

Abstract
Mutations in the intraflagellar transport-A (IFT-A) gene, THM1, have been identified in skeletal ciliopathies. Here, we report a genetic interaction between Thm1, and its paralog, Thm2, in postnatal skeletogenesis. THM2 localizes to primary cilia, but Thm2 deficiency does not affect ciliogenesis and Thm2-null mice survive into adulthood. However, by postnatal day 14, Thm2−/−; Thm1aln/+ mice exhibit small stature and small mandible. Radiography and microcomputed tomography reveal Thm2−/−; Thm1aln/+ tibia are less opaque and have reduced cortical and trabecular bone mineral density. In the mutant tibial growth plate, the proliferation zone is expanded and the hypertrophic zone is diminished, indicating impaired chondrocyte differentiation. Additionally, mutant growth plate chondrocytes show increased Hedgehog signaling. Yet deletion of one allele of Gli2, a major transcriptional activator of the Hedgehog pathway, exacerbated the Thm2−/−; Thm1aln/+ small phenotype, and further revealed that Thm2−/−; Gli2+/- mice have small stature. In Thm2−/−; Thm1aln/+ primary osteoblasts, a Hedgehog signaling defect was not detected, but bone nodule formation was markedly impaired. This indicates a signaling pathway is altered, and we propose that this pathway may potentially interact with Gli2. Together, our data reveal that loss of Thm2 with one allele of Thm1, Gli2, or both, present new IFT mouse models of osteochondrodysplasia. Our data also suggest Thm2 as a modifier of Hedgehog signaling in postnatal skeletal development.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-03806-w.
Keywords: Ttc21a, Ttc21b, Knock-out, CRISPR, Cell-specific
Introduction
Skeletal dysplasias affect 1:5000 births and range in severity from perinatal lethality to craniofacial dysmorphogenesis and short stature. These disorders are often heritable, and identifying the causative mutations has been instrumental in improving diagnosis, understanding the mode of inheritance, and identifying therapeutic targets.
Conversely, genetic disorders that cause skeletal dysplasias provide valuable insight into underlying molecular and cellular mechanisms. One such class of genetic disorders are ciliopathies, which result from mutation and dysfunction of primary cilia. The primary cilium is a solitary, sensory organelle that protrudes from the surface of most mammalian cells, including bone and cartilage cells [1, 2]. As such, ciliopathies affect multiple organ systems, and a proportion of ciliopathies manifest osteochondrodysplasias [3, 4]. These skeletal ciliopathies include Short-Rib Polydactyly Syndromes (I–IV), Jeune Syndrome, Oro-facial-digital syndrome type 1, Ellis van Creveld syndrome, and cranioectodermal dysplasia (also known as Sensenbrenner Syndrome), which manifest to varying degrees, shortened long bones, narrow rib cage, polydactyly and craniofacial defects. Fibrocystic diseases of the kidney, liver and pancreas can also arise. These ciliopathies reveal that primary cilia are potent modifiers of skeletal growth.
Primary cilia detect and transduce extracellular mechanical and chemical cues into signaling cascades to ultimately affect cellular behavior [5, 6]. These antenna-like organelles are dynamic structures, formed and maintained by intraflagellar transport (IFT), which is the bi-directional transport of cargo (structural and signaling proteins) by multiprotein complexes along a microtubular axoneme. IFT-B proteins, together with the kinesin motor, transport cargo from the base to the tip of the cilium in anterograde IFT, while IFT-A proteins and the dynein motor are required to return proteins from the ciliary tip to the base in retrograde IFT. IFT-A proteins also mediate ciliary entry of membrane-associated and signaling proteins [7–10]. Primary cilia transduce the Hedgehog (Hh) signal [11]. The presence of Hh ligand induces ciliary enrichment of the Smoothened signal transducer and culminates in activation of the Gli transcription factors, of which GLI2 is the primary transcriptional activator [12, 13]. The differing roles of IFT-B versus -A can result in contrasting ciliary and Hh signaling phenotypes in Ift mutant mouse embryos. Loss of IFT-B or kinesin, generally results in loss of cilia and Hh activity, and mid-gestational lethality [14–16], while deletion of IFT-A causes shortened primary cilia with protein accumulation in a bulbous distal tip. Further, two IFT-A mouse embryonic mutants show overactivation of the Hh pathway, and perinatal lethality [17, 18]. Yet in contrast to these earlier reports, deletion of kinesin subunit, Kif3a, in neural crest cells increased GLI activity in the frontonasal prominence of the developing face [19], while deletion of IFT-A gene, Thm1, in glial cells decreased Hh signaling in the developing cerebellum [20]. Thus, whether an IFT protein positively or negatively regulates Hh or GLI activity is cell-specific and/or context-dependent.
In patients with skeletal ciliopathies, mutations have been identified in all of the six characterized IFT-A components (IFT43 [21, 22], IFT121 [22], IFT122 [23, 24], WDR19/IFT144 [25], IFT140 [26], and THM1 [27]) and in 4 of the 14 IFT-B components (IFT80 [28], IFT52 [29, 30], IFT172 [4] and IFT56 [31]. In accordance with the human genetics studies, many of the reported IFT-A mouse mutants have exhibited polydactyly and abnormal development of the thoracic ribs during embryogenesis [32–34]. Additionally, Ift80-hypomorphic mice that survive past birth, and mice with conditional deletion of IFT-B gene, Ift88, in the limb bud and craniofacial mesenchyme, exhibit polydactyly, shortened long bones, and disorganization of the growth plate [35, 36]. Thus, both IFT-B and -A are essential for mammalian skeletal growth, although the human genetics studies may suggest that IFT-A mutations are more compatible with life.
Previously, we identified THM1 (TPR-containing Hh modulator 1; also known as TTC21B) as an IFT-A component and regulator of Hh signaling [17]. A germ-line mutation resulting in THM1 loss in mouse causes shortened long bones, split and fused ribs, and polydacytyly. THM1 mutations have also been identified in patients with Jeune Syndrome (JATD) [18], which is characterized by shortened long bones, craniofacial defects, and a narrow rib cage that often inhibits lung development and causes 60–80% mortality among infants and children [37]. We also identified a paralog of THM1, which we call THM2 (also known as TTC21A) [17]. THM1 and THM2 are orthologues of the IFT-A protein, IFT139, in Chlamydomonas reinhardtii, and have similar levels of homology to IFT139/FLA16. Thm1 and Thm2 have similar predicted protein structures with multiple TPR domains and similar RNA expression patterns during mouse development. A recent report revealed that THM2 is required for male fertility [38]. However, a role for Thm2 in early postnatal development has not been elucidated. Here by generating in vitro and in vivo models of Thm2 deficiency and implementing genetic crosses, we reveal a role for THM2 in postnatal skeletogenesis.
Methods
Generation of THM2 knockdown cell line
Lentiviral transfection was used to generate a human embryonic kidney (293T) THM2 knockdown cell line. Briefly, viral particles were created by transfecting three plasmids, 4.2 µg vector (Thm2 shRNA sequence GACTTTGATTAATTACTAT), 7.4 µg delta 8.2 and 0.4 µg VSVG into 293T packaging cells using the Fugene transfection reagent (Promega, E2691) into 293T cells. After 48 h, media was collected and filtered to obtain viral particles. This media containing virus was placed onto 293T target cells for 6 h and then replaced by fresh media. Infected cells were selected using 1 µg/ml puromycin. After reaching confluence, cells were trysinized, resuspended, then seeded sparsely onto 25 cm dishes to allow individual clones to grow. Clones were selected using cloning disks immersed in Trypsin. Clones were then expanded.
Western blot
293T cells and mouse tissues were collected. Protein was extracted by resuspending cells or homogenizing tissue in passive lysis buffer (Promega, E1941) containing protease inhibitors (Thermo Scientific, 88669), and rocking samples for 15 min at room temperature. Lysed cells and tissues were pelleted and supernatants were collected. Protein concentration of lysates was determined using the Pierce BCA Protein Assay Kit (ThermoFisher, 23227). Lysates with loading dye were boiled for 5 min, cooled on ice, and loaded onto a 4–20% SDS-gel (BioRad, 456-8094). Gels were run for approximately 3.5 h at 90 V to adequately separate protein. Membranes were incubated with THM2 antibody using Western blot as described [17]. THM2 antibody was targeted to N′ Cys-RRQNYETAINLYHQVLEK, 963-980aa, in exon 22. (Proteintech S4132-2, 1:5000).
Immunofluorescence
Retinal pigment epithelial (RPE) cells, 293T cells, and primary chondrocytes and osteoblasts were seeded onto poly-l-lysine coated coverslips in 24-well plates. Confluent RPE and 293 T cells were serum-starved overnight to increase cilia lengths. All post-confluent cells were fixed in 4% PFA in PBS containing 0.1% Triton x-100 for 10 min at room temperature, then blocked in 1% Bovine Serum Albumin (Sigma, A9647) for 1 h. Primary antibodies against Arl13b (Proteintech, 17711-1-AP, 1:300), acetylated α-tubulin (Sigma T6557, 1:4000), THM2 (Custom-made Antibody, Proteintech S4132-2, 1:50), IFT81 (Proteintech, 11744-1-AP, 1:200), SMO (Santa Cruz, sc-166685, 1:50) were incubated with cells in 1% BSA overnight at 4°C. Cells were washed three times in PBS, then incubated with secondary antibody conjugated to Alexa Fluor 594 or Alexa Fluor 488 [Life Technologies, A-11005 (anti-mouse) and A-11008 (anti-rabbit), 1:500] for 1 h at room temperature. Cells were washed three times in PBS. Coverslips were inverted and mounted onto slides using DAPI Fluoromount-G (Electron Microscopy Services, 17984-24). RPE cells were imaged using a Nikon Eclipse TiE attached to an A1R-SHR confocal with an A1-DU4 detector and LU4 laser launch. All other cells were imaged using a Nikon 80i or Nikon TiS attached to a Nikon DS-Fi1 or QICAM FAST1934 camera, respectively.
Generation of Thm2 knockout mouse
Thm2 knockout-first C57BL/6J embryonic stem cells were obtained from the NIH Knockout Mouse Project (KOMP) Repository (www.komp.org). Embryonic stem cells were confirmed to contain the correct Thm2 knockout construct showing evidence of homologous recombination and correct karyotype. Subsequently, cells were injected into C57BL/6J-Tyrc−2 J (albino) blastocysts and implanted into female mice by Dr. Melissa Larson at the University of Kansas Medical Center Transgenic and Gene Targeting Facility. Resulting male chimeras were mated to multiple females. Genomic DNA from tails of resulting pups with black coat color was genotyped by polymerase chain reaction (PCR) and PCR amplicons were sequenced to confirm the presence of the construct in the DNA. The KOMP allele contains a LacZ neo construct flanked by flippase recombinase (FLP) recombinase target (FRT) sites. Mice containing the KOMP construct were mated to a mouse carrying a FLP recombinase (Jackson Laboratories, 009086) to excise the LacZ-Neo cassette. Resulting mice were then mated to mice expressing cytomegalovirus (CMV) Cre recombinase (Jackson Laboratories, 006064) to excise exon 6, which created a premature stop codon and a Thm2 null allele. All animal procedures were conducted in accordance with KUMC-IACUC and AAALAC rules and regulations.
Genotyping of Thm2 knockout mouse
Genotyping primers were designed flanking the loxP sites surrounding exon 6. Primers Thm2-KO-F—5′ CAG ATA TCT CCC CAC TTG TTA ACG 3′ and Thm2-KO-R—5′ GTG TCA GAT ACC CTG GAA CCA GAG 3′ amplify a WT band of 1086 bp and a knockout band of approximately 400 bp. Since at times, this PCR reaction favors amplification of the smaller knockout band, making the WT band difficult to see, additional PCR primers, Thm2-WT-F-5′AAC TTC CTG CCC GCT TTA GT 3′ and Thm2-WT-R-5′ GTG TCA GAT ACC CTG GAA CCA GAG 3′ are used to amplify only a WT band of 461 bp.
Generation of Thm2−/−;Thm1aln/+ mice
Thm2−/−;Thm1aln/+ mice were generated by crossing Thm2+/- mice on a C57BL/6 background with Thm1aln/+ mice on an FVB background. Thm2+/-;Thm1aln/+ mice were then backcrossed five generations onto a C57BL6/J background.
Weight and length measurements
Total mouse body weight was measured using a standard laboratory weighing scale. Crown-to-rump measurements, from the tip of the nose to the end of the rump, were obtained using a standard ruler.
Skeletal preparations
Alizarin red and alcian blue staining of skeletons was performed as described [32]. Briefly, P14 mice were eviscerated and fixed in 95% ethanol for 1–2 days, then stained with alcian blue (Acros Organics 33864-99-2) for 14 days. Subsequently, skeletons were fixed in 95% ethanol for 1–2 days and cleared with potassium hydroxide for 2–5 days, or until the tissue was cleared. Skeletons were stained with 1% alizarin red (Acros Organics 130-22-3) in potassium hydroxide for 1–2 days or until staining was complete. Skeletons were then cleared in glycerol for imaging and long-term storage. Skeletal preparations were imaged using a dissecting scope (Leica M165C) and bone lengths were quantified using Image J software.
Microcomputed tomography and Faxitron
Whole mice and tibias were radiographed at 1X and 3X magnifications, respectively, using a LX-60 DC12 Cabinet X-ray System (Faxitron) at 26kVP for 10 s. Images were analyzed using ImageJ to calculate the length of each mineralized caudal vertebrae.
Ethanol-fixed tibia from control and triple allele mutant mice were scanned in a Skyscan 1174 micro-computed tomography system (Bruker) at a resolution of 14 μm3 with a voltage of 50kVp using a 0.5 mm aluminum filter. Tibias were wrapped in gauze soaked with 70% ethanol and placed in a low-density polyethylene tube for scanning. Camera pixel binning was not applied and the integration time was set to 3000 ms. The scan orbit was 180°/360° with a rotation step of 0.3°. Reconstruction was carried out with a modified Feldkamp algorithm using the SkyScanTM NRecon software accelerated by GPU3. Gaussian smoothing, ring artifact reduction, and beam hardening correction were applied. Reconstructed images were analyzed by CTAn software. Irregular, anatomic regions of interest (ROIs) were selected for both trabecular and cortical bone analysis. For trabecular bone analysis, slices from the proximal tibia starting at the distal end of the growth plate to the end of the trabeculae were analyzed for bone mineral density (BMD), bone volume fraction (BV/TV), bone surface per BV (BS/BV), trabecular thickness (Tb.Th), separation (Tb.Sp), and number (Tb.N). For cortical bone analysis, two sets of ROIs were analyzed for BMD, total cross-sectional bone area (Tt.Ar), cortical bone area (Ct.Ar), fraction (Ct.Ar/Tt.Ar), perimeter (Ct.Pm), and thickness (Ct.Th). The first was from mid-diaphysis and the second started at the end of the trabeculae at the proximal end of the tibia. To control for length differences, the same percentage of slices were analyzed in mutants as relative length for both trabecular and cortical analysis. The minimum and maximum thresholds were set to 45 and 95 for trabecular bone analysis and 55 and 255 for cortical bone analysis.
Dissection, embedding and sectioning of tibias and soft organs
Tibias were dissected and placed in Cal-Ex, a decalcifying and fixing solution (Fisher C5511-1D), for 3 days. Kidney, heart and lung were dissected and weighed. Kidneys were fixed in Bouin’s fixative (Polysciences, Inc. 16045) overnight. Following fixation, tibias and kidneys were placed into 70% ethanol. Tissues were processed and dehydrated through an ethanol series and embedded in paraffin wax. Paraffin blocks were sectioned at 10 μm and 7 μm for tibias and soft organs, respectively.
Hematoxylin and eosin and safarnin O and fast green staining
Tibia and kidney sections were de-paraffinized and rehydrated through an ethanol series. Sections were stained with Hematoxylin (Sigma HHS32) and Eosin (Sigma HT110216) or with Weigert’s Hematoxylin, 0.2% Fast Green, and Safranin O. Sections were dehydrated through an ethanol series and mounted with Permanent Mounting Media (Fisher, SP15-500). Sections were imaged using a light microscope (Nikon 80i) attached to a Nikon DS-Fi1 camera.
Antigen retrieval
Prior to immunostaining, tissue sections were deparaffinized, rehydrated, and subjected to antigen retrieval. Tissue sections were steamed for 25 min in Sodium Citrate Buffer (10 mM Sodium Citrate, 0.05% Tween 20, pH 6.0). Sections were cooled and rinsed in distilled water ten times.
LacZ staining
Gli1-LacZ reporter mice (Jackson Laboratories, stock 008211) were crossed into the Thm2; Thm1 colonies. Tibias were dissected, fixed in acetone for 48 h at 4°C as described [39]. Fixed tissues were immersed in wash buffer (100 mM sodium phosphate buffer (pH 7.3), 2 mM magnesium chloride, 0.02% Nonidet P-40, and 0.01% sodium deoxycholate) for 15 min, three times. Tibia were stained with X-gal solution (Teknova X1220; 1 mg/ml X-gal in N,N-dimethylformamide) in wash buffer containing 5 mM potassium ferrocyanide (Sigma P-9387) and 5 mM potassium ferricyanide (Sigma P-8131) protected from light overnight or until desired color intensity was obtained. Stained tibia were fixed and demineralized in CalEx, processed, embedded in paraffin, and sectioned as described above. Sections were rehydrated through an ethanol series, counterstained with nuclear fast red, and mounted. Stained sections were imaged using a Nikon 80i attached to a Nikon DS-Fi1 camera. Using ImageJ, colored images were converted to monochrome images using RGB stack, thresholds were obtained, and growth plates were demarcated for measurements of area, mean and integrated density to quantify LacZ staining of litter-matched sections of control and triple allele mutant growth plates.
Generation of primary chondrocytes and osteoblasts
Primary chondrocytes were derived from P10 mouse tibial growth plates, as described [40]. Briefly, tibial growth plates were dissected and digested in 10 cm plates containing DMEM containing 3 mg/ml Collagenase Type 4 (Worthington Biochemical Corporation, CLS-4) for 90 min in the cell incubator at 37°C. Plates were swirled every 30 min. Following the 90 min, media was replaced with DMEM containing 5 mg/ml Collagenase Type 4 for 3 h in the cell incubator at 37°C. Plates were swirled every 60 min. Media containing digested growth plates was filtered through a 70 mm pore size cell strainer, then centrifuged to pellet the cells. Cells were resuspended in DMEM containing 10% FBS and penicillin/streptomycin and plated.
Primary osteoblasts were derived from P10 mouse calvaria, as described [41]. Briefly, calvaria was dissected in αMEM, rinsed in PBS, then digested in αMEM containing 2 mM l-glutamine and 0.2% collagenase, Type I (Sigma, T1005)/0.05% trypsin (Sigma, C9891) for 20 min in the cell incubator at 37°C. Media from this first digestion was discarded and the calvaria were subjected to an additional four rounds of digestion in αMEM containing 2 mM l-glutamine and 0.2% collagenase, Type I/0.05% trypsin each for 20 min in the cell incubator at 37°C. Following each digestion, media was collected and fetal bovine serum (FBS) was added to obtain 10% final concentration and kept at 4°C. Following the 5th digestion, all media containing digested growth calvaria with 10% FBS was filtered through a 70 mm pore size cell strainer, then centrifuged to pellet the cells. Cells were resuspended in αMEM containing 10% FBS, 2 mM l-glutamine and penicillin/streptomycin, and plated.
Phase-contrast images of primary chondrocytes and osteoblasts were obtained using an EVOS® FL Auto system (ThermoFisher) with an integrated environmental control system (air-CO2 premix) and CCD monochrome camera.
SAG treatment and qPCR
Confluent cells were treated with 0.1% DMSO or 500 nM SAG for 24 h prior to immunostaining for SMO or for 48 h prior to immersing in Trizol (ThermoFisher) for RNA extraction. Cells were stored in Trizol at − 80 °C until RNA extraction. RNA was extracted according to the manufacturer’s protocol. One microgram of RNA was converted into cDNA using Quanta Biosciences qScript cDNA mix (VWR International). qPCR was performed in duplicate using Quanta Biosciences Perfecta qPCR Supermix (VWR International) and a BioRad CFX Connect Real-Time PCR Detection System. Primers used were Thm2 (Forward: 5′‐CCC CAC AAT CCA AAC CTA CA‐3′; reverse: 5′‐GCT CAC AAG CCG ATG GAC‐3′); Gli1 (forward: 5′‐CTG ACT GTG CCC GAG AGT G‐3′; Reverse: 5′‐CGC TGC TGC AAG AGG ACT‐3′); mGli2 (Forward: 5′ GCA GAC TGC ACC AAG GAG TA 3′; reverse: CGT GGA TGT GTT CAT TGT TGA 3′); mGli3 (forward: 5′ CAC CAA AAC AGA ACA CAT TCC A 3′; Reverse: 5′ GGG GTC TGT GTA ACG CTT G 3′) and housekeeping gene mOaz1 (forward: 5′—GCC TGA GGG CAG TAA GGA C-3′; reverse: 5′-GGA GTA GGG CGG CTC TGT-3′).
Osteoblast differentiation assay
Osteoblasts were plated in duplicate into 24-well plates in αMEM containing 10% FBS, 2 mM l-glutamine and penicillin/streptomycin. Upon confluency, cells were cultured in differentiation media, consisting of αMEM, 10% FBS, 2 mM l-glutamine, penicillin/streptomycin and 50 mg/ml ascorbic acid and 5 mM b-glycerophosphate, which was refreshed every 3 days over 2–4 weeks. Cells were fixed with 4% paraformaldehyde for 15 min, then stained with 2% alizarin red solution for 10–15 min in the dark. Once cells reached desired color intensity, cells were washed 4 times with distilled water, then immersed in PBS. Cells were imaged immediately using an EVOS® FL Auto system (ThermoFisher Scientific) with CMOS color camera. Bone nodule formation was quantified as the percentage of the area of redbone nodules/total image area, which were measured using Image J.
Statistics
Unpaired, two-tailed t tests for comparison of two groups and one-way ANOVA followed by Tukey’s test for comparison of multiple groups were used to evaluate statistical significance (p < 0.05) using Graphpad Prism 9.0 software.
Results
THM2 localizes to primary cilia
To characterize the subcellular localization of THM2, an antibody recognizing a unique amino acid sequence encoded within exon 22 was generated (Proteintech). To determine the specificity of the antibody, we generated human embryonic kidney (293 T) THM2 knock-down (kd) clonal cell lines using lentiviruses expressing THM2 shRNA. Western blot analysis revealed THM2 kd, clone 2–1, showed the most effective knockdown (Fig. 1a). We analyzed cilia length in EV 2–5 (empty vector control) and Thm2 kd 2–1 clonal cell lines by immunostaining for ARL13B, a ciliary membrane marker (Fig. 1b). Quantification of ciliary lengths demonstrated similar lengths between EV and THM2 kd cell lines (Fig. 1c), indicating THM2 is not required for ciliogenesis. Immunostaining of retinal pigment epithelial (RPE) cells for THM2 showed co-localization with acetylated α-tubulin, a marker of the ciliary axoneme (Fig. 1d), revealing THM2 localizes to primary cilia.
Fig. 1.

THM2 is dispensable for ciliogenesis but localizes to the primary cilium. a Western blot for THM2 on cellular extracts of 293T EV (control) and THM2 kd clonal lines. b Immunostaining for Arl13B (green) in 293T EV and THM2 kd (clone 2–1) cells. Scale bar—5 μm. c Quantification of cilia lengths. Each data point represents an individual cilium. Graphs represent mean ± SD. d Immunostaining for THM2 (green) and acetylated α-tubulin (red) in RPE cells. Scale bar—10 μm
Generation of Thm2 knockout mouse model
Using a Thm2 knock-out construct from the Knockout Mouse Project (KOMP) repository, we generated a Thm2 null allele, in which exon 6 was excised, creating a premature stop codon (Fig. 2a). Primers flanking exon 6 were designed to amplify a WT band of 1086 bp and a recombined band at approximately 400 bp (Fig. 2b). Additionally, since this PCR reaction favors amplification of the smaller recombined (Thm2-null) allele, we designed additional primers to amplify only the WT allele, generating a 461 bp product. In accordance with the amplification of the mutant band, Western blot analysis confirmed the loss of THM2 protein in testis protein extracts of a Thm2-null mouse (Fig. 2c).
Fig. 2.

Thm2 knock-out mouse was generated using a KOMP construct. a Schematic diagram of the KOMP constructs in genomic DNA of Thm2. Exons are represented by black boxes and introns by the black line. Lox P sites surrounding exon 6 are depicted as red triangles. Genomic primers are indicated in blue (WT and recombination band) and purple (WT band). Splicing of exon 6 creates a premature stop codon. b PCR analysis of WT, Thm2+/-, and Thm2−/− genotypes using primers indicated in the blue and additional confirmation of the WT allele in WT and Thm2+/- mice using primers indicated in purple. c Western blot analysis of P14 testis extracts confirms the loss of THM2 in Thm2 ko mice
Thm2−/−; Thm1aln/+ mice are smaller than control littermates
Unlike most ciliary gene knock-out mice, which are embryonic lethal [17, 33, 34, 42], Thm2-null mice are born, phenotypically indistinguishable from their littermates, and reach adulthood with seemingly normal health. Since paralogs can have redundant functions [43, 44], we introduced a mutant allele of Thm1 (aln) onto the Thm2-null background. The aln allele harbors a missense mutation that results in absence of THM1 protein [17]. We backcrossed the colony five generations onto a C57BL6/J background. At P14, Thm2−/−; Thm1aln/+ mice were noticeably smaller than their control littermates with reduced body weight and shorter crown-to-rump length (Fig. 3a, b). This phenotype was also observed at P21 (Fig. 3c), and we noted that a proportion of triple allele mutant mice (7/25 = 28%) do not survive to weaning age.
Fig. 3.

Thm2−/−; Thm1aln/+ mice exhibit a runted phenotype. Following five generations of backcrossing onto a C57BL6/J background, triple allele mutants showed a runted phenotype. a Images of mice at P14. b Body weights and crown-to-rump lengths at P14 and c at P21. Error bars represent mean ± SD. Statistical significance was determined using one-way ANOVA and Tukey’s test. ****p < 0.0001
Thm2−/−; Thm1aln/+ lung weight/body weight ratio is increased
Since renal cysts are among the most common clinical manifestations of ciliopathies [3], we analyzed the kidneys of P14 Thm2−/−; Thm1aln/+ mice. Histological analysis revealed normal kidney morphology of Thm2−/−; Thm1aln/+ mice (Fig. 4a). Total kidney weights were smaller in Thm2−/−; Thm1aln/+ mice, but this was proportional to body size since kidney weight/body weight ratios of Thm2−/−; Thm1aln/+ mice were similar to those of control littermates. Similarly, heart weight was lower in triple allele mutant mice, but heart weight/body weight ratios were comparable between control and Thm2−/−; Thm1aln/+ mice (Fig. 4b). In contrast, although lung weight was lower in triple allele mutant mice, lung weight/body weight ratio was elevated in triple allele mutant mice relative to control mice (Fig. 4c).
Fig. 4.

Thm2−/−; Thm1aln/+ mice have increased lung/bodyweight ratios. a Haematoxylin and eosin staining of kidney sections from P14 mice. Scale bars—1 mm and 500 μm, upper and lower panels. Two kidney weights and % kidney weight/body weight ratios. b Heart weights and % heart weight/body weight ratios. c Lung weights and % heart weight/body weight ratios. Graphs represent mean ± SD. Statistical significance was determined using one-way ANOVA and Tukey’s test. *p < 0.05; **p < 0.005
Thm2−/−;Thm1aln/+ mice exhibit shortened and less dense bone
We next performed skeletal preparations to examine the skeletal phenotype of Thm2−/−; Thm1aln/+ mice (Fig. 5a). At P14, Thm2−/−; Thm1aln/+ mice exhibited shorter long bones, including tibia, femur, humerus, radius and ulna (Fig. 5b). The sternum and ribcage diameter of Thm2−/−; Thm1aln/+ mice were also smaller than those of control littermates. Additionally, cranial length, as measured from the tip of the nose to back of the head, was shorter in Thm2−/−; Thm1aln/+ mice. To determine if these reduced bone lengths reflect the overall dwarfing of the tripe allele mutants, we examined bone length/crown-to-rump length ratios. Long bone, sternum and ribcage/crown-to-rump ratios were similar between control and triple allele mutants, but the skull/crown-to-rump ratio was slightly increased in triple allele mutants (Fig. 5c). Moreover, triple allele mutants showed small mandible/skull ratios, indicative of craniofacial defects (Fig. 5d).
Fig. 5.

Thm2−/−; Thm1aln/+ mice have shorter bones. a Skeletal preparations at P14 using alizarin red (bone) and alcian blue staining (cartilage). b Bone lengths. c Bone/Crown-to-Rump (C-to-R) length ratios. d Mandibular height/cranial length. Graphs represent mean ± SD of n = 4 ctrl and n = 4 triple allele mutant mice. Statistical significance was determined using two-tailed, unpaired t test. *p < 0.05; **p < 0.005
We further analyzed the Thm2−/−; Thm1aln/+ skeleton using radiography (Fig. 6a and b). Individual caudal vertebra length was decreased in Thm2−/−; Thm1aln/+ mice (Fig. 6c), but in proportion to crown-to-rump length (Fig. 6d). We also noted that the hindlimb and tibia of Thm2−/−; Thm1aln/+ mice were less opaque than those of control mice using both radiography and micro-computed topography (Fig. 6e and f). This was particularly noticeable when tibias were rotated (Movies S1 and S2). Consistent with this, Thm2−/−; Thm1aln/+ tibia showed reduced cortical bone mineral density (BMD), cortical thickness, and cortical bone area at mid-diaphysis and proximal end of the tibia (Figs. 6g-6h). Additionally, cortical area fraction (cortical bone area/total area) at the proximal end of the tibia, as well as trabecular BMD were reduced (Fig. 6i, j). Other cortical and trabecular bone properties of control and Thm2−/−; Thm1aln/+ tibia were similar (Fig. S1). These data demonstrate that Thm2, together with Thm1, is required for postnatal skeletal growth.
Fig. 6.

Thm2−/−; Thm1aln/+ tibia have less dense bone. a Radiographs of P14 skeleton and b vertebra. Scale bars—1 cm and 500 μm, respectively. c Caudal vertebrae lengths by number. d Caudal vertebrae/Crown-to-Rump (C-to-R) length ratios. Graphs represent mean ± SD of n = 3 ctrl and n = 3 triple allele mutant mice. Statistical significance was determined using two-tailed, unpaired t-test. *p < 0.05; **p < 0.01 e Radiographs of hindlimbs and f microCT imaging of tibias. Scale bars—5 mm and 1 mm, respectively. g Cortical bone mineral density (BMD), thickness and area at mid-diaphysis and h at the proximal end of tibia. i Cortical bone area/total bone area (fraction) at the proximal end of tibia. j Trabecular BMD at proximal end of tibia. Graphs represent mean ± SD of n = 3 ctrl and n = 3 triple allele mutant mice. Statistical significance was determined using a two-tailed, unpaired t test. *p < 0.05; **p < 0.005
Growth plates of Thm2−/−;Thm1aln/+ show aberrant architecture
To determine the cause of the tibial skeletal defects, we next examined chondrocyte organization in the tibial growth plate. Broadly, the growth plate can be divided into two major regions: the proliferative zone and the hypertrophic zone [45]. As part of endochondral ossification, chondrocyte differentiation occurs. In this process, early chondroblasts are stimulated to proliferate, then become pre-hypertrophic, hypertrophic, and finally undergo cell death and calcification of cartilage. Capillaries and osteoblasts invade the cartilage matrix and form new bone which replaces cartilage. H&E staining, as well as Safranin O and fast green staining, which marks cartilage and bone, respectively, of P14 Thm2−/−; Thm1aln/+ tibial growth plates revealed an expanded proliferation zone and a reduced hypertrophic zone (Fig. 7). We also observed a more basophilic H&E staining and more intense safranin-O staining in the Thm2−/−; Thm1aln/+growth plate cartilage proliferation zone. This may indicate a change in the proteoglycan content of the extracellular matrix. Together, these defects suggest impaired chondrocyte hypertrophy and endochondral ossification, which could contribute to the shortened long bones seen in Thm2−/−;Thm1aln/+ mice.
Fig. 7.
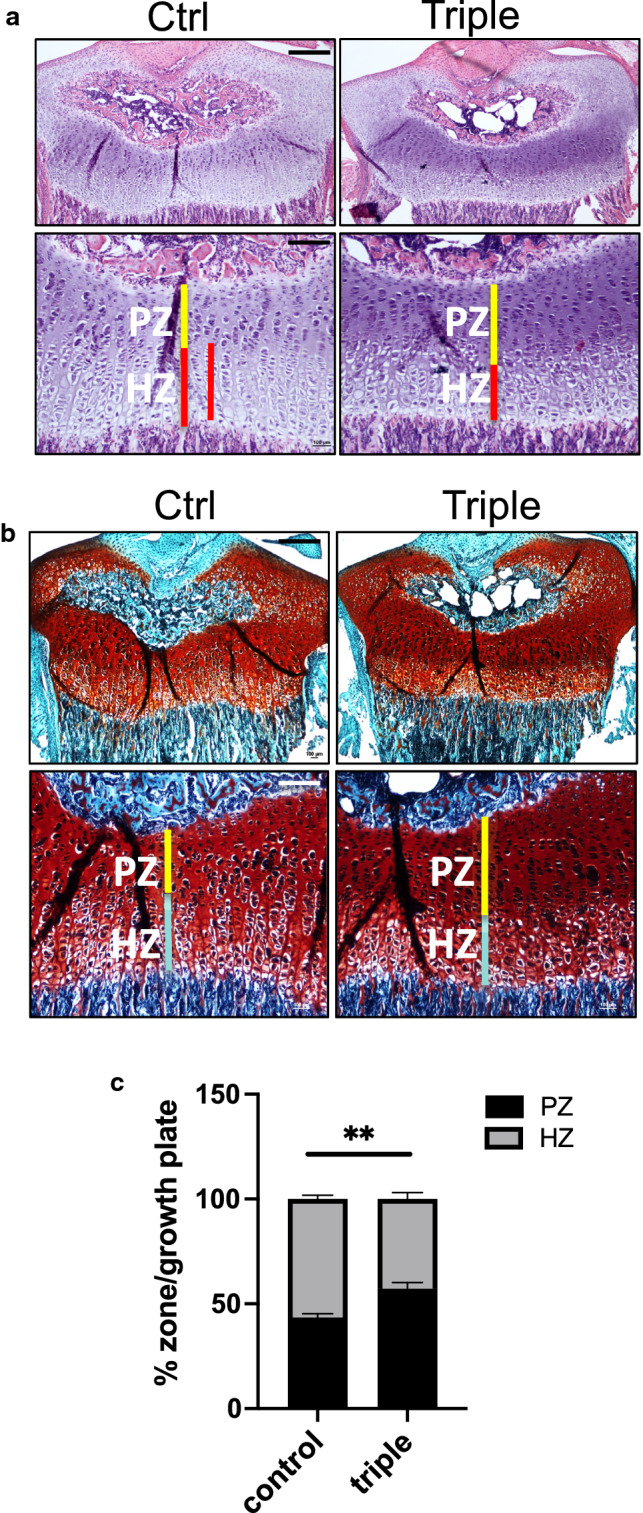
Thm2−/−; Thm1aln/+ mice have altered tibial growth plates. a Haematoxylin and eosin (H&E) and b Safranin O and fast green staining of growth plates at P14. Scale bars—500 μm and 200 μm, upper and lower panels. PZ - proliferation zone, HZ - hypertropic zone c Quantification of % PZ/growth plate and % HZ/growth plate from H&E images. Error bars represent SD of n = 3 ctrl and n = 3 triple allele mutant growth plates. Statistical significance was determined using two-tailed, unpaired t test. **p ≤ 0.005
Thm2−/−; Thm1aln/+chondrocytes show increased Hh response
Chondrocyte differentiation in the growth plate is regulated by ciliary-mediated canonical Hedgehog (Hh) signaling [46]. We, therefore, examined primary cilia and Hh signaling in triple allele mutant chondrocytes. Immunostaining for the ciliary marker, Arl13B, on chondrocytes of the growth plate suggested similar ciliary lengths between control and Thm2−/−; Thm1aln/+ tissue (Fig. 8a). Incorporation of a Gli1-lacZ allele, which acts as a reporter of the Hh pathway, revealed increased Gli1 expression in chondrocytes of the triple allele mutant growth plate (Figs. 8b and c). To further examine Hh signaling, we generated primary chondrocytes from P10 tibial growth plates (Fig. 8d). Cilia lengths were similar between control and triple allele mutant primary cells (Fig. 8e, f). In response to Hh stimulation, the Smoothened (SMO) signal transducer translocates to the primary cilium [12]. Accordingly, treatment of cells with Smoothened Agonist (SAG) caused the enrichment of SMO in cilia (Fig. 8g). SAG-induced increase in ciliary SMO was similar between control and triple allele mutant chondrocytes (Fig. 8h). qPCR revealed reduced Thm2 transcripts in triple allele mutant cells (Fig. 8i). In both control and triple allele mutant chondrocytes, SAG treatment caused an increase in Gli1 and Gli2 transcripts and did not affect Gli3 (Fig. 8j–l). However, SAG induced a greater increase in Gli1 transcription in triple allele mutant chondrocytes relative to control cells, suggesting the triple allele mutation causes an increased Hh response in chondrocytes.
Fig. 8.

Hedgehog signaling is increased in Thm2−/−; Thm1aln/+ chondrocytes. a Immunostaining of P14 tibial growth plates for ciliary membrane marker, Arl13B (green). Scale bar—10 µm. b Gli-lacZ staining of growth plates. Scale bar—100 µm. c Quantification of Gli1-lacZ staining. Error bars represent mean ± SEM from n = 4 ctrl and n = 5 triple allele mutant mice. d Phase contrast images of tibial growth plate-derived primary chondrocytes. Scale bar—100 µm. e Immunostaining for ciliary markers, acetylated α-tubulin (red) and IFT81 (green). Scale bar—10 µm. f Quantification of ciliary lengths. Error bars represent mean ± SD from n = 3 ctrl and n = 3 triple allele mutant mice. g Ciliary localization of SMO (red) and ARL13B (green) in response to vehicle (DMSO) or SAG. Scale bar—10 µm. h Quantification of percentage of cells with ciliary SMO i qPCR for Thm2; j Gli1; k Gli2; and l Gli3 relative to Oaz1. Bar graphs represent mean ± SEM from cell cultures of n = 4 ctrl and n = 4 triple allele mutant mice. Statistical significance was determined using unpaired t test for c and i and one-way ANOVA and Tukey’s test for h–l. *p < 0.05; **p < 0.01, ****p < 0.001
Reducing Gli2 dosage in Thm2-null mice results in a small phenotype
To determine whether Hh signaling contributes to the skeletal phenotype, we introduced a null allele of Gli2, the main transcriptional activator of the Hh pathway, into the Thm2; Thm1 colony. Since chondrocytes show an elevated Hh response, we anticipated that reducing Hh signaling would correct the runted phenotype. While Gli2−/− mice show skeletal defects and perinatal lethality, the Gli2 ± genotype does not result in a phenotype [47]. Yet the loss of one allele of Gli2 in Thm2−/−; Thm1aln/+; Gli2+/-mice further decreased body weight and crown-to-rump length relative to triple allele mutant mice (Fig. 9a–c). Additionally, loss of one allele of Gli2 in Thm2−/−; Gli2 +/- mice resulted in a small phenotype, with reduced body weight and crown-to-rump length relative to control and Thm2−/− mice. Thus, reducing Hh signaling uncovers a role for Thm2 in postnatal skeletal growth. Further, the exacerbated runted phenotype of triple allele mutants by Gli2 deficiency could suggest that Thm2, together with Thm1, positively regulates the Hh pathway in cells that are not chondrocytes.
Fig. 9.

Gli2 deficiency causes small phenotype in Thm2 ko mice and exacerbates small phenotype in Thm2−/−; Thm1aln/+ mice. a Images of P14 littermates. b Body weight and crown-to-rump lengths at P14 and c at P21. Graphs represent mean ± SD. Statistical significance was determined using one-way ANOVA and Tukey’s test. #p < 0.05; ##p < 0.01; ###p < 0.005; ####p < 0.0001 relative to Ctrl. *p < 0.05
Thm2−/−; Thm1aln/+osteoblasts show defective differentiation
We, therefore, derived primary osteoblasts from calvaria of P10 mice (Fig. 10a). We immunostained osteoblasts for primary cilia (Fig. 10b), which revealed similar cilia lengths between control and mutant osteoblasts (Fig. 10c). SAG treatment resulted in normal ciliary localization of SMO in mutant osteoblasts (Fig. 10d, e). qPCR showed reduced Thm2 transcripts in triple allele mutant osteoblasts (Fig. 10f). SAG treatment-induced increases in Gli1 transcripts that were similar between control and mutant osteoblasts (Fig. 10g), while Gli2 and Gli3 transcripts were not altered (Fig. 10h, i). However, the formation of bone nodules by the triple allele mutant osteoblasts was markedly impaired (Fig. 10j, k), indicating the triple allele mutation impairs differentiation of osteoblasts.
Fig. 10.

Thm2−/−; Thm1aln/+ osteoblasts show defective differentiation. a Phase contrast images of calvarial-derived primary osteoblasts. Scale bar—100 µm. b Immunostaining for acetylated α-tubulin (red) and IFT81 (green). Scale bar—10 µm. c Quantification of ciliary lengths. Error bars represent mean ± SD from n = 3 ctrl and n = 3 triple allele mutant mice. d Ciliary localization of SMO (red) and ARL13B (green) in response to vehicle (DMSO) or SAG. Scale bar—10 µm. e Quantification of proportion of cells with ciliary SMO. Bar graphs represent mean ± SEM from cell cultures of n = 3 ctrl and n = 3 triple allele mutant mice. f qPCR for Thm2; g Gli1; h Gli2; i Gli3 relative to Oaz1. Bar graphs represent mean ± SEM from cell cultures of n = 4 ctrl and n = 4 triple allele mutant mice. j Alizarin red staining following bone nodule formation assay. Scale bar—500 µm. k Quantification of bone nodule formation of cell cultures from n = 4 ctrl and n = 4 triple allele mutant mice. Statistical significance was determined using one-way ANOVA and Tukey’s test for e, g–i, and using two-tailed, unpaired t test for f and k. *p < 0.05; **p < 0.01
Discussion
This study establishes THM2 as a ciliary protein. Thm2 depletion results in normal cilia length, but the loss of Thm2, together with loss of one allele of either Thm1 or Gli2, impairs postnatal skeletal growth, resulting in a small phenotype. The small phenotype in Thm2−/−; Thm1aln/+ mouse mutants is accompanied by small mandible and architectural defects of the long bone (tibia), including decreased bone mineral density. Additionally, the Thm2−/−; Thm1aln/+ genetic interaction is important for both chondrocyte and osteoblast differentiation, and for negative regulation of Hh signaling downstream of SMO ciliary localization in chondrocytes. Our findings expand the IFT genes and genetic network that regulate skeletal development. The Thm2−/−; Thm1aln/+ mouse is also the first global Ift-null mouse model surviving embryogenesis and exhibiting postnatal skeletal abnormalities.
Although Thm2 and Thm1 genetically interact in skeletal development, notably, the skeletal and ciliary phenotypes differ between Thm1aln/aln and Thm2−/−; Thm1aln/+ mouse mutants. The Thm1aln/aln mouse mutant exhibits shortened long bones and ribs, with normal crown-to-rump length [32]. Additionally, mutations in THM1 have been identified in patients with Jeune Syndrome, a skeletal ciliopathy characterized by shortened long bones and ribs relative to body trunk length [18]. In contrast to this disproportionate dwarfism, the Thm2−/−; Thm1aln/+ mice are small in stature overall. There are other IFT mouse mutants that also display an overall runted phenotype. This includes the Ift140 conditional knock-out mouse, in which Ift140 is deleted in osteoblasts using the Osx-Cre [48]. Additionally, Ift80gt/gt hypomorphic mutant mice are also small in stature [35]. Proportionately small mice may reflect that the mutation affects the growth of most or all bones, instead of primarily affecting the long bones and ribs. In the human population, proportionate dwarfism causes small stature. In patients with proportionate dwarfism, mutations have been identified in centriole-related genes, such as pericentrin [49], CEP152 [50], and CENPJ4 [51]. Interestingly, defects in centriolar satellites have been observed in cells depleted of certain IFT-A subunits [52], raising the possibility that a centriole-related component might also be defective in the small IFT mouse mutants and thus may warrant investigation.
Further contrasting with Thm1aln/aln mouse embryos [17, 32], Thm2−/−; Thm1aln/+ mice are not polydyactylous. Preaxial polydactyly is caused by disrupted GLI3A:GLI3R ratios along the anterior–posterior axis of the developing limb bud [53]. Since Thm2 and Thm1 show very similar expression patterns in E10.5 whole-mount mouse embryos [17], the absence of polydactyly in Thm2−/−; Thm1aln/+ mice indicates divergent mechanisms of Hh regulation between the Thm2;Thm1 genetic interaction and Thm1 alone. Thm1 loss disrupts GLI3 processing [17], while the Thm2−/−; Thm1aln/+ mutation likely does not. Finally, while Thm2−/−; Thm1aln/+ cilia lengths and kidneys appear normal, loss of Thm1 causes shortened primary cilia with a bulbous distal tip and cystic kidneys [54]. Together, these differences reflect the unique and redundant roles of the Thm1 and Thm2 paralogs.
In the growth plate, Parathyroid hormone-related protein (PTHrP) is expressed by perichondral cells and chondrocytes to promote chondrocyte proliferation [45]. When PTHrP levels reach below a certain threshold, pre-hypertrophic and early hypertrophic cells produce Indian Hedgehog (Ihh), which effects various skeletal processes, including increased chondrocyte proliferation, stimulation of PTHrP production, and differentiation of perichondral cells to osteoblasts of the bone collar [45]. In E14.5 Ihh-null mice, Hh signaling is decreased in growth plates and the proliferation zone is reduced [55]. Conversely, in mice harboring a Col2a1-specific knock-out of Suppressor of Fused (Sufu), a negative regulator of Hh signaling, Hh signaling is increased in growth plates, causing an increased proliferation zone and a reduced hypertrophic zone [56]. Similarly, in triple allele mutant growth plates, Hh signaling is increased in chondrocytes, and the proliferation zone is increased while the hypertrophic zone is reduced. Thus, deficiency of Hh signaling causes chondrocytes to undergo premature hypertrophy, while inappropriate activation of Hh signaling causes chondrocytes to remain in the proliferation zone[57]. Both alterations impair endochondral bone formation.
In triple allele mutant osteoblasts, a defect in Hh response to (500 nM) SAG was not observed. The differential Hh response in chondrocytes and osteoblasts indicates cell-specific regulation of Hh signaling by the triple allele mutation. Cell-specific Hh regulation by Thm1 and other ciliary genes has been reported [17, 20, 58]. In other experiments, we have observed that SAG-treated Thm2−/− mouse embryonic fibroblasts (MEF) have normal SMO ciliary localization and Gli1 transcription, while SAG-treated triple allele mutant MEF shows normal SMO ciliary localization but reduced Gli1 transcription [59]. Collectively, our data suggest that the triple allele mutation alters Hh signaling in a cell-specific manner, downstream of SMO ciliary localization, possibly at the level of SMO or GLI activation.
Although a defect in Hh signaling was not detected in triple allele mutant primary osteoblasts, in vitro differentiation was impaired. This could indicate two possibilities: (1) that the in vitro Hh assays were not sensitive enough to detect a more subtle Hh signaling defect and/or (2) that another signaling pathway is perturbed. Misregulation of several pathways, including TGF-β, Notch and Wnt, hampers in vitro bone nodule formation, and knockout mice of components of these pathways exhibit skeletal defects [41, 60–63]. Thus, future experiments will examine these two possibilities. Since Gli2 deficiency in the triple allele mutant exacerbated the runted phenotype, rather than rescuing it, we propose that the chondrocyte defect may be superseded by the osteoblast defect in this global knock-out. In endochondral ossification, osteoblasts act downstream of chondrocytes, replacing the extracellular matrix laid down by the chondrocytes with bone. Additionally, deletion of IFT-A gene, Ift140, in osteoblasts causes a small phenotype [48], demonstrating that loss of an IFT gene in osteoblasts can result in small stature. Since the loss of just one allele of Gli2 is sufficient to worsen the triple allele mutant small phenotype, we propose that the altered pathway in osteoblasts may potentially interact with Gli2.
Skeletal ciliopathies manifest varying degrees of severity, and this characteristic is present in triple allele mutant mice, in which almost a third of triple allele mutants do not survive to weaning. In addition to levels of Gli2, genetic background modifies the triple allele mutant small phenotype, since the phenotype was apparent only once the colony was backcrossed from a mixed FVB/C57BL6/J strain background to C57BL6/J over five generations. Taken together, cell type, genetic interaction, and strain background are important determinants of cilia dysfunction and ciliopathy phenotypes. Additionally, the mechanisms by which ciliary defects cause developmental defects can differ due to variations in the function of individual ciliary proteins. For example, germline deletion of almost all reported Ift genes causes misregulation of the Hh pathway, but not all Ift gene deletions affect ciliary structure [11, 64].
Direct evidence of a ciliopathy in Thm2−/−; Thm1aln/+ mice is still lacking. Finding a ciliary defect (structural and/or ciliary mislocalization of proteins) may require high-resolution imaging. For instance, Ift25−/− cilia appear normal using conventional fluorescence microscopy [64] but show abnormal structure using super-resolution microscopy [65]. Additionally, Ift56hop/hop cilia also appear grossly normal but have defects of the axonemal microtubules revealed by transmission electron microscopy [66]. Alternatively, Thm2 together with Thm1 may play a role outside the cilium. Future experiments will explore both possibilities.
Thm2 is the least explored IFT gene, but roles for THM2 are emerging. Mutations in THM2 have been identified in male patients with infertility [38]. In contrast, our Thm2−/− mice do not show evidence of fertility defects, which could suggest there is redundancy in the mouse or that Thm2 functions as part of a genetic network to regulate fertility. Other studies also suggest that THM2 expression may be misregulated in certain cancers and in neuronal degeneration [67–69]. For the first time, our study shows that THM2 can localize to primary cilia and that Thm2 regulates postnatal skeletal development. Thus, THM2 could be an additional locus to query in patients with skeletal dysplasia. This work generates the first global IFT gene knock-out mouse that allows the study of skeletal development postnatally. This model will enable future biomechanical studies, including identification of mechanical or chemical stimuli that are received and transduced by primary cilia to regulate bone development and homeostasis.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank members of the KUMC Dept. of Anatomy and Cell Biology, the Jared Grantham Kidney Institute, and the UMKC Dept. of Oral and Craniofacial Sciences for helpful discussions. We thank Ms. Jing Huang of the KUMC Histology Core and Drs. Vivian and Larson of the KUMC Gene Targeting Institution Facility and acknowledge the support of these cores (Intellectual and Developmental Disabilities Research Center NIH U54 HD090216; KU Cancer Center NIH P30 CA168524; COBRE NIH P30 GM122731). This work was also supported by a KUMC Biomedical Research Training Fellowship, a pilot grant from the Kansas City Consortium on Musculoskeletal Diseases, and the National Institutes of Health (P20 GM14936; R01DK103033).
Author contributions
BAA, WW, TSP, HM, BMJ, HHW, LMS, DTJ, JW, EEB, and PVT performed experiments and analyzed data. BAA, JW, EEB and PVT designed research. BAA, HM, JW, EEB and PVT wrote the manuscript. All authors read and accepted the final manuscript.
Funding
We thank members of the KUMC Dept. of Anatomy and Cell Biology, the Jared Grantham Kidney Institute, and the UMKC Dept. of Oral and Craniofacial Sciences for helpful discussions. We thank Jing Huang of the KUMC Histology Core and Drs. Vivian and Larson of the KUMC Gene Targeting Institution Facility and acknowledge the support of these cores (Intellectual and Developmental Disabilities Research Center NIH U54 HD090216; KU Cancer Center NIH P30 CA168524; COBRE NIH P30 GM122731). This work was also supported by a KUMC Biomedical Research Training Fellowship, a pilot grant from the Kansas City Consortium on Musculoskeletal Diseases, and the National Institutes of Health (P20 GM14936; R01DK103033).
Code availability
Not applicable.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
All animal procedures were conducted in accordance with KUMC IACUC and AAALAC rules and regulations.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, Dallas SL, Maser R, Calvet JP, Bonewald L, Quarles LD. Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J Biol Chem. 2006;281(41):30884–30895. doi: 10.1074/jbc.M604772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol. 2007;305(1):202–216. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinlan RJ, Tobin JL, Beales PL. Modeling ciliopathies: primary cilia in development and disease. Curr Top Dev Biol. 2008;84:249–310. doi: 10.1016/s0070-2153(08)00605-4. [DOI] [PubMed] [Google Scholar]
- 4.Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, Krug P, Filhol E, Davis EE, Airik R, Czarnecki PG, Lehman AM, Trnka P, Nitschke P, Bole-Feysot C, Schueler M, Knebelmann B, Burtey S, Szabo AJ, Tory K, Leo PJ, Gardiner B, McKenzie FA, Zankl A, Brown MA, Hartley JL, Maher ER, Li C, Leroux MR, Scambler PJ, Zhan SH, Jones SJ, Kayserili H, Tuysuz B, Moorani KN, Constantinescu A, Krantz ID, Kaplan BS, Shah JV, Consortium UK. Hurd TW, Doherty D, Katsanis N, Duncan EL, Otto EA, Beales PL, Mitchison HM, Saunier S, Hildebrandt F. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93(5):915–925. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111(3):p39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valente EM, Rosti RO, Gibbs E, Gleeson JG. Primary cilia in neurodevelopmental disorders. Nat Rev Neurol. 2014;10(1):27–36. doi: 10.1038/nrneurol.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24(19):2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liem KF, Jr, Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, Anderson KV. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol. 2012;197(6):789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirano T, Katoh Y, Nakayama K. Intraflagellar transport-A complex mediates ciliary entry and retrograde trafficking of ciliary G protein-coupled receptors. Mol Biol Cell. 2017;28(3):429–439. doi: 10.1091/mbc.E16-11-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Picariello T, Brown JM, Hou Y, Swank G, Cochran DA, King OD, Lechtreck K, Pazour GJ, Witman GB. A global analysis of IFT-A function reveals specialization for transport of membrane-associated proteins into cilia. J Cell Sci. 2019 doi: 10.1242/jcs.220749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 13.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1(4):e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 15.Arsov T, Silva DG, O'Bryan MK, Sainsbury A, Lee NJ, Kennedy C, Manji SS, Nelms K, Liu C, Vinuesa CG, de Kretser DM, Goodnow CC, Petrovsky N. Fat aussie—a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20(7):1610–1622. doi: 10.1210/me.2005-0494. [DOI] [PubMed] [Google Scholar]
- 16.Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102(32):11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008;40(4):403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Gyapay G, Rieger S, Tonshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H, Gallagher TE, Lewis RA, Bergmann C, Otto EA, Saunier S, Scambler PJ, Beales PL, Gleeson JG, Maher ER, Attie-Bitach T, Dollfus H, Johnson CA, Green ED, Gibbs RA, Hildebrandt F, Pierce EA, Katsanis N. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43(3):189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CF, Chang YT, Millington G, Brugmann SA. Craniofacial ciliopathies reveal specific requirements for GLI proteins during development of the facial midline. PLoS Genet. 2016;12(11):e1006351. doi: 10.1371/journal.pgen.1006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Driver AM, Shumrick C, Stottmann RW. Ttc21b is required in bergmann glia for proper granule cell radial migration. J Dev Biol. 2017 doi: 10.3390/jdb5040018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers JH, de Leeuw N, Cormier-Daire V, Brunner HG, Knoers NV, Roepman R. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48(6):390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 22.Duran I, Taylor SP, Zhang W, Martin J, Qureshi F, Jacques SM, Wallerstein R, Lachman RS, Nickerson DA, Bamshad M, Cohn DH, Krakow D. Mutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive campomelia. Cilia. 2017;6:7. doi: 10.1186/s13630-017-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW. Cranioectodermal dysplasia, sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet. 2010;86(6):949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alazami AM, Seidahmed MZ, Alzahrani F, Mohammed AO, Alkuraya FS. Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol Genet Genomic Med. 2014;2(2):103–106. doi: 10.1002/mgg3.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbo M, Filhol E, Bole-Feysot C, Nitschke P, Gilissen C, Haugen OH, Sanders JS, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BC, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rodahl E, Veltman JA, Knappskog PM, Knoers NV, Roepman R, Arts HH. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89(5):634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90(5):864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci USA. 2011;108(11):4482–4487. doi: 10.1073/pnas.1018217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39(6):727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 29.Girisha KM, Shukla A, Trujillano D, Bhavani GS, Hebbar M, Kadavigere R, Rolfs A. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016;90(6):536–539. doi: 10.1111/cge.12762. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Taylor SP, Nevarez L, Lachman RS, Nickerson DA, Bamshad M, University of Washington Center for Mendelian Genomics C. Krakow D, Cohn DH. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet. 2016;25(18):4012–4020. doi: 10.1093/hmg/ddw241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaheen R, Alsahli S, Ewida N, Alzahrani F, Shamseldin HE, Patel N, Al Qahtani A, Alhebbi H, Alhashem A, Al-Sheddi T, Alomar R, Alobeid E, Abouelhoda M, Monies D, Al-Hussaini A, Alzouman MA, Shagrani M, Faqeih E, Alkuraya FS. Biallelic mutations in tetratricopeptide repeat domain 26 (Intraflagellar Transport 56) cause severe biliary ciliopathy in humans. Hepatology. 2020;71(6):2067–2079. doi: 10.1002/hep.30982. [DOI] [PubMed] [Google Scholar]
- 32.Herron BJ, Lu W, Rao C, Liu S, Peters H, Bronson RT, Justice MJ, McDonald JD, Beier DR. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat Genet. 2002;30(2):185–189. doi: 10.1038/ng812. [DOI] [PubMed] [Google Scholar]
- 33.Miller KA, Ah-Cann CJ, Welfare MF, Tan TY, Pope K, Caruana G, Freckmann ML, Savarirayan R, Bertram JF, Dobbie MS, Bateman JF, Farlie PG. Cauli: a mouse strain with an Ift140 mutation that results in a skeletal ciliopathy modelling Jeune syndrome. PLoS Genet. 2013;9(8):e1003746. doi: 10.1371/journal.pgen.1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashe A, Butterfield NC, Town L, Courtney AD, Cooper AN, Ferguson C, Barry R, Olsson F, Liem KF, Jr, Parton RG, Wainwright BJ, Anderson KV, Whitelaw E, Wicking C. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum Mol Genet. 2012;21(8):1808–1823. doi: 10.1093/hmg/ddr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rix S, Calmont A, Scambler PJ, Beales PL. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum Mol Genet. 2011;20(7):1306–1314. doi: 10.1093/hmg/ddr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134(2):307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 37.Drera B, Ferrari D, Cavalli P, Poggiani C. A case of neonatal Jeune syndrome expanding the phenotype. Clin Case Rep. 2014;2(4):156–158. doi: 10.1002/ccr3.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, He X, Yang S, Zouari R, Wang J, Wu H, Kherraf ZE, Liu C, Coutton C, Zhao R, Tang D, Tang S, Lv M, Fang Y, Li W, Li H, Zhao J, Wang X, Zhao S, Zhang J, Arnoult C, Jin L, Zhang Z, Ray PF, Cao Y, Zhang F. Bi-allelic Mutations in TTC21A induce asthenoteratospermia in humans and mice. Am J Hum Genet. 2019;104(4):738–748. doi: 10.1016/j.ajhg.2019.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada A, Komatsu K, Nakashima K, Poschl E, Nifuji A. Improved methods for detection of beta-galactosidase (lacZ) activity in hard tissue. Histochem Cell Biol. 2012;137(6):841–847. doi: 10.1007/s00418-012-0936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belluoccio D, Etich J, Rosenbaum S, Frie C, Grskovic I, Stermann J, Ehlen H, Vogel S, Zaucke F, von der Mark K, Bateman JF, Brachvogel B. Sorting of growth plate chondrocytes allows the isolation and characterization of cells of a defined differentiation status. J Bone Miner Res. 2010;25(6):1267–1281. doi: 10.1002/jbmr.30. [DOI] [PubMed] [Google Scholar]
- 41.Dallas SL, Miyazono K, Skerry TM, Mundy GR, Bonewald LF. Dual role for the latent transforming growth factor-beta binding protein in storage of latent TGF-beta in the extracellular matrix and as a structural matrix protein. J Cell Biol. 1995;131(2):539–549. doi: 10.1083/jcb.131.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci USA. 2011;108(4):1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127(8):1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- 44.Abbasi AA, Goode DK, Amir S, Grzeschik KH. Evolution and functional diversification of the GLI family of transcription factors in vertebrates. Evol Bioinform Online. 2009;5:5–13. doi: 10.4137/ebo.s2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 46.Yuan X, Yang S. Primary cilia and intraflagellar transport proteins in bone and cartilage. J Dent Res. 2016;95(12):1341–1349. doi: 10.1177/0022034516652383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, Joyner AL, Hui C. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124(1):113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- 48.Tao D, Xue H, Zhang C, Li G, Sun Y. The role of IFT140 in osteogenesis of adult mice long bone. J Histochem Cytochem. 2019;67(8):601–611. doi: 10.1369/0022155419847188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dorfler A, Kinning E, Megarbane A, Meinecke P, Semple RK, Spranger S, Toutain A, Trembath RC, Voss E, Wilson L, Hennekam R, de Zegher F, Dorr HG, Reis A. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 50.Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tuysuz B, Nurnberg G, Kiess W, Koegl M, Baessmann I, Buruk K, Toraman B, Kayipmaz S, Kul S, Ikbal M, Turner DJ, Taylor MS, Aerts J, Scott C, Milstein K, Dollfus H, Wieczorek D, Brunner HG, Hurles M, Jackson AP, Rauch A, Nurnberg P, Karaguzel A, Wollnik B. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43(1):23–26. doi: 10.1038/ng.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47(6):411–414. doi: 10.1136/jmg.2009.076646. [DOI] [PubMed] [Google Scholar]
- 52.Fu W, Wang L, Kim S, Li J, Dynlacht BD. Role for the IFT-A complex in selective transport to the primary cilium. Cell Rep. 2016;17(6):1505–1517. doi: 10.1016/j.celrep.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. doi: 10.1016/S0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 54.Tran PV, Talbott GC, Turbe-Doan A, Jacobs DT, Schonfeld MP, Silva LM, Chatterjee A, Prysak M, Allard BA, Beier DR. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol. 2014;25(10):2201–2212. doi: 10.1681/ASN.2013070735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsu SH, Zhang X, Cheng S, Wunder JS, Hui CC, Alman BA. Suppressor of fused (Sufu) mediates the effect of parathyroid hormone-like hormone (Pthlh) on chondrocyte differentiation in the growth plate. J Biol Chem. 2012;287(43):36222–36228. doi: 10.1074/jbc.M112.382275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J, Andre P, Ye L, Yang YZ. The Hedgehog signalling pathway in bone formation. Int J Oral Sci. 2015;7(2):73–79. doi: 10.1038/ijos.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schock EN, Struve JN, Chang CF, Williams TJ, Snedeker J, Attia AC, Stottmann RW, Brugmann SA. A tissue-specific role for intraflagellar transport genes during craniofacial development. PLoS ONE. 2017;12(3):e0174206. doi: 10.1371/journal.pone.0174206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Allard BA, Pottorf TS, Wang HH, Vivian JL, Tran PV. Genetic interaction of mammalian IFT-A paralogs regulates cilia disassembly, ciliary entry of membrane protein, Hedgehog signaling, and embryogenesis. FASEB J. 2020 doi: 10.1096/fj.201902611R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124(13):2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ji Y, Ke Y, Gao S. Intermittent activation of notch signaling promotes bone formation. Am J Transl Res. 2017;9(6):2933–2944. [PMC free article] [PubMed] [Google Scholar]
- 62.Tao J, Chen S, Lee B. Alteration of notch signaling in skeletal development and disease. Ann N Y Acad Sci. 2010;1192:257–268. doi: 10.1111/j.1749-6632.2009.05307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong Z, Ethen NJ, Williams BO. WNT signaling in bone development and homeostasis. Wiley Interdiscip Rev Dev Biol. 2014;3(6):489–500. doi: 10.1002/wdev.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev Cell. 2012;22(5):940–951. doi: 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon J, Comerci CJ, Weiss LE, Milenkovic L, Stearns T, Moerner WE. Revealing nanoscale morphology of the primary cilium using super-resolution fluorescence microscopy. Biophys J. 2019;116(2):319–329. doi: 10.1016/j.bpj.2018.11.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xin D, Christopher KJ, Zeng L, Kong Y, Weatherbee SD. IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development. 2017;144(8):1544–1553. doi: 10.1242/dev.143255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dmitriev AA, Rudenko EE, Kudryavtseva AV, Krasnov GS, Gordiyuk VV, Melnikova NV, Stakhovsky EO, Kononenko OA, Pavlova LS, Kondratieva TT, Alekseev BY, Braga EA, Senchenko VN, Kashuba VI. Epigenetic alterations of chromosome 3 revealed by NotI-microarrays in clear cell renal cell carcinoma. Biomed Res Int. 2014;2014:735292. doi: 10.1155/2014/735292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Ren S, Wang Z, Zhang C, Huang J. Increased expression of TTC21A in lung adenocarcinoma infers favorable prognosis and high immune infiltrating level. Int Immunopharmacol. 2020;78:106077. doi: 10.1016/j.intimp.2019.106077. [DOI] [PubMed] [Google Scholar]
- 69.Li S, Weinstein G, Zare H, Teumer A, Volker U, Friedrich N, Knol MJ, Satizabal CL, Petyuk VA, Adams HHH, Launer LJ, Bennett DA, De Jager PL, Grabe HJ, Ikram MA, Gudnason V, Yang Q, Seshadri S. The genetics of circulating BDNF: towards understanding the role of BDNF in brain structure and function in middle and old ages. Brain Commun. 2020;2(2):fcaa176. doi: 10.1093/braincomms/fcaa176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.