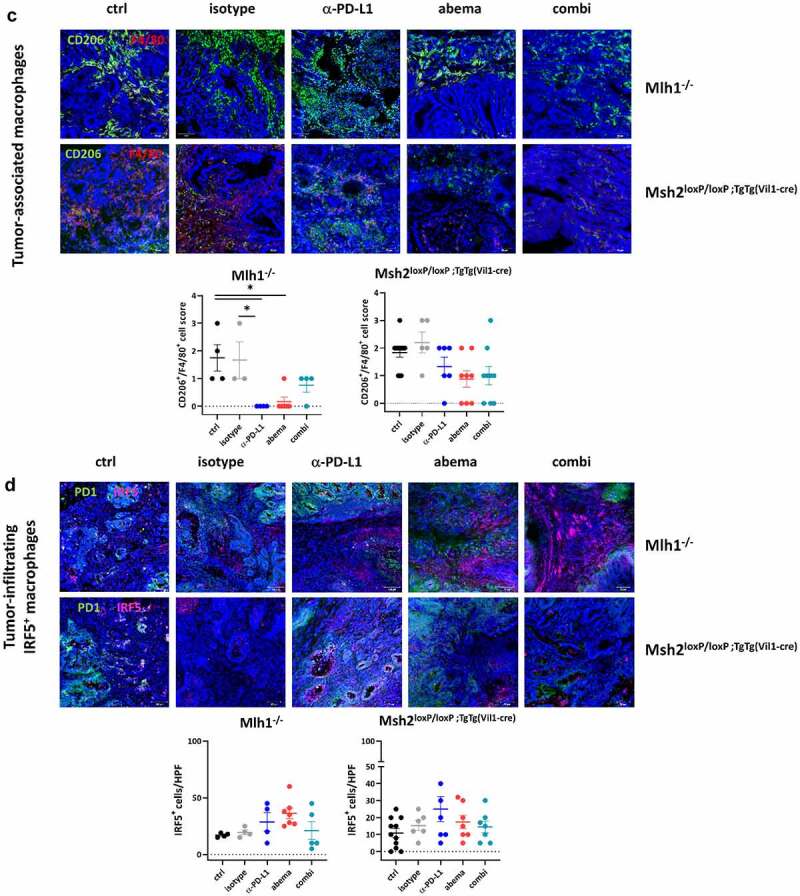
(Continued)
Abemaciclib and α-PD-L1 monotherapy triggered focal CD3+CD4+ and CD3+CD8+ T-cell infiltration, especially in Mlh1−/− mice (Figure 5a). Cytotoxic T cell numbers were even significantly elevated under abemaciclib and α-PD-L1 monotherapy in MLH1−/− mice, whereas in the combination treatment, this massive immune stimulation was partially abrogated (Figure 5a). In Msh2loxP/loxP;TgTg(Vil1−cre) mice, infiltrating T cell numbers increased upon treatment (Figure 5a). The overall higher infiltration with T cells was accompanied by decreased numbers of regulatory granulocytes and TAMs in both mouse strains (Figure 5b, c). Quite in line, IRF5, which leads to M1 polarization, was more abundant in abemaciclib-treated tumors and additionally slightly higher upon α-PD-L1 blockade (Figure 5d). This positive immunomodulatory effect was negated almost completely in both mouse strains receiving the combination.
Supplemental flow cytometric assessment of MLH1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) tumors identified decreased numbers of Tregs in the combination groups (supplementary Figure S3). The amount of CTLA-4+ T cells increased in tumors of Mlh1−/− mice upon abemaciclib mono- and combination therapy, but remained unchanged in Msh2loxP/loxP;TgTg(Vil1−cre) tumors.
To get a detailed overview of the tumor-infiltrating leukocyte (TIL) compartments and identify treatment-related pathway alterations in-depth, the PanCancer IO 360 Gene Expression Panel was applied (Figure 6 and supplementary Figure S4). B cell levels decreased under all therapies in both mouse strains. In Mlh1−/− mice, abemaciclib increased the level of cytotoxic T cells within the TIL compartment. Dendritic cell (DC) and neutrophil levels were contrary between the two mouse strains. Abemaciclib mono- and combination therapy increased the level of both cell types in Mlh1−/− mice, while in Msh2loxP/loxP;TgTg(Vil1−cre) mice, it was the other way round.
Figure 6.
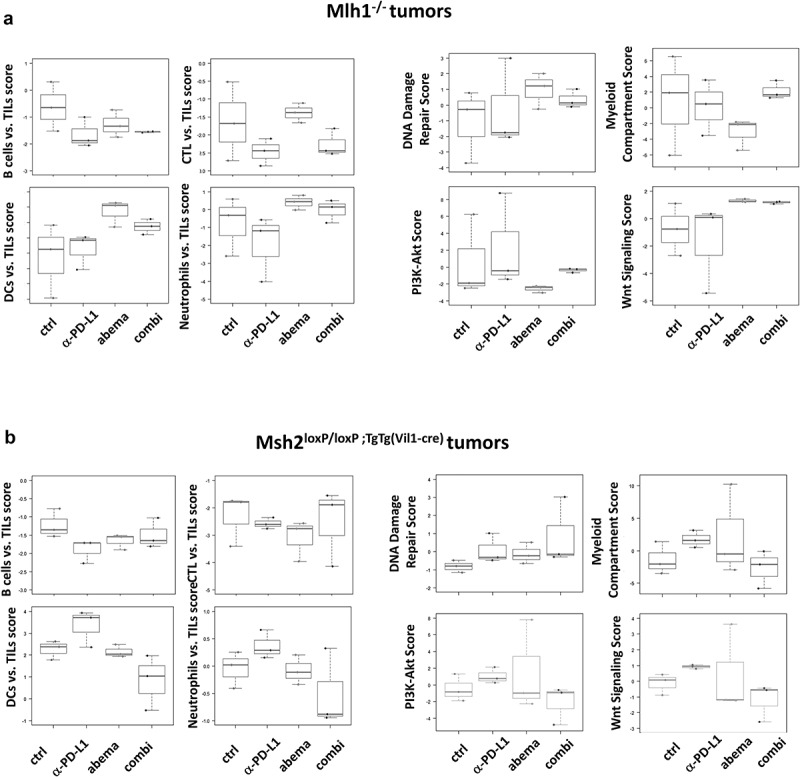
Nanostring gene expression analysis of tumors from Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. The PanCancer IO 360 Gene Expression Panel was applied. Relative abundances measuring various contrasts between cell types reported for each group. Data result from n = 3 samples/group.
DNA damage repair genes, such as Rad51, MGMT, and Exo1 were higher expressed under abemaciclib mono- and combination therapy in both mouse lines (Figure 6). A comparable effect was seen on Wnt signaling, which was highly activated in these two treatment groups, especially in Mlh1−/− mice. Vice versa, the myeloid compartment score as well as genes related to PI3K/Akt or Jak/STAT signaling were reduced (Figure 6, and supplementary Figure S4). Additionally altered genes included those involved in epigenetic regulation and hypoxia. Here again, Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice responded contrarily (supplementary Figure S4A,C). In the combination, these strong immune-modulatory effects were almost completely neutralized as illustrated in a heatmap (supplementary Figure S4B, D). Here, the contradictory effects of both monotherapies (abemaciclib, α-PD-L1) on cellular pathways are shown. In Mlh1−/− mice, abemaciclib led to a significant downregulation of most pathways (depicted in blue), but the α-PD-L1 antibody activated them (depicted in red). In the combination, these opposite effects were blunted. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, the differences were weaker, still, a comparable trend was seen for most pathway alterations, providing a likely explanation for the missing benefit in vivo.
Treatment-associated gene expression changes in the tumor microenvironment and spleen
The above findings indicated Wnt activation by abemaciclib as well as epigenetic modulation by either treatment. Since these mechanisms are drivers of epithelial-mesenchymal transition (EMT), we determined whether this may also play a role here and provide another explanation for the different treatment responses seen under mono- and combination therapy. Therefore, the expression levels of the EMT markers Tgfb1, Vimentin, N-Cadherin, and Fpr2 were studied by qPCR (Figure 7).
Figure 7.
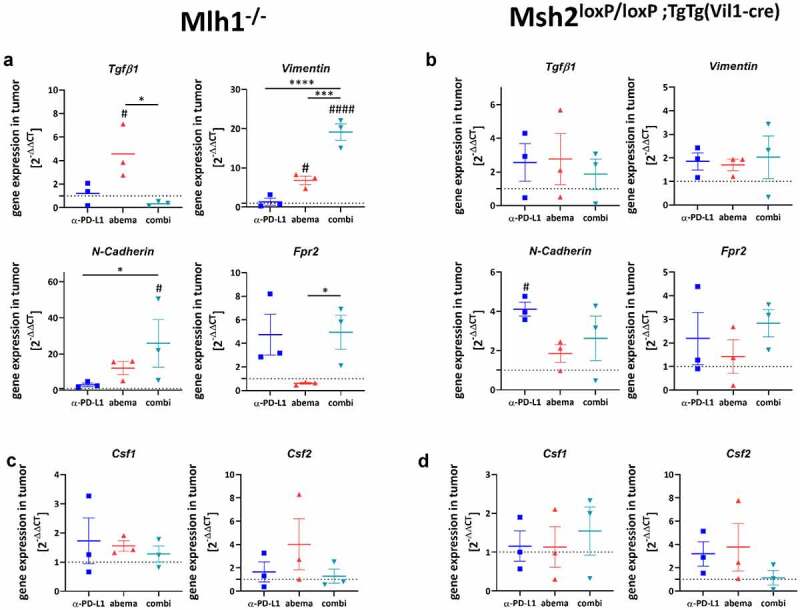
Expression levels of selected genes and functional immunological analysis. (a-f) Total RNA from tumors (A-F) and spleens (g, h) was reverse transcribed into cDNA and qPCR was done as described in material and methods. All data are given as 2-ΔΔCT values + SEM. Analysis was done in triplicates with n = 3 mice/group, respectively, * p < .05; *** p < .001; **** p < .0001; # p < .05; ## p < .01; #### p < .0001 vs. control. One-way ANOVA (Tukey’s multiple comparisons test) or Kruskal-Wallis test (Dunn’s multiple comparisons test).
Figure 7.
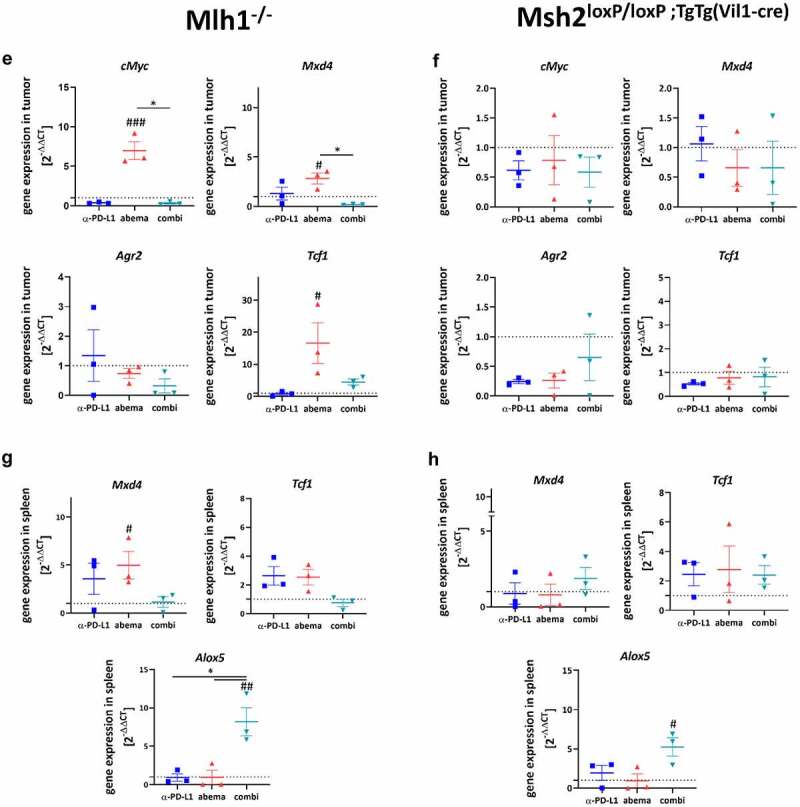
(Continued)
Monotherapy with abemaciclib upregulated Tgfb1, Vimentin, and N-Cadherin, but effectively suppressed Fpr2 in Mlh1−/− mice (Figure 7a). The α-PD-L1 antibody had no or opposite effects on gene expression. In mice receiving the combination, Vimentin, N-Cadherin, and Fpr2 were highly upregulated in residual tumors. The latter is a chemoattractant receptor of G-protein-coupled receptors, which, in conjunction with the EMT effectors Vimentin and N-Cadherin, triggers cancer invasion.30
In Msh2loxP/loxP;TgTg(Vil1−cre) mice, no such clear correlations were seen. Abemaciclib alone had no impact on EMT markers (Figure 7b). By contrast, α-PD-L1 treatment upregulated N-Cadherin and Fpr2. Heterogenous effects were seen in the combination group, showing slightly elevated expression levels of Vimentin, N-Cadherin, and Fpr2.
Then, the impact on macrophages was studied (Figure 7c, d). Csf1 and Csf2 were used as markers for macrophage polarization. Although statistical significance was not reached, we observed a trend toward a higher expression of Csf2 vs. Csf1 in both mouse strains treated with abemaciclib or α-PD-L1. Csf2 is associated with an M1-like phenotype, which supports our above findings on M1-polarization upon monotherapy. Notably, in both mouse models receiving the combination, Csf1 and Csf2 were comparable to controls.
Next, we checked genes related to cancer immunity and T cell activation (cMyc, Mxd4, Agr2, Tcf-1).18,31 Abemaciclib induced a significant upregulation of Myc and Mxd4 in Mlh1−/−, but not Msh2loxP/loxP;TgTg(Vil1−cre) tumors. Also, Tcf1, a transcription factor of the Wnt signaling pathway and Treg suppressor,32 was highly upregulated. Tcf-1 was recently identified in intratumoral memory CD8+ T cells with stem cell-like properties.31 Notably, no such changes were seen upon α-PD-L1 mono- or combination therapy (Figure 7e). The tumor-promoting anterior gradient-2 (Agr2) was found reduced in all mice (Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre)) receiving abemaciclib alone or in combination with α-PD-L1 (figure 7f).
These results prompted us to check for the expression of immunologically and functionally relevant genes in the spleen (Figure 7g, h). Abemaciclib, but not the other treatment regimens, induced Mxd4 and Tcf1 in spleens of Mlh1−/− mice. As anticipated, no significant alterations were detectable in Msh2loxP/loxP;TgTg(Vil1−cre) mice in either treatment. By contrast, Alox5, a neutrophil and macrophage marker with pro-inflammatory and tumor-promoting activity,33 was highly elevated in both mouse lines receiving the combination.
Finally, mutations in dMMR-specific target genes were examined (supplementary Figure S5). The overall mutation frequency in the tumor compared to normal tissue was slightly elevated under abemaciclib therapy in Mlh1−/− mice (supplementary Figure S5). In addition, abemaciclib was the only treatment that led to mutations in Mdm2 and Ncapd2 and also in combination therapy to mutations in Spen and FAS. For Taf1b, Kcnma1, and Rfc3 nearly all treatments triggered mutations. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, α-PD-L1 mono- and combination therapy led to slightly decreased mutation frequency, whereas for the displayed genes, all treatments induced mutations.
Discussion
Using two clinically relevant mouse models of spontaneous dMMR-driven tumorigeneses, we report that low-dose abemaciclib treatment is as effective as immune-checkpoint blockade, while the combination is not superior to either monotherapy.
Abemaciclib is approved for high-risk early and advanced/metastatic breast cancer.34 The underlying mode of action includes decreased cell proliferation and induction of senescence.13,35,36 Here, we also found increased numbers of apoptotic and necrotic cells, a G1-arrest, and impaired colony formation abilities of murine dMMR tumor cells. The latter effect was evident after several days of treatment rest, which is in line with abemaciclibs’ ability to suppress DNA synthesis even after drug removal.13 In a co-culture system of Mlh1−/− tumor and semi-autologous immune cells, abemaciclib boosted cytotoxic effects to an amount much higher than the therapeutic α-PD-L1 antibody. Hence, we confirm the strong immune-stimulating potential of this CDKI.14,15,37 This finding may expand the spectrum of tumors eligible for CDK inhibition.38
In a proof-of-concept in vivo trial, abemaciclib was given therapeutically to tumor-bearing Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. While both mouse strains responded to CDK4/6 inhibition, disease control was better in the former. Notably, 80% of mice underwent partial remission, finally resulting in significantly prolonged overall survival. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, the overall survival was similarly extended, but longitudinal PET/CT imaging yielded heterogeneous effects on tumor sizes. The survival benefit was comparable to PD-L1 blockade, which improved the outcome by several weeks. Noteworthy, in this context, is the fact that treatment was given once a week as opposed to most preclinical studies in which a daily treatment regimen is applied.12,36,37,39 The rationale for this dose reduction is based on the fact that dMMR tumors are highly immunogenic per se and pre-formed immune responses may exist.19,20,40 Accordingly, the primary aim was to re-activate the immune system rather than inducing de novo T cell immune responses.14,16,18,29 With this reduced dosing schedule, abemaciclib still triggered immune modulation, characterized by enhanced secretion of Th1 and Th2-specific cytokines. This was accompanied by reduced numbers of circulating, and to some degree also splenic, CD4+CD25+FOXP3+ regulatory T cells – likely due to the CDKI-mediated repression of DNA methyltransferase 115. Tregs express CDK6 at higher levels than effector T cells, making them more vulnerable to CDK inhibition.15,41 This fact explains why abemaciclib does not impair effector T cell functions. Although these immunological changes emerged in both models, we identified striking differences in the response profile between Mlh1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) mice. Given the fact that both strains develop dMMR-related tumors in the gastrointestinal tract (jejunum) spontaneously, this finding is intriguing. However, the mutational driver (Mlh1 vs. Msh2) and the resulting tumor microenvironment (T cells vs. TAMs vs. MDSCs vs. PD-L1 positivity) varies. This is consistent with the human counterpart, e.g. in the tumor mutational burden.42
Recent studies describe the restoration of the T cell function by CDK4/6 blockade.15,18,37,38,43 By dissecting the local immune response in detail, we found significantly increased numbers of cytotoxic T cells and DCs within the TIL compartment, especially in Mlh1−/− mice. Vice versa, numbers of TAMs decreased, accompanied by rising numbers of IRF5+ cells, reduced levels of the myeloid compartment as well as genes related to PI3K/Akt signaling. Higher expression of IRF5 leads to M1 polarization and the formation of a pro-inflammatory, antitumoral phenotype.44 Comparable positive immune-modulating side-effects were recently reported for dinaciclib, turning the microenvironment of immunologically ‘cold’ pancreatic cancers into a ‘hot’ one.45 Yet, the plasticity of macrophages may result in dual activation or a mixed M1/M2-like phenotype46 as seen here upon abemaciclib treatment via upregulation of CSF1 and CSF2. The latter supports the differentiation of hematopoietic myeloid cells47 and plays an important role in macrophage polarization by enhancing antigen presentation and DC formation.48 Here, abemaciclib-treated Mlh1−/− tumors had elevated numbers of DCs, likely because of CSF2-driven M1 polarization. In the combination, the beneficial effects of the monotherapies were blunted by suppressing activated pathways. Such striking differences in specific subpopulations were primarily detectable in tumors from Mlh1−/− mice, while Msh2loxP/loxP;TgTg(Vil1−cre) mice responded differently and effects were often weaker. The exact underlying reason for this model-individual response is not known. A recent study classified inherited and sporadic human dMMR endometrial tumors into distinct immunological entities.49 Apart from this, dMMR-related malignancies are often grouped together and responses compared to proficient MMR-tumors, without MMR-subclassification, i.e. MLH1, MSH2, MSH6, or PMS2.50 Hence, we can only speculate on the cause for the different responses seen here, such as: (I) these two dMMR models were created by different methods (Cre-lox System vs. constitutional knock out), the impact of either method on antitumoral immune function is inestimable; (II) Msh2loxP/loxP;TgTg(Vil1−cre) mice are bred homozygous, while Mlh1−/− mice are offsprings of heterozygous littermates; (III) the mutational load within coding microsatellites and the loci affected by a specific mutation within tumors differs; and (IV) the tumor microenvironment is heterogeneous.
When looking at regulatory granulocytes, another immune-suppressive subpopulation, only marginal changes were seen. Likewise, circulating and splenic MDSCs increased during after-care and may eventually have contributed to relapse. In support of this, gene expression analysis identified higher amounts of neutrophils within MLH1−/− and Msh2loxP/loxP;TgTg(Vil1−cre) tumors, together with high levels of DNA damage repair genes as part of the global stress-response. Another interesting finding of our preclinical in vivo trial is the activation of the Wnt pathway, which was again higher in MLH1−/− mice. This effect has been described before as a result of GSK3β inhibition by abemaciclib.51 GSK3β is an integral kinase within the β-catenin destruction complex. Its specific inhibition by abemaciclib does not apply to other CDK4/6 inhibitors and prospective studies will have to show whether WNT/β-catenin activation constitutes a potentially harmful side effect. One of the conceivable effector mechanisms is the induction of epithelial-mesenchymal transformation (EMT), characterized by invasion and enhanced motility.52 Indeed, abemaciclib triggered vimentin and N-cadherin expression, especially in Mlh1−/− mice, but effectively suppressed Fpr2, which is also involved in invasion and metastasis.53,54 We, therefore, conclude a compensatory mechanism to counteract abemaciclib-driven EMT. In the combination, no such “protective” effects were seen, with expression levels of EMT-markers being equal to or higher than in either monotherapy. Therefore, we propose EMT-driven tumor progression as one of the mechanisms that contribute to treatment failure. This hypothesis is supported by comparable findings in Msh2loxP/loxP;TgTg(Vil1−cre) mice. Although the effects were weaker, we observed a trend toward a higher expression of EMT markers in the combination group.
Another positive effect of CDK4/6 inhibition was recently described on the transcription factor Mxd4, a negative regulator of MYC.18 Interestingly, we also found the upregulation of Mxd4 after abemaciclib therapy in tumors and spleens of Mlh1−/− mice, but in contrast to Heckler et al.,18 we additionally detected higher expression levels of cMyc. Furthermore, the previously described formation of CD8+ effector memory cells18 was not confirmed by us (not shown). Despite effective anti-tumor treatment, we conclude a missing long-term immunity, quite possibly attributable to the low-dose and short-term therapy. The fact that neither tumors nor spleens from Msh2loxP/loxP;TgTg(Vil1−cre) mice showed any changes in these two genes confirm the better outcome of Mlh1−/− mice functionally.
Considering the effectiveness of PD-L1 blockade, overall survival was comparable between both mouse strains. Msh2loxP/loxP;TgTg(Vil1−cre) mice tended to benefit more from ICI, likely because of the higher PD-L1 abundance within the tumor stroma. The immunological effects of PD-L1 blockade can be summarized as follows: ICI-monotherapy triggered IL-6 release in both mouse strains, accompanied by reduced levels of exhausted T cells in the blood and slightly elevated levels of tumor-infiltrating T helper and cytotoxic cells. In the TIL compartment, regulatory granulocytes and TAMs faintly decreased. Tregs were only lower in the circulation of both mouse strains. Vice versa, splenic or tumor-infiltrating Treg numbers marginally changed. Although we detected higher expression levels of genes involved in antigen presentation, apoptosis, and interferon signaling in tumors of Msh2loxP/loxP;TgTg(Vil1−cre) mice, the overall immune stimulation was lower as seen for abemaciclib and the beneficial effects were consistently attenuated in the combination. With regard to EMT, ICIs are thought to have a minor direct impact.55 This is in line with our findings, in which N-cadherin was the only elevated gene after α-PD-L1 blockade in Msh2loxP/loxP;TgTg(Vil1−cre) mice. Apart from this, no impact was seen on either marker, suggesting no interference with EMT at least in these models.
Finally, the question remains why the combination failed to surpass the respective monotherapy. Preexisting effector T cell levels and dynamic changes in circulating myeloid cells were previously identified as decisive factors for response to combined CDK4/6-immune-checkpoint inhibition (palbociclib + pembrolizumab) in metastatic breast cancer.43 Here, we did not see such beneficial effects under combination therapy. Tumor growth control and overall survival were not better than in the respective monotherapy. Immunologically, both mouse strains responded with elevated levels of IL-4 in the plasma. IL-4 activates T helper cells, indicating at least partial maintenance of immune stimulation. In support of this, circulating, splenic, and tumor-infiltrating numbers of exhausted T cells were reduced to a degree comparable to abemaciclib monotherapy. In contrast, Mlh1−/− tumors had reduced cytotoxic T cell levels, which were replaced by TAMs. The latter may have stimulated EMT, characterized by high vimentin, N-cadherin, and Fpr2 expression. In Msh2loxP/loxP;TgTg(Vil1−cre) mice, overall effects were weaker, still, a comparable trend toward neutralizing beneficial effects of either monotherapy, such as apoptosis, co-stimulation, immune cell adhesion, and migration, was seen. Although this finding is somehow underwhelming, it supports recent findings, in which no significant difference in the anti-tumor response was seen compared with the activity of abemaciclib monotherapy.29,37 By applying a simultaneous setting, the strong immune-modulatory effects of abemaciclib are not boosted and in some cases, they are even dismantled. This finding complies with results from the phase Ia/Ib PACT study in which patients with advanced, refractory solid tumors received an α-PD-L1 inhibitor as monotherapy or in combination with abemaciclib.56 Lead-in CDK inhibition was not feasible due to hepatotoxicity.56 Detailed immunological analyses were not done in this clinical trial leaving the impact on the immune system unanswered.
To the best of our knowledge, this is the first comprehensive preclinical study reporting the immune-modulatory and therapeutic activity of abemaciclib on dMMR tumors. We not only provide another piece of evidence for the broad entity-overlapping potential of this selective CDKI but additionally propose an interesting option for dMMR patients not eligible for ICI treatment. However, caution is given when abemaciclib is used as lead-in therapy in combination with α-PD-L1, and follow-up studies are warranted to identify ideal combination partners for CDKI as an immunotherapy backbone.
Supplementary Material
Acknowledgments
We gratefully thank Genentech, a subsidiary of Roche, South San Francisco, USA, for providing the clone 6E11 for in vivo experiments. We additionally thank Mrs. Ilona Klamfuss and Ms. Chantal von Hoersten for breeding mice, Brigitte Vollmar and Bernd Krause for their continuous support in their efforts of chairing the Core Facility of Multimodal Small Animal Imaging. We also gratefully acknowledge the excellent technical assistance of Mrs. Joanna Förster. Furthermore, we thank Carina Bergner and Anja Gummesson, radiopharmacy team of the Department of Nuclear Medicine of the University Medical Centre Rostock, for providing 18F-FDG for the small animal PET/CT experiments. We likewise thank the Core Facility for Cell Sorting & Cell Analysis, Laboratory for Clinical Immunology, University Medical Center Rostock (Mrs. Wendy Bergmann, Prof. Brigitte Müller-Hilke) for their continuous support and providing access to the technical devices. Moreover, we thank Prof. Winfried Edelmann and Prof. Christoph Gasche for providing Msh2loxP/loxP;TgTg(Vil1-cre) breeding pairs and giving the permission to breed mice in our facility.
Funding Statement
This work was supported by grants from the German research foundation [DFG grant number MA5799/2-1 and MA5799/2-2] and the Brigitte und Dr. Konstanze Wegener-Stiftung N/A to C; Deutsche Forschungsgemeinschaft.
List of abbreviations
abema – abemaciclib
cMS – coding microsatellite
combi – combination
ctrl – control
dina – dinaciclib
dMMR – Mismatch repair-deficient
ICI – immune checkpoint inhibitors
MDSC – myeloid-derived suppressor cell
ROS – reactive oxygen species
TAM – tumor-associated macrophages
Treg – regulatory T cells
Authors’ contributions
IS – performed in vivo experiments, flow cytometry, analyzed data and participated in writing, LE, JH and PK – performed ex vivo analyses (staining of blood, spleen, and tumor samples, immunofluorescence, fragment length), BS – analyzed fragment length analyses; CR – performed Nanostring analysis, HL – provided assistance in confocal laser scanning microscopy, LH and CJ – critically revised the manuscript, CM – conducted the study and applied for grants, participated in the experiments, analyzed data, and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
The German local authority approved all animal experiments: Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg‐Vorpommern (7221.3‐1‐062/19; −025/20), under the German animal protection law and the EU Guideline 2010/63/EU. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Data availability statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2094583
References
- 1.Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, Deng L, Wang Y, Zhang X, Jiang T, et al. Targeting PD-1 and Tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J ImmunoTher. 2016;39(4):171–20. doi: 10.1097/CJI.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 2.Tan E, Sahin IH.. Defining the current role of immune checkpoint inhibitors in the treatment of mismatch repair-deficient/microsatellite stability-high colorectal cancer and shedding light on future approaches. Expert Rev Gastroenterol Hepatol. 2021. Feb 4;15(7):735–742. doi: 10.1080/17474124.2021.1886077. [DOI] [PubMed] [Google Scholar]
- 3.Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site — when a biomarker defines the indication. N Engl J Med. 2017;377(15):1409–1412. doi: 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- 4.Sousa Linhares A D, Battin C, Jutz S, Leitner J, Hafner C, Tobias J, Wiedermann U, Kundi M, Zlabinger GJ, Grabmeier-Pfistershammer K, et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci Reports. 2019;9(1):1–9. doi: 10.1038/s41598-019-47910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JH, Kim SY, Baek JY, Cha YJ, Ahn JB, Kim HS, Lee K-W, Kim J-W, Kim T-Y, Chang WJ, et al. A phase II study of avelumab monotherapy in patients with mismatch repair-deficient/microsatellite instability-high or POLE-Mutated metastatic or unresectable colorectal cancer. Cancer Res Treat. 2020. Apr 24. doi: 10.4143/crt.2020.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taïeb J, André T, El Hajbi F, Barbier E, Toullec C, Kim S, Bouche O, Di Fiore F, Chauvenet M, Perrier H, et al. Avelumab versus standard second line treatment chemotherapy in metastatic colorectal cancer patients with microsatellite instability: the SAMCO-PRODIGE 54 randomised phase II trial. Dig Liver Dis. 2020;53(3):318–323. doi: 10.1016/j.dld.2020.11.031. [DOI] [PubMed] [Google Scholar]
- 7.Salewski I, Henne J, Engster L, Schneider B, Lemcke H, Skorska A, Berlin P, Henze L, Junghanss C, Maletzki C. Combined gemcitabine and immune-checkpoint inhibition conquers anti-PD-L1 resistance in low-immunogenic mismatch repair-deficient tumors. Int J Mol Sci. 2021;22(11):5990. doi: 10.3390/IJMS22115990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salewski I, Kuntoff S, Kuemmel A, Feldtmann R, Felix SB, Henze L, Junghanss C, Maletzki C. Combined vaccine-immune-checkpoint inhibition constitutes a promising strategy for treatment of dMMR tumors. Cancer Immunol Immunother. 2021;70(12):3405–3419. doi: 10.1007/s00262-021-02933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sahin IH, Akce M, Alese O, Shaib W, Lesinski GB, El-Rayes B, Wu C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer. 2019;121(10):809–818. doi: 10.1038/s41416-019-0599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen SC, Manso L. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol. 2017 Nov 10;35(32): 3638–3646. doi: 10.1200/JCO.2017.75.6155. [DOI] [PubMed] [Google Scholar]
- 11.Hino H, Iriyama N, Kokuba H, Kazama H, Moriya S, Takano N, Hiramoto M, Aizawa S, Miyazawa K. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020;111(6):2132–2145. doi: 10.1111/cas.14419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naz S, Sowers A, Choudhuri R, Wissler M, Gamson J, Mathias A, Cook JA, Mitchell JB. Abemaciclib, a selective CDK4/6 inhibitor, enhances the radiosensitivity of non–small cell lung cancer in vitro and in vivo. Clin.Cancer Res. 2018;24(16):3994–4005. doi: 10.1158/1078-0432.CCR-17-3575/73825/AM/ABEMACICLIB-A-SELECTIVE-CDK4-6-INHIBITOR-ENHANCES. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres-Guzmán R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, McNulty AM, Gong X, Boehnke K, Du J, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget. 2017;8(41):69493–69507. doi: 10.18632/ONCOTARGET.17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang QF, Li J, Jiang K, Wang R, Ge JL, Yang H, Liu SJ, Jia LT, Wang L, Chen BL. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics. 2020;10(23):10619–10633. doi: 10.7150/THNO.44871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petroni G, Formenti SC, Chen-Kiang S, Galluzzi L. Immunomodulation by anticancer cell cycle inhibitors. Nat. Rev. Immunol. 2020;20(11):669–679. doi: 10.1038/S41577-020-0300-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat. Rev. 2022;22(6):356–372. doi: 10.1038/S41568-022-00456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurvitz SA, Martin M, Press MF, Chan D, Fernandez-Abad M, Petru E, Rostorfer R, Guarneri V, Huang CS, Barriga S, et al. Potent cell-cycle inhibition and upregulation of immune response with abemaciclib and anastrozole in Neomonarch, phase II neoadjuvant study in HR+/HER2- Breast cancer. Clin Cancer Res. 2020;26(3):566–580. doi: 10.1158/1078-0432.CCR-19-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heckler M, Ali LR, Clancy-Thompson E, Qiang L, Ventre KS, Lenehan P, Roehle K, Luoma A, Boelaars K, Peters V, et al. Inhibition of CDK4/6 Promotes CD8 T-cell Memory Formation. Cancer Discov. 2021;11(10):2564–2581. doi: 10.1158/2159-8290.CD-20-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roudko V, Cimen Bozkus C, Greenbaum B, Lucas A, Samstein R, Bhardwaj N. Lynch syndrome and MSI-H cancers: from mechanisms to “Off-The-Shelf” cancer vaccines. Front Immunol. 2021:12. doi: 10.3389/FIMMU.2021.757804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A, Doeberitz MVONK. BASIC – ALIMENTARY TRACT immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134(4):988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Kloor M, Von Knebel Doeberitz M. The immune biology of microsatellite-unstable cancer. Trends in Cancer. 2016;2(3):121–133. doi: 10.1016/j.trecan.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Kortüm B, Campregher C, Lang M, Khare V, Pinter M, Evstatiev R, Schmid G, Mittlböck M, Scharl T, Kucherlapati MH, et al. Mesalazine and thymoquinone attenuate intestinal tumour development in Msh2loxP/loxP Villin-Cre mice. Gut. 2015;64(12):1905–1912. doi: 10.1136/gutjnl-2014-307663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kucherlapati MH, Lee K, Nguyen AA, Clark AB, Hou HJ, Rosulek A, Li H, Yang K, Fan K, Lipkin M, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology. 2010;138(3):993–1002.e1. doi: 10.1053/j.gastro.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AMC, Lipkin M, Kucherlapati R. Tumorigenesis in Mlh1 and Mlh1/Apc1638N Mutant Mice. Cancer Res. 1999;59:1301–1307. [PubMed] [Google Scholar]
- 25.Maletzki C, Beyrich F, Hühns M, Klar E, Linnebacher M. The mutational profile and infiltration pattern of murine MLH1-/- tumors: concurrences, disparities and cell line establishment for functional analysis. Oncotarget. 2016;7(33):53583–53598. doi: 10.18632/oncotarget.10677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gladbach YS, Wiegele L, Hamed M, Merkenschläger AM, Fuellen G, Junghanss C, Maletzki C. Unraveling the heterogeneous mutational signature of spontaneously developing tumors in MLH1 -/- Mice. Cancers (Basel). 2019;11(10):1485. doi: 10.3390/CANCERS11101485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maletzki C, Huehns M, Bauer I, Ripperger T, Mork MMMM, Vilar E, Klöcking S, Zettl H, Prall F, Linnebacher M. Frameshift mutational target gene analysis identifies similarities and differences in constitutional mismatch repair-deficiency and Lynch syndrome. Mol Carcinog. 2017;56(7):1753–1764. doi: 10.1002/mc.22632. [DOI] [PubMed] [Google Scholar]
- 28.Lee K, Tosti E, Edelmann W. Mouse models of DNA mismatch repair in cancer research. DNA Repair (Amst). 2015:1–7. doi: 10.1016/j.dnarep.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usman S, Waseem NH, Nguyen TKN, Mohsin S, Jamal A, Teh MT, Waseem A. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers. 2021;13(19):4985. doi: 10.3390/CANCERS13194985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen S, Lu H, Wang D, Guo J, Dai W, Wang Z. TCF-1 maintains CD8 + T cell stemness in tumor microenvironment. J Leukoc Biol. 2021;110(3):585–590. doi: 10.1002/JLB.5MR1120-778R. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Lyu T, Cao Y, Feng H. Role of TCF-1 in differentiation, exhaustion, and memory of CD8+ T cells: a review. FASEB J. 2021;35(5):e21549. doi: 10.1096/FJ.202002566R. [DOI] [PubMed] [Google Scholar]
- 33.Weigert A, Strack E, Snodgrass RG, Brüne B. mPGES-1 and ALOX5/-15 in tumor-associated macrophages. Cancer Metastasis Rev. 2018;37(2):317–334. doi: 10.1007/S10555-018-9731-3. [DOI] [PubMed] [Google Scholar]
- 34.Groenland SL, Martínez-Chávez A, van Dongen MGJ, Beijnen JH, Schinkel AH, Huitema ADR, Steeghs N, van Dongen MGJ. Clinical pharmacokinetics and pharmacodynamics of the cyclin-dependent Kinase 4 and 6 inhibitors palbociclib, ribociclib, and abemaciclib. Clin Pharmacokinet. 2020;59(12):1501–1520. doi: 10.1007/s40262-020-00930-x. [DOI] [PubMed] [Google Scholar]
- 35.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34(1):9. doi: 10.1016/J.CCELL.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK, Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK. Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget. 2017;8(27):43678–43691. doi: 10.18632/ONCOTARGET.18435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018;22(11):2809–2817. doi: 10.1016/j.celrep.2018.02.053. [DOI] [PubMed] [Google Scholar]
- 38.Lelliott EJ, Sheppard KE, McArthur GA. Harnessing the immunotherapeutic potential of CDK4/6 inhibitors in melanoma: is timing everything? NPJ Precis Oncol. 2022;6(1):26. doi: 10.1038/S41698-022-00273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dowless M, Lowery CD, Shackleford T, Renschler M, Stephens J, Flack R, Blosser W, Gupta S, Stewart J, Webster Y, et al. Abemaciclib is active in preclinical models of ewing sarcoma via multipronged regulation of cell cycle, DNA methylation, and interferon pathway signaling. Clin Cancer Res. 2018;24(23):6028–6039. doi: 10.1158/1078-0432.CCR-18-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwitalle Y, Linnebacher M, Ripberger E, Gebert J, von Knebel Doeberitz M. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immun. 2004;4:14. [PubMed] [Google Scholar]
- 41.Rowell EA, Wang L, Chunder N, Hancock WW, Wells AD. Regulation of T cell differentiation and alloimmunity by the cyclin-dependent kinase inhibitor p18ink4c. PLoS One. 2014;9(3). doi: 10.1371/JOURNAL.PONE.0091587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salem ME, Bodor JN, Puccini A, Xiu J, Goldberg RM, Grothey A, Korn WM, Shields AF, Worrilow WM, Kim ES, et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int J Cancer. 2020;147(10):2948–2956. doi: 10.1002/ijc.33115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egelston C, Guo W, Yost S, Lee JS, Rose D, Avalos C, Ye J, Frankel P, Schmolze D, Waisman J, et al. Pre-existing effector T-cell levels and augmented myeloid cell composition denote response to CDK4/6 inhibitor palbociclib and pembrolizumab in hormone receptor-positive metastatic breast cancer. J Immunother Cancer. 2021;9(3):e002084. doi: 10.1136/JITC-2020-002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 45.Huang J, Chen P, Liu K, Liu J, Zhou B, Wu R, Peng Q, Liu Z-X, Li C, Kroemer G, et al. CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer. Gut. 2021;70(5):890–899. doi: 10.1136/gutjnl-2019-320441. [DOI] [PubMed] [Google Scholar]
- 46.Veremeyko T, Yung AWY, Anthony DC, Strekalova T, Ponomarev ED. Early growth response gene-2 is essential for M1 and M2 macrophage activation and plasticity by modulation of the transcription factor CEBPβ. Front Immunol. 2018;9(NOV):2515. doi: 10.3389/FIMMU.2018.02515/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114(7):1289–1298. doi: 10.1182/BLOOD-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai H, Zhang Y, Wang J, Gu J. Defects in macrophage reprogramming in cancer therapy: the negative impact of PD-L1/PD-1. Front Immunol. 2021:12. doi: 10.3389/FIMMU.2021.690869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramchander NC, Ryan NAJ, Walker TDJ, Harries L, Bolton J, Bosse T, Evans DG, Crosbie EJ. Distinct immunological landscapes characterize inherited and sporadic mismatch repair deficient endometrial cancer. Front Immunol. 2020;11:10. doi: 10.3389/FIMMU.2019.03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Willvonseder B, Stögbauer F, Steiger K, Jesinghaus M, Kuhn PH, Brambs C, Engel J, Bronger H, Schmidt GP, Haller B, et al. The immunologic tumor microenvironment in endometrioid endometrial cancer in the morphomolecular context: mutual correlations and prognostic impact depending on molecular alterations. Cancer Immunol Immunother. 2021;70(6):1679–1689. doi: 10.1007/S00262-020-02813-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cousins EM, Goldfarb D, Yan F, Roques J, Darr D, Johnson GL, Major MB. Competitive kinase enrichment proteomics reveals that abemaciclib inhibits GSK3β and activates WNT signaling. Mol Cancer Res. 2018;16(2):333–344. doi: 10.1158/1541-7786.MCR-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang Z, Zhang Z, Zhou C, Liu L, Huang C. Epithelial-mesenchymal transition: the history, regulatory mechanism, and cancer therapeutic opportunities. MedComm. 2022;3(2). doi: 10.1002/MCO2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu J, Zhao J, Jia C, Zhou L, Cai Y, Ni J, Ma J, Zheng M, Lu A. FPR2 enhances colorectal cancer progression by promoting EMT process. Neoplasma. 2019;66(5):785–791. doi: 10.4149/NEO_2018_181123N890. [DOI] [PubMed] [Google Scholar]
- 54.Xie X, Yang M, Ding Y, Yu L, Chen J. Formyl peptide receptor 2 expression predicts poor prognosis and promotes invasion and metastasis in epithelial ovarian cancer. Oncol Rep. 2017;38(6):3297–3308. doi: 10.3892/OR.2017.6034/HTML. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K, Hamanishi J, Baba T, Matsumura N, Mandai M. Tumor immune microenvironment during epithelial- mesenchymal transition. Clin Cancer Res. 2021;27(17):4669–4679. doi: 10.1158/1078-0432.CCR-20-4459/672159/AM/TUMOR-IMMUNE-MICROENVIRONMENT-DURING-EPITHELIAL. [DOI] [PubMed] [Google Scholar]
- 56.Patnaik A, Yap TA, Chung HC, de Miguel MJ, Bang Y-J, Lin -C-C, W-C S, Italiano A, Chow KH, Szpurka AM, et al. Safety and clinical activity of a new anti-PD-L1 antibody as monotherapy or combined with targeted therapy in advanced solid tumors: the PACT phase Ia/Ib Trial. Clin Cancer Res. 2021;27(5):1267–1277. doi: 10.1158/1078-0432.CCR-20-2821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].