

Abstract
For decades, the marine snail Aplysia has proven to be a powerful system for analyzing basic neurobiological mechanisms, particularly cellular and molecular mechanisms of neural plasticity. Three new findings on Aplysia may be relevant for the understanding and treatment of chronic human disorders. This research on this simple molluscan nervous system may lead to new therapeutic approaches for spinal cord injury, Fragile X syndrome, and genetic learning deficits more generally.
The relative simplicity of Aplysia’s nervous system and the ease with which changes in its identified neurons can be shown to participate in specific behavioral changes have enabled a rich series of discoveries about fundamental mechanisms of neuronal plasticity. In addition, the clear, direct link between neuronal plasticity in Aplysia and behavior frequently enables insights into functional roles of cellular and molecular processes that would otherwise be missed. Researchers studying simpler model systems such as Aplysia frequently put their research in the context of the challenges of understanding human diseases and suggest that insights gained in these more readily analysable systems may ultimately lead to advances in the treatment of clinical disorders. For example, in describing a novel molecular mechanism that underlies an attention-like process in Aplysia, which was understood precisely because of the direct link between synaptic plasticity and behavior, we recently suggested that this mechanism may also contribute to attentional processes in mammals [1]. The link between basic biology and disease is epitomized by the transcription factor CREB. The initial discovery that CREB plays a critical role in initiating persistent plasticity during learning was made in Aplysia [2]; subsequently, this finding led to an understanding of the role of CREB-binding protein (CBP) in memory and of the importance of the CBP mutation in Rubenstein-Taybi syndrome.
Typically, the actual relevance of such findings in Aplysia for human disease is entirely unanticipated at the time the novel findings are made, and may take some years to be appreciated. Perhaps because the immediate relevance for human disease is unclear, some researchers discount the power of this non-genetic system for gaining relevant fundamental insights. Contrary to the typical progression where the relevance of discoveries emerges some years later, three recent studies presented at the 2012 Molluscan Neuroscience Meeting at Scripps, Florida suggest that investigations in Aplysia may more directly lead to improved strategies for treatment of human disorders.
The history of these three studies is quite disparate. Only one began with an initial translational focus on a disease model, chronic pain. Another study has provided a remarkable insight into how Fragile X Mental Retardation Protein (FMRP) may mediate coupling between neuronal activity and local protein synthesis. A third advance emerged out of computational studies of the kinetics of signaling pathways involved in memory, which are now leading to new strategies for improving learning in genetic disorders that disrupt these biochemical pathways.
Can computational models of signaling pathways be used to ameliorate genetic cognitive disorders?
In a recent paper, Jack Byrne and colleagues [3] described an innovative modeling approach in which they utilized information about the dynamics of activation of molecular signaling pathways in an attempt to optimize training protocols for behavioral learning experiments in Aplysia. Typically, computational models are used to make predictions on the analytical level at which they are developed. In contrast, Byrne and colleagues took the bold step of attempting to predict optimal cellular and behavioral training protocols based on their understanding of the temporal dynamics of the molecular signaling pathways that initiate long-term synaptic plasticity and learning. As emphasized above, what is exceptional about the simple Aplysia nervous system is that links between molecular and cellular processes and behavior are unusually direct; this simple, straight-forward relationship encouraged the use of simulations of biochemical processes to generate testable predictions about cellular and behavioral outcomes.
It has long been known by both educators and researchers studying learning that when studying efforts or learning trials are ‘massed’ — that is, when all the training occurs in a single long session — the resulting long-term memory is poor compared with spaced learning trials. Nevertheless, little is known about how the precise timing of training trials impacts learning because, traditionally, training involves regularly spaced trials. In Aplysia, most behavioral and cellular studies of long-term learning use four or five training trials at regular intervals of 20 or 30 minutes. In behavioral long-term sensitization training, animals are given four to five trials with tail shocks; when animals are tested one day later, there is long-term enhancement of their defensive withdrawal reflexes, known as sensitization. In a cellular analog, presynaptic sensory neurons and postsynaptic motor neurons, which have been paired in dissociated cell co-culture, are given a series of five minute exposures to serotonin, the facilitatory transmitter released by tail shock. One day later, the sensory-motor synapses display long-term strengthening or facilitation, which in the intact animal underlies behavioral sensitization. It is well known in these paradigms in Aplysia that spaced trials are superior to massed trials for initiating both long-term synaptic facilitation and long-term memory; however, it was not known whether the pattern for the spaced trials used in these experiments is optimal.
Cyclic AMP-dependent protein kinase (or PKA) and mitogen activated protein kinase (MAPK) are both required for initiation of long-term synaptic facilitation at sensory-motor synapses in the circuits for the defensive withdrawal reflexes of Aplysia. Since the perfused membrane adenylyl cyclase assays of Yovell, Dudai, Kandel and Abrams in the 1980s, it has been known that synthesis of cAMP and PKA-dependent phosphorylation closely follow the time course of serotonin release (for example [4]). More recently, Philips, Carew and colleagues [5] found that MAPK activation develops much more gradually: after a single tail shock, which releases serotonin for several minutes, the peak of MAPK activation is delayed, occurring as much as ~45 minutes later. Thus, the timing of PKA and MAPK stimulation, at least after a single exposures to serotonin, is not well synchronized. Zhang, Byrne and colleagues [3] asked: could initiation of long-term memory be improved if the disparate kinetics of the initiating biochemical processes were taken into account?
In order to predict optimal training protocols that would enhance the interactions between the PKA and MAPK signaling pathways, Zhang et al. [3] developed a computational model that incorporated what was known about the kinetics of activation of these two kinases (Figure 1A). One major challenge is that the interactions between the cAMP/PKA pathway and the MAPK pathway are complex, and not fully understood. PKA may promote the translocation of activated MAPK into the nucleus, as shown by early studies of Martin, Kandel and colleagues [6]. Schacher, Hu and colleagues [7] have found that PKA is required for release of the neuropeptide sensorin, which activates MAPK. cAMP may also more directly stimulate the MAPK pathway by activating a small G protein, Ras or Rap1, either via PKA or EPAC. Given the gradual rise in MAPK phosphorylation [5], a mechanism involving cAMP-dependent secretion of a signaling molecule seems more likely to mediate MAPK activation.
Figure 1.
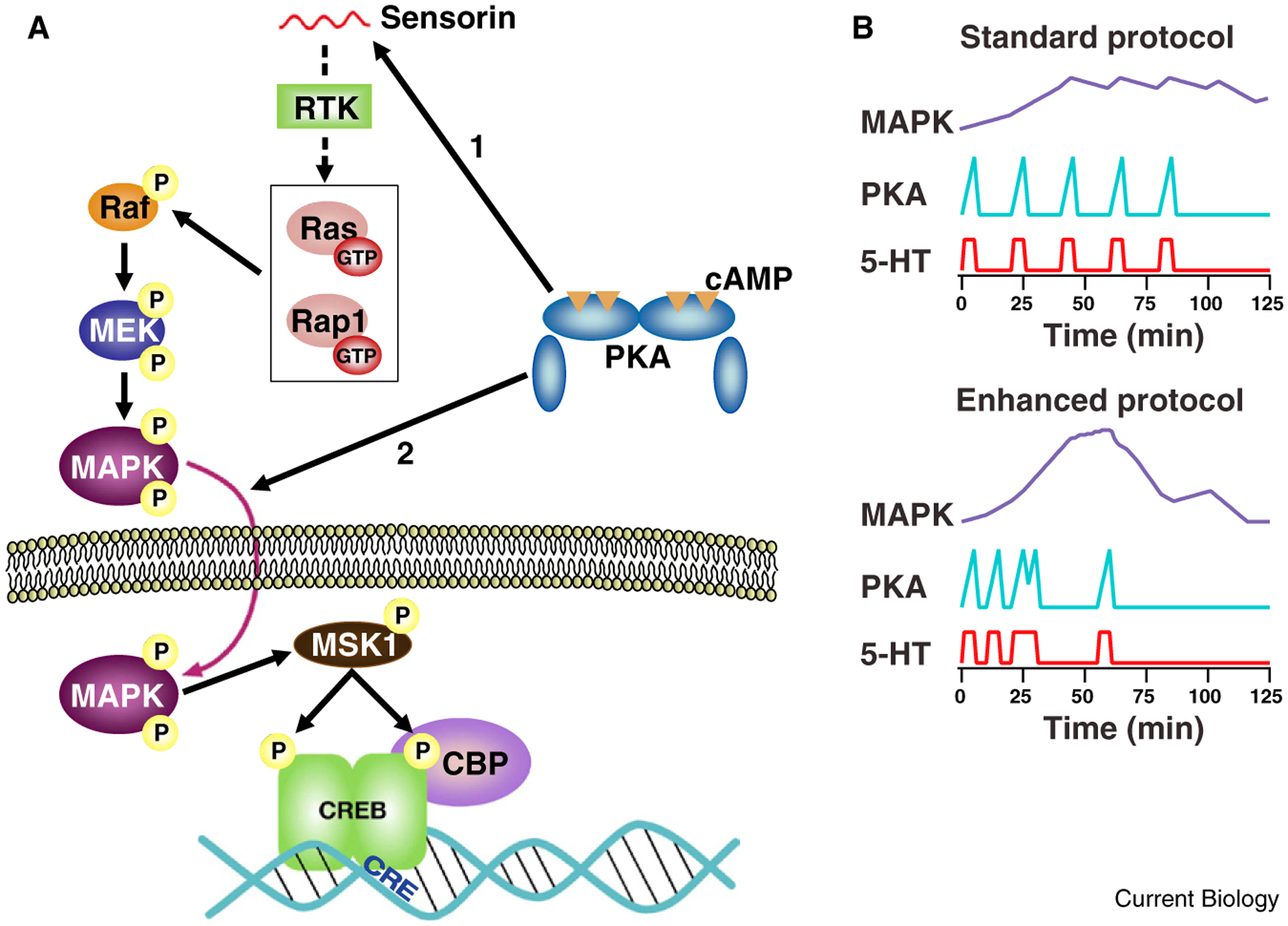
PKA and MAPK signaling pathways activated with enhanced training protocol. (A) Convergence of PKA and MAPK upstream of CREB transcription factor. The cartoon illustrates two possible loci of convergence. Tail shock releases serotonin (not shown), which stimulates adenylyl cyclase, activating PKA. PKA is shown as triggering release of the neuropeptide sensorin from sensory neurons (arrow #1) [7], which acts by binding to a receptor tyrosine kinase (RTK) to activate the MAPK pathway, via either Ras or Rap1. MAPK translocates into the nucleus where it stimulates phosphorylation of CREB by activating MSK1. PKA is also shown enhancing the translocation of MAPK (arrow #2) [6]. (Other mechanisms of interaction between the PKA and MAPK pathways, not shown here, have also been described.) CREB-binding protein (CBP) is recruited by activated CREB; CBP promotes transcription due to it’s histone acetyl transferase activity. (B) Enhanced training results in improved coincidence of activation of PKA and MAPK. Curves are simple schematized examples of activation profiles similar to those generated by the computational model of Zhang et al. [3]. Note, PKA activation is tightly temporally linked to serotonin exposure. MAPK activation develops gradually, peaking at about 45 min. With the enhanced protocol, more powerful peak activation of MAPK is achieved, which is available to interact with PKA activated by the final pulse of serotonin.
To minimize assumptions about the precise interactions between these signaling pathways, Zhang et al. [3] used a greatly simplified model in which the initiation of long-term synaptic facilitation and long-term memory was mediated by a symbolic variable, denoted ‘inducer’. The instantaneous activation of inducer was proportional to the product of the activation of PKA and MAPK at each time point. To avoid assumptions about non-linearities in this initiating process, the value used to predict initiation of long-term memory was simply the peak level of inducer reached during a given simulated training protocol. To determine optimal spacing of training trials, the four intervals between the training trials were varied independently and systematically between 5 and 50 minutes.
In simulations of 104 possible stimulus patterns, the maximal activation of ‘inducer’ was observed with three short intervals between serotonin exposures, 10, 10 and 5 minutes, followed by a 30 minute interval (Figure 1B). To empirically test their prediction that this protocol, which optimizes the temporal overlap between activation of PKA and MAPK, would enhance learning, Zhang et al. [3] examined phosphorylation of the transcription factor CREB1, synaptic facilitation and behavioral sensitization. With the optimized, ‘enhanced’ training protocol, the increase in phosphorylation of CREB1 in sensory neurons was more than five-fold greater than with the traditional, standard protocol. The consequences of following the optimized training protocol for both synaptic facilitation and long-term learning were dramatic. Synaptic facilitation persisted at five days after training with the enhanced protocol, by which time facilitation had decayed completely with the traditional training protocol. With the enhanced training protocol, long-term behavioral sensitization was actually 60% stronger at five days after training than at one day, whereas with the traditional, ‘standard’ training protocol, sensitization had disappeared by five days [3].
Zhang et al. [3] suggested that similar computational approaches could be used to improve training protocols in memory systems important for human cognitive performance. Is it plausible that such models of biochemical signaling would be useful in studies of learning involving the far more complex mammalian CNS? The beneficial effects of optimizing interactions between molecular cascades must actually be quite robust, because Zhang et al. [3] used greatly simplified parameters for their modeling. For example, the data on MAPK activation were for a single tail shock, rather than a series of training trials [3]. Schwartz and colleagues [8] had found that repeated serotonin exposures result in persistent activation of PKA, which was not included in this simplified model. Signaling in synaptic regions was also not taken into account, nor was signaling in postsynaptic motor neurons. Given the remarkably strong benefits to learning with the enhanced training protocol, together with the limitations in the parameters used to generate the model, the impact of enhancing interactions between the activating signaling cascades must be very powerful. This suggests that a similar computational approach could yield substantial improvement in learning, even with more complex nervous systems.
In an extension of this approach described at the 2012 Molluscan Neuroscience Meeting, the Byrne lab is currently asking whether this same simulation strategy might be used to benefit individuals with genetic disorders that affect learning and memory. By knocking down expression of specific genes in the PKA–MAPK–CREB pathway in Aplysia neurons, Byrne and colleagues are generating models of human disorders that affect this pathway. One example is Rubinstein-Taybi syndrome, where there is a mutation in CREB-Binding Protein (CBP). Optimizing training protocols at synapses where CBP has been knocked down may provide a novel approach for improving learning and memory in patients with the disease. A related strategy is a hybrid pharmacological-behavioral therapy, in which optimized training protocols are combined with pharmacological manipulation of the PKA–MAPK–CREB pathway. Byrne and colleagues are exploring whether such a combination of enhanced training protocols with pharmacological treatment can substantially improve outcomes compared with drug therapy alone.
FMRP and Slack K+ channels: a bidirectional regulatory complex
Typically when work in an invertebrate model neural system proves to be more broadly influential, the initial observation is made in this simpler model system and then subsequently followed up in studies on the mammalian nervous system. The recent investigation by Len Kaczmarek and colleagues of interactions between FMRP and the Slack K+ channel followed the reverse sequence. Nevertheless, the novel observations about FMRP function made recently in Aplysia are very likely to be relevant for understanding how FMRP-gated protein synthesis is regulated by neuronal activity in mammalian neurons. Full understanding of the functional role of FMRP may have important clinical implications for efforts to restore normal synaptic plasticity in Fragile X syndrome individuals and possibly for treatment of other disorders in which FMRP is involved, as suggested in the third section below on chronic pain.
Fragile X syndrome is a major genetic cause of mental retardation and the most common identified single gene cause of autism. Fragile X syndrome is associated with cognitive deficits in short-term memory and speech. The genetic basis of Fragile X syndrome is the loss of expression of FMRP, an 80 kDa protein, most commonly due to serial expansion of a tandem trinucleotide repeat located ~70 base pairs upstream of the coding region; this expanded trinucleotide repeat results in local hypermethylation and silencing of the FMR1 gene.
FMRP binds specific mRNAs and also has a protein-interacting domain. It acts to inhibit translation of a subset of mRNAs [9]. The mechanisms by which FMRP represses translation and how this repression is regulated are not fully understood. Some of these FMRP-regulated transcripts code for proteins that are important in the endocytosis of AMPA glutamate receptors, which is stimulated by activation of metabotropic (G-protein-coupled) glutamate receptors. Normally, the inhibition of protein translation by FMRP acts as a brake on this metabotropic glutamate receptor-initiated protein synthesis and the resulting downregulation of these AMPA receptors. In contrast, when FMRP is not expressed, the metabotropic glutamate receptor stimulation of AMPA receptor endocytosis is exaggerated, enhancing the normal process of long-term synaptic depression. Based on this ‘metabotropic glutamate receptor theory of fragile X’ and promising experiments in several model systems, the leading candidates for treatment of fragile X syndrome patients are antagonists of these receptors [10]. FMRP is conserved in invertebrates, including Aplysia, where it is known to regulate long-term synaptic plasticity [11].
A novel role for FMRP was recently suggested by Brown, Kaczmarek and colleagues [12], who found that FMRP binds to the unusually long carboxy-terminal tail of the Slack B Na+-activated K+ channel subunit from mouse. Single channel recordings and whole cell recordings confirmed that this interaction was functionally important, by demonstrating that FMRP plays a translation-independent role in directly increasing the open probability of these Na+-activated K+ channels. However, the functional role of this modulation of Slack channels by FMRP was unclear.
In order to analyse how FMRP interactions with Slack channels affect the firing properties of neurons, Kaczmarek and colleagues turned to an extensively studied model system, the neuroendocrine bag cells of Aplysia. During egg-laying behavior, the bag cells initiate bursting behavior lasting tens of minutes, known as afterdischarge, which triggers release of egg-laying hormone and other neuropeptides. Zhang et al. [13] found that bag cells express a Slack ortholog with similar properties, including the Na+ dependence. To test if there was also regulation by FMRP, they injected the protein-binding amino-terminal domain of mammalian FMRP, which increased the Slack-like outward current. This FMRP-enhanced current was dependent on the presence of extracellular Na+. In patch clamp recordings, application of the protein-binding domain of FMRP to the cytoplasmic face of the membrane substantially increased the open probability of these large conductance channels, much as with mammalian Slack channels.
What is remarkable in the Zhang et al. [13] study is the key role of the Slack channels in protein synthesis-dependent recovery from long-term refractoriness. One characteristic of bag cells that is not well understood is that, once an afterdischarge is completed, the bag cells remain refractory until the next day (they are unable to initiate another full afterdischarge). The recovery from refractoriness requires protein synthesis. Blocking protein synthesis with anisomycin had no effect on an initial afterdischarge, but dramatically inhibited the bag cells’ recovery from refractoriness [13]. Surprisingly, bag cells treated with siRNA for Slack initiated normal afterdischarge, but showed little or no afterdischarge one day later, revealing that normal recovery from refractoriness also requires the Slack channel [13]. One interpretation is that prolonged activation of Slack during bag cell afterdischarge triggers release of the Slack-associated FMRP from the polyribosome, thereby derepressing local translation, which is required for recovery from refractoriness (Figure 2). There are other possible interpretations, but the FMRP link to the translation machinery is the simplest explanation for the requirement for Slack in recovery from refractoriness.
Figure 2.
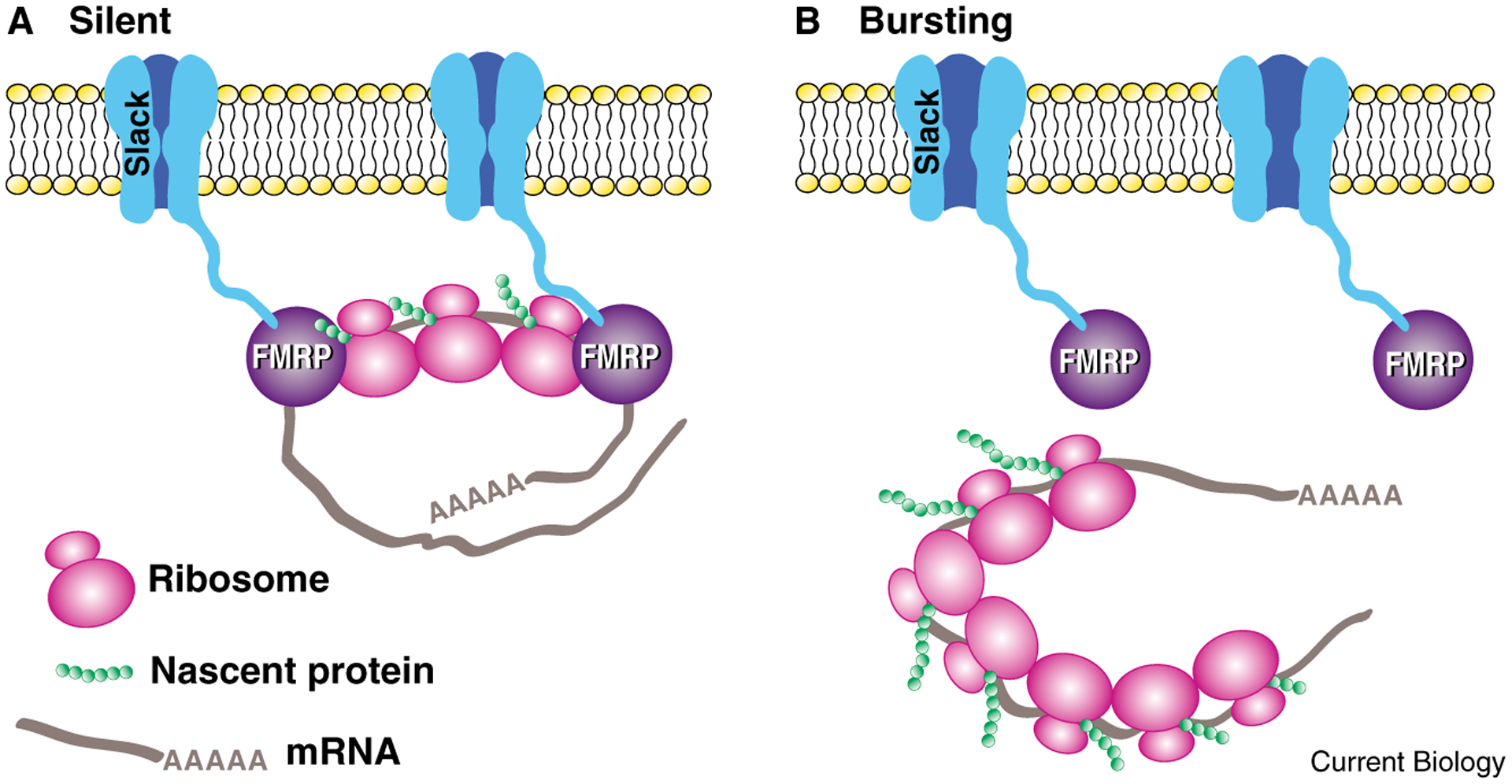
The hypothesized coupling between activity and local translation mediated by the interaction between Slack K+ channels and FMRP in the neurosecretory bag cells of Aplysia. (A) Silent neurons. FMRP inhibits local translation by binding to polyribosomes. (B) Bursting neurons. During bursting behavior (prolonged activity known as afterdischarge), Slack channels activated by depolarization trigger dissociation of FMRP from polyribosomes and mRNA, permitting initiation of translation. This locally initiated translation may mediate recovery from refractoriness at the end of afterdischarge. (Model based on results from [9].)
Traditionally, ion channels were believed to be modulated indirectly by kinases and phosphatases, and interaction of channels with downstream signaling cascades has been thought to be mediated primarily by Ca2+ influx. (The direct ‘membrane-delimited’ modulation of K+ and Ca2+ channels by heterotrimeric G proteins has been known for a few decades, but considered an exception.) The new findings by Kaczmarek and colleagues on Slack channels in Aplysia bag cells suggest that the Slack interaction with FMRP may provide a mechanism for direct, activity-dependent regulation of local translation. More generally, one can speculate that activity may regulate local translation independent of Ca2+ influx by direct interactions between ion channels and RNA-binding proteins.
The relevance of such activity-dependent Slack-mediated regulation of local protein synthesis for cognitive deficits in Fragile X syndrome individuals is not yet clear. It has not yet been demonstrated that Slack channels regulate message translation locally in mammalian neurons. However, it is tempting to speculate that the Slack–FMRP interaction is important in cognitive function, because local protein synthesis plays a central role in initiation of long-term memory. It seems likely that Slack–FMRP-mediated activation of translation is particularly important when there are high levels of activity or with prolonged depolarization, such as discussed in the following section. These results suggest that, although treatment of Fragile X syndrome individuals with inhibitors of metabotropic glutamate receptors may be beneficial for a number of cognitive and neurological deficits, these antagonists may not restore key aspects of FMRP-regulation of translation at synaptic sites. Restoring activity-dependent activation of local translation in Fragile X syndrome individuals would likely require a gene therapy approach.
Long-term memory mechanisms in nociceptive sensory neurons may contribute to chronic pain after spinal cord injury
The nervous systems of most animals must adapt to changes in the environment; this adaptive plasticity is the fundamental basis of learning. In addition to adaptating to environmental changes, the nervous system must be able to adapt to changes that animals experience with injury. During sensitization and classical conditioning protocols, Aplysia are typically trained with noxious stimuli delivered to the skin, such as tail shocks, which cause facilitation of the synaptic connections from the mechanosensory neurons that mediate the defensive withdrawal reflexes. In the mid 1980s, Walters and Byrne and Klein, and Hochner and Kandel found that these noxious peripheral stimuli and serotonin released by these stimuli also result in a dramatic increase in excitability in these sensory neurons. Walters appreciated that these high-threshold, broad dynamic range mechanoreceptors that mediate the defensive withdrawal reflexes in Aplysia had properties reminiscent of mammalian nociceptors. He further understood that the noxious stimulus, tail shock, was a signal representing peripheral injury. Moreover, Walters and Byrne [14] found that the sensory neurons that underwent the most dramatic increase in excitability during training were those that were themselves activated by the peripheral noxious stimulus; these were precisely those sensory neurons that would experience damage to their peripheral fields during an actual injury (for example, when encountering a predator). This observation led Walters to propose that enhanced excitability in these high-threshold mechanoreceptors could serve an adaptive function, leading injured snails to protect vulnerable, damaged parts of their body against further trauma.
Over the subsequent twenty-five years, Walters and colleagues have explored the adaptive role of injury-triggered increases in excitability in the high threshold mechanoreceptors of Aplysia. A wide variety of signals are capable of initiating persistent increases in sensory neuron excitability, including serotonin. After peripheral nerve injury, serotonin may be released by inflammatory cells that respond to crush. Hyperexcitability produced by applications of serotonin is local; the increase in excitability is restricted to a short axonal segment where serotonin is applied (Figure 3). Moreover, this long-term increase in excitability is blocked by local application of protein synthesis inhibitors. Thus, long-term hyperexcitability involves local protein synthesis in the axon. Another novel study by Lewin and Walters implicated the nitric oxide-cyclic GMP pathway in long-term hyperexcitability initiated by either serotonin or peripheral injury. Persistent hyperexcitability also involves transcription and the transcription factor CREB.
Figure 3.
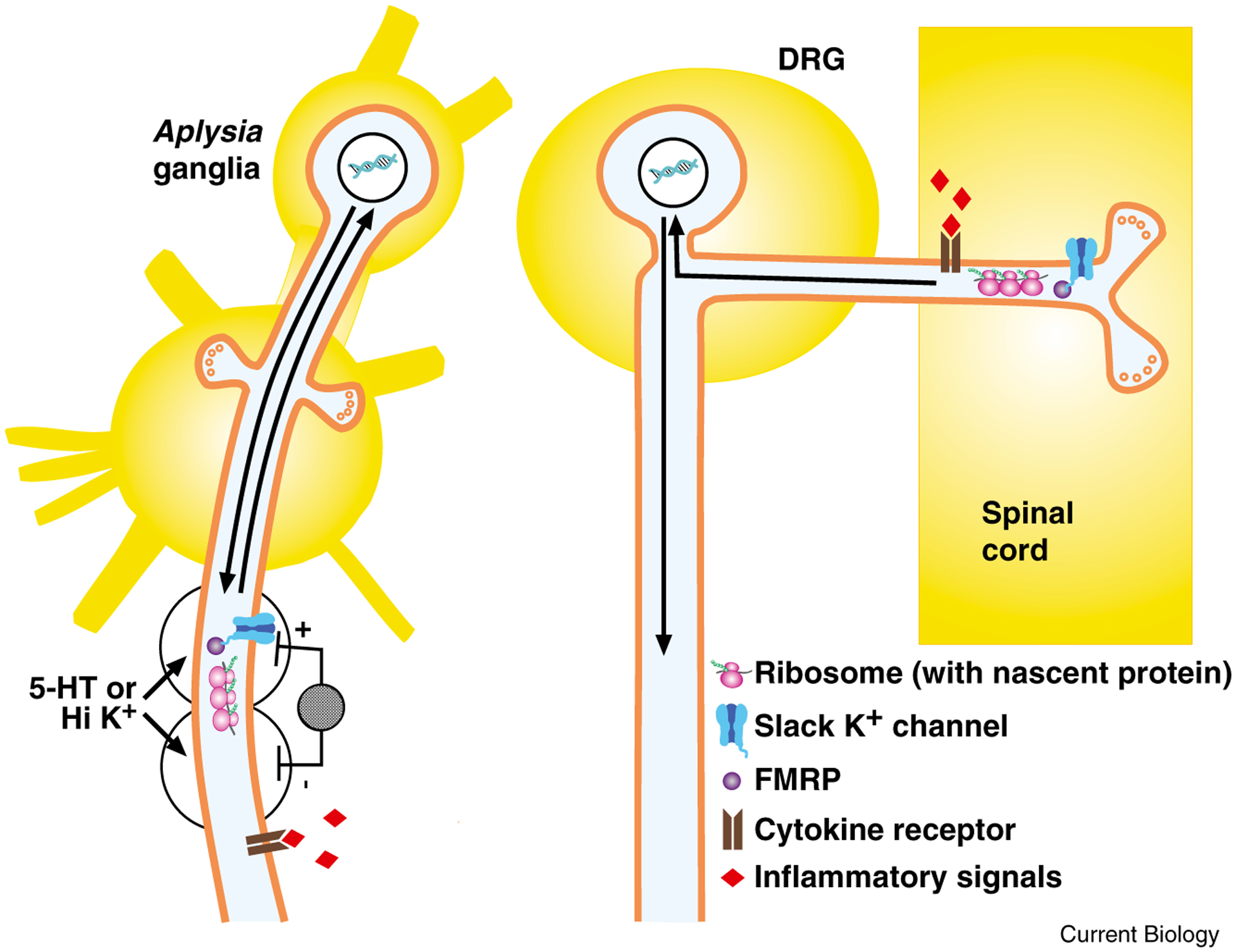
After injury, injury-activated signals trigger local translation and transcription-dependent changes to increase excitability and spontaneous activity in Aplysia sensory neurons and mammalian nociceptors.
Left: sensory neuron in Aplysia. After either nerve crush or application of loose ligature, hemocytes accumulate and release inflammatory signals, such as cytokines and serotonin; these signals initiate long-term hyperexcitability in sensory neuron axons. Alternatively, local application of the inflammatory signal serotonin (5-HT) to, or depolarization with high K+ solution of a small region of axon isolated in wells produces local increase in excitability, assessed by electrical stimulation within wells, with no increase in excitability in other axonal regions. This local increase in excitability requires local protein synthesis (as indicated by ribosome, with mRNA and nascent protein) and transcription (indicated by arrows representing signal to nucleus and new gene products transported from nucleus to axon), but does not require Ca2+ influx or Ca2+ release for initiation. One possible mechanism for initiating local protein synthesis independent of Ca2+ increase is Slack K+ channel regulation of FMRP-gated translation (indicated by Slack channel bound to FMRP). Right: dorsal root ganglion (DRG) nociceptor. After spinal cord contusion, DRG nociceptors at segments above and below crush exhibit increased frequency of spontaneous activity, which is evident both in the peripheral axon and in the isolated cell body. Signaling pathways have not yet been identified, but as suggested here, inflammatory signals from microglia in spinal cord several segments away from injury may initiate the response. Because the increase in excitability is expressed in isolated cell bodies in dissociated cell culture, it is likely to involve changes in transcription (as symbolized by arrows to and from nucleus). Depolarization-induced Slack K+ channel-FMRP triggering of local translation may also contribute to increased excitability and spontaneous activity in sensory neurons; local depolarization is probable after spinal cord injury due to massive release of the excitatory neurotransmitter glutamate.
More recently, Walters and colleagues examined the role of Ca2+ in initiating long-term hyperexcitability. Intracellular Ca2+ in axons is elevated in response to injury, so it is reasonable to expect that Ca2+ plays a critical role in this plasticity. However, removing 99% of extracellular Ca2+ did not reduce the hyperexcitability triggered by serotonin or nerve crush. In axons loaded locally with the Ca2+ chelator BAPTA-AM and bathed in EGTA saline, depolarization by a two minute exposure of a nerve segment to high K+ (Figure 3) induced intermediate-term and long-term hyperexcitability lasting at least 24 hours [15]. This prolonged exposure to Ca2+ chelators is very harsh treatment even for rugged Aplysia neurons, which survive a wide variety of insults; unfortunately, it has not been possible to do parallel experiments with mammalian sensory neurons, which do not survive very low Ca2+ conditions.
These results on initiation of persistent hyperexcitability by brief depolarization raise the fascinating question of how the sensory neurons detect the depolarization if not through Ca2+ influx. For the initiation of a long-term increase in excitability by depolarizing stimuli, which depends on local protein synthesis, a possible mechanism is suggested by the work by Kaczmarek and colleagues on Slack K+ channels and FMRP [13]. If Slack channels couple activity to initiation of translation of specific transcripts, as the experiment on the protein synthesis-dependent recovery from refractoriness in bag cells suggests, perhaps depolarization acts via Slack–FMRP-gated local protein synthesis to produce long-term increases in excitability in Aplysia sensory neuron axons (Figure 3). If this mechanism operates in Aplysia, it would be interesting to test whether it also contributes to persistent increases in excitability in mammalian dorsal root ganglion (DRG) neurons.
In the early 1990s Walters, Clatworthy and colleagues [16] explored the effects of a ligature applied to a peripheral Aplysia nerve in inducing long-term hyperexcitability. These investigators intentionally applied the cotton gauze ligature sufficiently loosely so as not to produce any damage to axons, which they confirmed with electron microscopy. They observed that this loose ligature stimulated the recruitment of hemocytes, which were later found by Ambron and colleagues to express serotonin, IL-6 and TGFb, all of which are inflammatory signals. These studies suggested a critical role of inflammatory signals, as opposed to nerve damage.
Walters then began a series of comparative experiments on excitability changes in rat DRG neurons, influenced by what he had learned about persistent changes in excitability of Aplysia mechanoreceptors after injury or inflammation. Clatworthy, Walters and their colleagues used a loose ligature approach similar to that developed by Bennett and Xie [17]; but contrary to the earlier approach which produced clear mechanical constriction of the nerve and disruption of axons, in these studies, as in Aplysia, the ligature did not produce mechanical injury [18]. Clatworthy et al. [18] directly tested whether the behavioral sensitization produced by the ligature was dependent on an inflammatory response, as the hemocyte aggregation in Aplysia suggested. First, they noticed the accumulation of inflammatory cells, including macrophages and neutrophils around the site of the ligature, without damage to the axons. Second, they found that application of Freund’s adjuvant to the ligature enhanced the behavioral effects: guarding behavior — protecting the paw — and enhanced thermal sensitivity. And third, blocking inflammation with dexamethasone completely eliminated the behavioral sensitization effects of the ligature. The dexamethasone also reduced the aggregation of inflammatory cells. Thus, in DRG neurons, inflammatory signaling plays a primary role in initiating persistent sensitization.
Research on chronic pain after spinal cord injury has focused primarily on central changes in the spinal cord, thalamus and cortex that could be triggered by inflammatory responses and glial activation near the injury. Walters was influenced by the extensive history of studies in Aplysia in which stable changes in primary sensory neurons lasting many days are initiated by central modulatory inputs. He speculated that central inflammatory signals could similarly initiate long-term changes in DRG nociceptors, and that after spinal cord injury, persistent hyperexcitability in DRG neurons might contribute to chronic pain. Independently, Carlton and colleagues [19] obtained evidence that, one month after spinal cord injury, there was enhanced spontaneous activity in sensory axons from the forepaw above the injury and enhanced sensitivity to mechanical and heat stimuli. Interestingly, these authors observed that activated microglia and astrocytes were present in thoracic and cervical cord rostral to the injury one month post-injury, suggesting a substantial rostral spread of glial activation. Their initial interpretation was that enhanced sensory neuron activity, which was driven centrally by inflammatory signals, generated peripheral inflammation (known as neurogenic inflammation).
Walters and Carlton then collaborated to test the alternative possibility that the memory that underlies the persistent increase in sensory neuron responsiveness is intrinsic to the sensory neurons themselves, analogous to what occurs in Aplysia sensory neurons [20]. Their experiments measured the firing properties of dissociated small DRG neurons at various times after thoracic spinal cord impact injury at T10. The sensory neurons were maintained in culture for one day prior to electrophysiological measurements to ensure that any altered properties reflected persistence in the isolated neurons. In sham control and naive animals, a low percentage (~15%) of small DRG neurons showed spontaneous activity. After injury, the frequency of spontaneously active DRG neurons increased by several fold, and this increase persisted for at least eight months both below and above the site of injury. All spontaneously active DRG neurons had properties characteristic of primary nociceptors [20]. These results on dissociated DRG nociceptor neurons suggest that, after spinal cord injury, local inflammatory signals or depolarization in the cord act on the central terminals of DRG nociceptors to trigger persistent increases in their excitability, thereby contributing to chronic hyperalgesia (Figure 3). In behavioral tests, increased sensitivity to peripheral thermal and mechanical stimuli was positively correlated with the number of spontaneously active DRG neurons, suggesting that spontaneously active nociceptors contribute to hyperalgesia after spinal cord injury.
As an additional test of the role of persistent enhanced excitability in DRG neurons in hyperalgesia after spinal cord injury, more recently, Walters and colleagues have targeted a Na+ channel selectively expressed in nociceptors, NaV1.8. Consistent with a contribution of primary sensory neurons to chronic pain after central injury, knockdown of NaV1.8 blocked the increase in spontaneous activity in these DRG neurons and behavioral signs of hyperalgesia. This suggests that pharmacologically targeting NaV1.8 may be therapeutically beneficial for spinal cord injury patients suffering with chronic pain. In summary, these recent studies on spinal cord injury have demonstrated that, as was found in Aplysia, central modulatory signals initiate very stable, long-term hyperexcitability in primary sensory neurons.
Conclusion
The three studies in Aplysia described here, on signaling cascades that initiate long-term memory, on Slack K+ channel regulation of FMRP-gated local translation, and on persistent hyperexcitability in sensory neurons initiated by inflammation, have all yielded unanticipated results with clear relevance for human diseases. Indeed, these novel insights may lead to innovative approaches for treatment of several chronic human disorders. What is perhaps surprising is that research on this simple invertebrate model system has the potential to benefit clinical treatments, even when the research would not, by some, be considered ‘translational’.
References
- 1.Wan Q, Jiang XY, Negroiu AM, Lu SG, McKay KS, and Abrams TW (2012). Protein kinase C acts as a molecular detector of firing patterns to mediate sensory gating in Aplysia. Nat. Neurosci 15, 1144–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dash PK, Hochner B, and Kandel ER (1990). Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature 345, 718–721. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Liu RY, Heberton GA, Smolen P, Baxter DA, Cleary LJ, and Byrne JH (2012). Computational design of enhanced learning protocols. Nat. Neurosci 15, 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarrard HE, Goldsmith BA, and Abrams TW (1993). In Aplysia sensory neurons, the neuropeptide SCPB and serotonin differ in efficacy both in modulating cellular properties and in activating adenylyl cyclase: implications for mechanisms underlying presynaptic facilitation. Brain Res. 616, 188–199. [DOI] [PubMed] [Google Scholar]
- 5.Philips GT, Tzvetkova EI, and Carew TJ (2007). Transient mitogen-activated protein kinase activation is confined to a narrow temporal window required for the induction of two-trial long-term memory in Aplysia. J. Neurosci 27, 13701–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, and Kandel ER (1997). MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18, 899–912. [DOI] [PubMed] [Google Scholar]
- 7.Hu JY, Glickman L, Wu F, and Schacher S (2004). Serotonin regulates the secretion and autocrine action of a neuropeptide to activate MAPK required for long-term facilitation in Aplysia. Neuron 43, 373–385. [DOI] [PubMed] [Google Scholar]
- 8.Hegde AN, Goldberg AL, and Schwartz JH (1993). Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. Proc. Natl. Acad. Sci. USA 90, 7436–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krueger DD, and Bear MF (2011). Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med 62, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Till SM, Li HL, Miniaci MC, Kandel ER, and Choi YB (2011). A presynaptic role for FMRP during protein synthesis-dependent long-term plasticity in Aplysia. Learn. Mem 18, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, and Kaczmarek LK (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci 13, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Brown MR, Hyland C, Chen Y, Kronengold J, Fleming MR, Kohn AB, Moroz L, and Kaczmarek LK (2012). Regulation of neuronal excitability by interaction of Fragile X Mental Retardation Protein with Slack potassium channels. J. Neurosci in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walters ET, and Byrne JH (1985). Long-term enhancement produced by activity-dependent modulation of Aplysia sensory neurons. J. Neurosci 5, 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunjilwar KK, Fishman HM, Englot DJ, O’Neil RG, and Walters ET (2009). Long-lasting hyperexcitability induced by depolarization in the absence of detectable Ca2+ signals. J. Neurophysiol 101, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clatworthy AL, Castro GA, Budelmann BU, and Walters ET (1994). Induction of a cellular defense reaction is accompanied by an increase in sensory neuron excitability in Aplysia. J. Neurosci 14, 3263–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett GJ, and Xie YK (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. [DOI] [PubMed] [Google Scholar]
- 18.Clatworthy AL, Illich PA, Castro GA, and Walters ET (1995). Role of peri-axonal inflammation in the development of thermal hyperalgesia and guarding behavior in a rat model of neuropathic pain. Neurosci. Lett 184, 5–8. [DOI] [PubMed] [Google Scholar]
- 19.Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, and Hulsebosch CE (2009). Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 147, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, and Walters ET (2010). Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J. Neurosci 30, 14870–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]