

Abstract
Checkpoint blockade-based immunotherapy has shown unprecedented effect in cancer treatments, but its clinical implementation has been restricted by the low host antitumor response rate. Recently, chemotherapy is well recognized to activate the immune system during some chemotherapeutics-mediated tumor eradication. The enhancement of immune response during chemotherapy might further improve the therapeutic efficiency through the synergetic mechanism. Herein, a synergistic antitumor platform (designated as BMS/RA@CC-Liposome) was constructed by utilizing CT26 cancer-cell-biomimetic nanoparticles that combined chemotherapeutic drug (RA-V) and PD-1/PD-L1 blockade inhibitor (BMS-202) to remarkably enhance antitumor immunity. In this study, the cyclopeptide RA-V as chemotherapeutic drugs directly killing tumor cells and BMS-202 as anti-PD agents eliciting antitumor immune responses were co-encapsulated in a pH-sensitive nanosystem. To achieve the cell-specific targeting drug delivery, the combination therapy nanosystem was functionalized with cancer cell membrane camouflage. The biomimetic drug delivery system perfectly disguised as endogenous substances, and realized elongated blood circulation due to anti-phagocytosis capability. Moreover, the BMS/RA@CC-Liposome also achieved the selective targeting of CT26 cells by taking advantage of the inherent homologous adhesion property of tumor cells. The in vitro and in vivo experiments revealed that the BMS/RA@CC-Liposome realized PD-1/PD-L1 blockade-induced immune response, RA-V-induced PD-L1 down-regulation and apoptosis in cancer cells. Such a system combining the advantages of chemotherapy and checkpoint blockade-based immunotherapy to create an immunogenic tumor microenvironment systemically, demonstrated improved therapeutic efficacy against hypoxic tumor cells and offers an alternative strategy based on the immunology of the PD-1/PD-L1 pathway.
KEY WORDS: Checkpoint blockade-based immunotherapy, Chemotherapy, Synergetic mechanism, Cancer-cell-biomimetic nanoparticles, Cyclopeptide RA-V, Antitumor immune response, Combination therapy nanosystem, PD-1/PD-L1 pathway
Graphical abstract
CT26 cancer-cell-biomimetic nanoparticles (BMS/RA@CC-Liposome) sensitized hypoxia tumor to checkpoint blockade-based immunotherapy in virtue of the synergistic effects between RA-V and BMS on PD-1/PD-L1 axis.
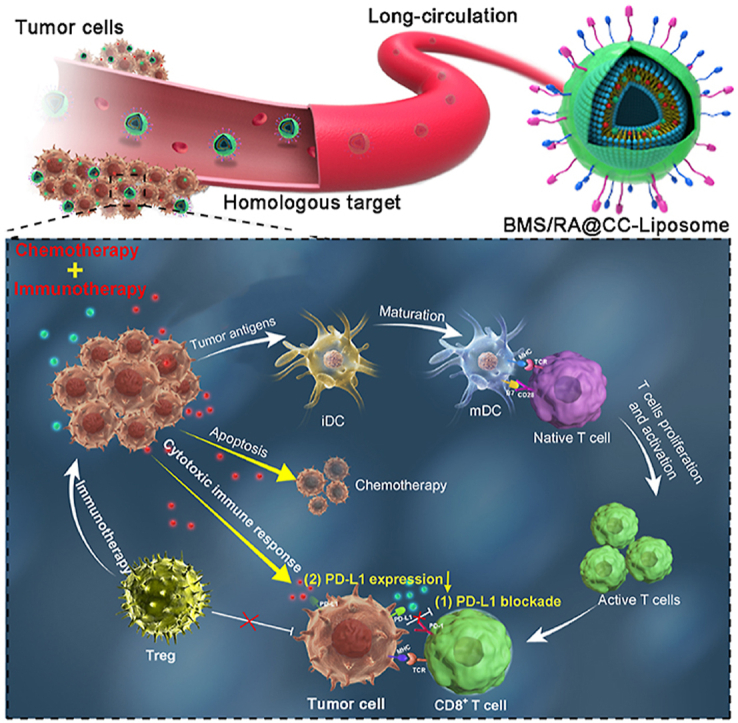
1. Introduction
Cancer immunotherapies (e.g., cancer vaccines therapy, cytokine therapy, chimeric antigen receptor (CAR)-T cell therapy)1, 2, 3, 4 have emerged as revolutionary cancer treatment from the last 30 years, with high overall response rates in clinical patients5. Among various cancer immunotherapies, immune checkpoint blockade therapy exhibited satisfactory clinical responses in certain types of tumors by reversing the immune tolerance6. Particularly, the PD-1/PD-L1 pathway is a valuable target of immune checkpoint blockade. The binding of PD-L1 with PD-1 brings about the immune evasion by suppressing the secretion of cytokines and inducing T cells apoptosis, which largely limited the actual efficacy of immune therapy7,8. Moreover, the high expression of PD-L1 on cancer cells restricts tumor cell killing by causing T cell exhaustion9,10. Therefore, blocking the PD-1/PD-L1 axis can recover exhausted T cells and boost the immune response against tumors11. However, in some patients, immune checkpoint PD-1/PD-L1 blockade has limited activity with low antitumor immune response. This is attributed to the complex intrinsic immunosuppressive mechanism, such as the absence of costimulation tumor microenvironment and abundance of immune suppressive cell populations12,13. Therefore, alternative approaches strengthening the effect of PD-1/PD-L1 axis blockade are urgent to be exploited. Recently, immune checkpoint inhibitor therapy in combination with chemotherapy has shown great prospect for highly efficient and selective treatment in multiple tumor types14,15.
Chemotherapy can enhance antitumor immune responses through a variety of mechanisms, such as stimulating the immune system through immunogenic cell death (ICD), and inhibiting immunosuppressive pathways in the tumor microenvironment (TME)16, 17, 18. Thus, searching for appropriate chemotherapeutics combined with immune checkpoint inhibitors seems to be a promising immunotherapeutic strategy. However, some chemotherapy agents would lead to up regulation of PD-L1 expression, which impair therapeutic effect. Different from conventional chemotherapeutics causing PD-L1 enrichment, cyclopeptide RA-V (deoxybouvardin), a unique natural cyclopeptide derived from the medical plant Rubia yunnanensis catches our attention because of its satisfactory activities in cancer cells19,20. According to our previous work, RA-V could induce cancer cells apoptosis through blocking the phosphatidylinositol 3-kinase (PI3K)/AKT pathway or down-regulating ERK1/2 phosphorylation21, 22, 23. Moreover, in our latest research, RA-V was found to effectively overcome hypoxia and induce apoptosis by down-regulating the expression of HIF-1α in tumor cells, in comparison to conventional MnO2 or catalase catalyst confined by low intracellular H2O2 concentration24. Many studies have demonstrated the crucial function of hypoxia to the acquisition of tumor immune escape including innate immunity and adaptive (T cell-mediated) immunity25,26. Especially, in adaptive immunity, HIF-1α increased the expression of PD-L1, leading to tumor-cell resistance to cytotoxic lymphocytes (CTLs)-mediated cytolysis27. Considering the intrinsic relation between hypoxia and tumor immune escape, we hypothesis that RA-V could effectively reduce the expression of PD-L1 in cancer cells and contribute to tumor immune response. Thus, RA-V is an ideal chemotherapeutic candidate for constructing combination therapy system to improve therapeutic outcomes through an attractive synergy between chemotherapy and immune checkpoint inhibition.
To further realize high cancer selectivity of the combination therapy system, biological functionalization of nanocarriers with natural membrane derived from cancer cells become a promising way28, 29, 30, 31. To date, a variety of cell membrane-derived nanoparticles have been developed for drug delivery systems, such as platelet membrane-camouflaged nanoparticles32, neutrophil membrane-coated liposome nanoparticles33, red blood cells membrane-coated nanozyme34, and cancer cell membrane-coated nanoparticles35. The cancer-cell-membrane-derived vesicles presents a unique top-down approach that could achieve the long-circulation efiect, as well as high specific accumulation in tumor, which could be attributed to the enhanced permeability and the cell membrane mediating homotypic targeting potential36,37.
Inspired by the remarkable performance of these cancer-cell-membrane-derived nanoparticles, we prepared CT26 colon cancer cells membrane-decorated liposomes as a carrier to construct a combination therapy nanosystem (BMS/RA@CC-Liposome) for highly effective therapy against hypoxic tumor (Scheme 1). In this system, the cyclopeptide RA-V and PD-1/PD-L1 inhibitor (BMS-202, “BMS” for short) were co-encapsulated in the biomimetic pH-sensitive liposome. The obtained cancer cell membrane coated-nanoparticles showed the abilities of anti-phagocytosis and homologous targeting. Moreover, the BMS/RA@CC-Liposome treatment could sensitize hypoxia tumors to checkpoint blockade therapy in virtue of the synergistic effects between RA-V and BMS on PD-1/PD-L1 axis. BMS intervened the binding of PD-1/PD-L1 and reinvigorated exhausted T cells, leading to the enhanced cancer immunotherapy. In addition, cyclopeptide RA-V effectively killed cancer cells and down-regulated the expression of PD-L1, synergistically enhancing cytotoxic immune responses of tumor-infiltrating lymphocytes. These findings indicated that the tumor inhibition could be substantially enhanced by combining PD-1/PD-L1 checkpoint blockade immunotherapy with chemotherapeutics such as RA-V.
Scheme 1.

Schematic illustration of the BMS/RA@CC-Liposome as the biomimetic nanoplatform for hypoxia tumor chemotherapy and immunotherapy.
2. Materials and methods
2.1. Reagents
Cholesteryl hemisuccinate (CHEMS) was purchased from Avanti Polar Lipids (USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy (polyethylene glycol)-2000] (DSPE-mPEG2000-COOH), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were obtained from Nanocs (New York, USA). HIF-1α antibody and HMGA1 antibody were purchased from Abcam (Cambridge, UK), phospho-Akt (Ser473) antibody, phospho-p44/42 MAPK (Erk1/2) antibody and E-cadherin antibody were obtained from Cell Signaling Technology (USA) and the other antibodies were purchased from Proteintech (Wuhan, China). Sulforhodamine B (SRB) was purchased from Sigma–Aldrich Inc. (St. Louis, MO, USA). Antibodies against immune cell surface markers for flow cytometry assay were purchased from BD Biosciences (San Jose, CA, USA). RA-V (purity>99%) was prepared in our laboratory, with identification by MS and NMR spectroscopy. PD-1/PD-L1 inhibitor (BMS-202) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Ultrapure water was obtained using a Millipore Simplicity System (Millipore, Bedford, USA).
2.2. Apparatus
Zeta potential and average hydrodynamic diameter measurement were carried out on Malvern Zetasizer nano instrument (Malvern Company, UK) at 25 °C. SDS-PAGE protein experiment and western blotting experiment were performed on Mini-Protean Tetra Cell (Bio-Rad, USA). Fluorescence imaging of cells was performed on a confocal laser scanning microscope (LSM700, Zeiss, Germany). And images were analyzed by a ZEN imaging software. Flow cytometric assay was performed with Attune NxT Flow Cytometer (Thermo Fisher Scientific, USA). Cytotoxicity assay was performed with microplate reader (Biotek, USA). The Liposome was sonicated using an ultrasonics processor (SCIENTZ, China).
2.3. Cell lines and animals
CT26 colorectal cancer cells and RAW264.7 macrophage cells were originally obtained from Shanghai Cell Bank, Chinese Academy of Sciences (CAS). CT26 cells were maintained 1640 medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL; Invitrogen), and streptomycin (100 U/mL; Invitrogen). RAW264.7 cells were cultured in DMEM medium containing 10% fetal bovine serum. The rest of colorectal cancer cells such as HCT116, HT29, SW480, RKO, SW620 cells, and other cancer cells such as A549 and NCM460 cells were also obtained from Shanghai Cell Bank, Chinese Academy of Sciences (CAS). All cells were cultured in an incubator at 37 °C with a humidified 5% CO2/95% air atmosphere.
BALB/c mice (female, 6–8 weeks old, 18–20 g) and nude BALB/c mice (female, 6–8 weeks old, 18–20 g) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China), raised with sterilized water in a controlled environment at China Pharmaceutical University. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University and were approved by the Animal Care Committee of China Pharmaceutical University (Nanjing, China).
2.4. Preparation and characterization of BMS/RA@CC-Liposome
To prepare BMS/RA@CC-Liposome, the top-down approach was used38. Firstly, Liposomes were prepared by the thin film hydration method39. Briefly, DOPE (34.74 mg), CHEMS (12.6 mg), DSPE-mPEG2000-COOH (3.2 mg), RA-V (2.68 mg) and BMS-202 (2.15 mg) were dissolved with chloroform (30 mL) and then evaporated by a rotary evaporator under vacuum at 45 °C to obtain a thin lipid film. Then, the lipid film was hydrated in aqueous solutions (10 mL). At last, the obtained liposome was further processed with ultrasonic treatment (total for 8 min at power of 100 W, with 2 s on/off pulse) and extrusion by a porous polycarbonate membrane (220 nm). Concurrently, CT26 cell membrane vesicles (CMVs) as the outer shells were prepared. Briefly, the harvested CT26 cells were emptied through mechanical membrane disruption and differential centrifugation. Firstly, CT 26 cells suspension (density of 24 × 106 cells/mL) with three freeze–thaw cycles, were sonicated in ice bath by a probe sonicator (total for 10 min at power of 100 W, with 2 s on/off pulse). Then, the obtained suspension was centrifuged to remove the intracellular contents (1000×g, 10 min). Then the supernatants were centrifuged (5000×g, 10 min) and then centrifuged again (22,000×g, 30 min) to get the cell membrane. Afterward, the cell membrane was washed with PBS and incubated for 1 h at 37 °C. The resulting cell membrane was sonicated again using probe sonicator (total for 1 min at power of 100 W, with 3 s on/off pulse) and extruded in sequence through 450 and 220 nm porous polycarbonate membrane. Finally, in order to coat the liposome cores with cancer cell membrane, the mixtures (membrane-to-liposome at a volume ratio of 1.5:1) were repeatedly coextruded through a porous polycarbonate membrane (220 nm). The resulting BMS/RA@CC-Liposome was stored at 4 °C for future use. Additionally, BMS/RA@RBC-Liposome (red blood cells membrane-camouflaged nanoparticles) and BMS/RA@Liposome (without membrane-coating nanoparticles) were introduced as controls. Similar procedures were used to prepare BMS/RA@RBC-Liposome, BMS@CC-Liposome, RA@CC-Liposome and BMS/RA@Liposome. Then the size and zeta potential of cancer cell membrane, BMS/RA@Liposome, and BMS/RA@CC-Liposome were measured using Malvern Zetasizer nano instrument (Malvern, UK).
The morphologies of cancer cell membrane, BMS/RA@Liposome, and BMS/RA@CC-Liposome were characterized using transmission electron microscope (TEM). In brief, cancer cell membrane, BMS/RA@Liposome, and BMS/RA@CC-Liposome were dropped onto a carbon-coated copper grid, with 1.0% (w/v) phosphotungstic acid for negative staining. For encapsulation efficiency determination (EE) study: acetonitrile (950 μL) was used for destruction of BMS/RA@CC-Liposome (50 μL) with maximal intense vortex. Then, the suspension was centrifugated (12,000 rpm, 30 min) for completely releasing RA-V and BMS-202. Liquid chromatography‒tandem mass spectrometry (LC‒MS/MS) was used to determine the concentrations of RA-V and BMS-202. Moreover, the in vitro release profiles of RA-V and BMS-202 in BMS/RA@Liposome and BMS/RA@CC-Liposome were further investigated. BMS/RA@CC-Liposome (3 mL) in dialysis bag (MWCO = 14 kDa, Millpore) were suspended in HEPES medium (20 mL) at 37 °C with shaken at 100 rpm. A total of 1 mL of the solution was collected at the predetermined time, followed by supplement with the same volume to maintain a stable volume. The percentage of drugs released was calculated using Eq. (1):
| (1) |
where V is the total volume; Ct is the concentration of drug at t time; Y is total amount of drug collected before t time; and M is original content for dialysis.
2.5. Investigation of the long-time stability of BMS/RA@CC-Liposome
To explore the effect of cell membrane coverage on liposome nanoparticles, the membrane-to-liposome volume ratios ranging from 6:1 to 0.25:1 were studied. Briefly, six aliquots of 330 μL liposome (1 mg) were mixed with 1980, 990, 495, 330, 165, and 82.5 μL cancer cell membrane, respectively. Then, the mixtures were repeatedly coextruded through porous polycarbonate membrane (220 nm) to obtain the liposome coated membrane. For stability studies, the obtained BMS/RA@CC-Liposome at membrane-to-liposome volume ratios stored after 0, 7 and 21 days were sent to analyze with Malvern Zetasizer nano instrument.
2.6. In vitro cytotoxicity assay
Sulforhodamine B (SRB) assay was used for evaluation cytotoxicity of CC-Liposome (without RA-V or BMS loading) on CT26, HCT116, HT29, RKO, SW620 and NCM460 cells. Briefly, cells were seeded in 96-well plates in 200 μL medium the day before incubation with a series of CC-Liposome (at doses of 55.5, 27.75, 13.87, 6.94, 3.46, and 1.73 μg/mL cancer cell membrane) concentrations for 48 h. Next, the cells were fixed for 1 h with 50 μL of 50% acetic acid. Then, after washing three times with water, 100 μL of 4% SRB was added to each well for staining 15 min. Next, 1% acetic acid was used to wash the cells to remove excess dye. At last, after dried in the oven, 100 μL of 10 mmol/L Tris [hydroxymethyl]aminomethane buffer (TRIS base) was used for dissolving the stain cells, which were measured at 540 nm using a microplate reader.
2.7. Colony forming assay
Cells were seeded in 6-well plates with a density of 1000 cells each well in 2 mL medium the day before incubation with CC-Liposome at dose of 55.5 μg/mL cancer cell membrane and liposome (without cancer cell membrane coating) for 48 h. The dose of liposome in cytotoxicity assay was equal to the amount of liposome in CC-Liposome. The medium was replaced every two days for adequate nutrition in the process of cell proliferation. At the eighth day, the cells were fixed for 1 h with 4% paraformaldehyde and then stained for 15 min with crystal violet. Next, after washing with PBS for three times, the stained cells were dried in the air and then photographed.
2.8. Cellular uptake of BMS/RA@CC-Liposome
For immune evasion ability study, RAW 264.7 cells were seeded at a density of 2 × 105 cells each dish before a day. Then, the cells were treated with Cy5.5-labeled BMS/RA@Liposome, BMS/RA@CC-Liposome and BMS/RA@RBC-Liposome for 1 and 2 h, next washed with PBS for three times, and later stained with Hoechst 33342 for 30 min. For homotypic targeting ability study, CT26 (2 × 105/mL), HCT116 (4 × 105/mL), SW620 (3 × 105/mL), RKO (3 × 105/mL) and A549 cells (2 × 105/mL) were firstly incubated with Cy5.5-labeled BMS/RA@CC-Liposome for 3 h, washed with PBS, and then stained with Hoechst 33342 for 30 min at room temperature. The cells were observed using ZEISS Laser Scanning Microscope (LSM700, Zeiss, Germany) with a 63 × oil-immersion objective.
2.9. Western blotting analysis
Cells were harvested and lysed in RIPA containing 1% protease and 2% phosphatase inhibitors. Protein lysates were equally subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF membrane (Millipore). After blocking with 5% milk in TBST, prepared in our laboratory, the membranes were first incubated with the primary antibodies overnight at 4 °C, and then incubated with an HRP-labeled secondary antibody at room temperature for 2 h. At last, the signal bands were visualized using enhanced chemiluminescence. GAPDH was chosen as the protein loading control.
2.10. In vivo tumor targeting
1 × 106 CT26 cells were subcutaneously injected into the right dorsal flank of 6–8 week-old female nude mice. After the tumor volume reached 300 mm3, CT26 tumor-bearing nude mice were intravenously injected with 200 μL Cy5.5-labeled BMS/RA@Liposome, BMS/RA@RBC-Liposome and BMS/RA@CC-Liposome. The fluorescence images were acquired by IVIS spectrum instrument with the mice anesthetized at various time points (1, 4, 8, 12 and 24 h). Ex vivo fluorescence bio-distribution image of organs and tumor tissue from CT26 tumor-bearing nude mice was taken at 24-h post injection of the three nanoparticles.
2.11. Pharmacokinetics in vivo
The CT26 tumor-bearing BALB/c mice were randomly divided into 3 groups (n = 3). Then, the mice were injected with free RA-V/BMS-202, BMS/RA@Liposome and BMS/RA@CC-Liposome at RA-V dose of 5 mg/kg and BMS-202 dose of 4 mg/kg through the tail vein. The blood samples were obtained at 0, 0.5, 1, 2, 4, 8 and 24 h after injection in each group, meanwhile the blood samples were centrifuged in the centrifuge (Eppendorf, Germany) (5000 rpm, 15 min). Then, plasma supernatant collected was mixed with methanol for protein precipitation. After concentration with nitrogen gas, the samples were dissolved in methanol and determined by LC‒MS/MS.
2.12. In vivo antitumor studies
The CT26 tumor-bearing BALB/c mice with the tumors volume reaching ∼200 mm3 were randomly divided into 4 groups (n = 8): control group (I); BMS@CC-Liposome group (II); RA-V@CC-Liposome group (III); BMS/RA@CC-Liposome group (IV). All mice were raised in a sterile environment. Mice were intravenously injected with physiological saline, BMS@CC-Liposome (at BMS-202 dose of 4 mg/kg), RA-V@CC-Liposome (at RA-V dose of 5 mg/kg) and BMS/RA@CC-Liposome (at RA-V dose of 5 mg/kg and BMS-202 dose of 4 mg/kg) every two days. Tumor size and body weight were monitored every two days for 22 days. The tumor volume was calculated as Eq. (2):
| (2) |
On Day 23, mice were euthanized. And tumors and the main organs were harvested for H&E staining, immunofluorescence staining and immunohistochemical analysis.
2.13. Cytokine determination
The expression of TNF-α, INF-γ and IL-1β in mice tumors and serum were determined using enzyme linked immunosorbent assay (ELISA) according to protocols.
2.14. Analysis of T cells in tumors using flow cytometry
At the end point, mice were sacrificed and tumors were collected and cut into pieces (3–4 mm3). These tumor pieces were then digested in medium (pH = 7.4) containing 1% fetal bovine serum and collagenase III (200 U/mL) for 3 h, with shaking the medium every 30 min. Next, the tissue suspension was filtrated through a cell strainer (40 μm) to obtain single cell suspensions. Then these cells were stained with fluorescence labeled antibodies for analysis of T cells with Attune NxT Flow Cytometer.
2.15. Statistical analysis
The mean ± SD were determined for all the treatment groups. Statistical analysis was performed by one-way analysis of variance (ANOVA). The difference between two groups is considered statistically significant for ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
3. Results and discussion
3.1. Synthesis and characterization of BMS/RA@CC-Liposome nanoparticles
The synthesis of BMS/RA@CC-Liposome is described in detail in the experimental section. Briefly, preparation of BMS/RA@CC-Liposome includes three steps: 1) synthesizing BMS/RA@Liposome with co-loading BMS and RA-V, 2) preparing CT26 cancer cell membrane vesicles, and then 3) coating CT26 cancer cell membrane vesicles onto BMS/RA@Liposome to obtain BMS/RA@CC-Liposome. As measured by Malvern Zetasizer nano instrument, the cell membrane vesicles were 271.5 ± 1.59 nm in hydrodynamic diameter. After the coverage of cell membrane vesicles on the BMS/RA@Liposome, the final BMS/RA@CC-Liposome were 192.46 ± 7.91 nm in size (Fig. 1A and B). Moreover, the zeta potential of BMS/RA@Liposome, BMS/RA@CC-Liposome and cell membrane vesicles were respectively determined to be −34.23 ± 1.35, −8.71 ± 0.60 and −10.38 ± 1.33 mV, demonstrating that the successful membrane vesicles coating, as the surface charge of the BMS/RA@CC-Liposome increased to approximately the level of the cell membrane vesicles (Fig. 1C). In addition, membrane coating around the BMS/RA@CC-Liposome was visualized using transmission electron microscopy (TEM). As shown in Fig. 1D, the obtained BMS/RA@CC-Liposome was averagely dispersed with spherical morphology and exhibited a core‒shell structure, confirming successful coating of the cell membrane vesicles on the surface of the liposome nanoparticles. Moreover, fluorescence resonance energy transfer (FRET) experiment was performed to further investigate whether cancer cells membranes were coated on liposomes or fused with liposomes. In the preparation of BMS/RA@CC-Liposome, the cancer cells membranes were fluorescently labeled with DiO, and the Black Hole Quencher-1 (BHQ-1) was doped into the liposome shell, serving as an ultra-efficient energy quencher of DiO. As the Supporting Information Fig. S1A shows, the fluorescence of DiO has hardly decreased along with the increasing proportions of BHQ-1, suggesting that there was not “FRET” effect between BHQ-1 and DiO due to the longer distance. In contrast, when the BHQ-1 and DiO were both doped into the liposome shell of BMS/RA@CC-Liposome, the fluorescence of DiO was decreased significantly as the proportions of BHQ-1 was increased (Fig. S1B), suggesting that “FRET” effect can be induced when BHQ-1 and DiO were both in the liposome shell. Therefore, it can be concluded that the cancer cell membranes did not fused with liposomes, because “FRET” was not observed in BMS/RA@CC-Liposome. LC‒MS/MS analysis showed that the encapsulation efficiency of RA-V and BMS in the obtained BMS/RA@CC-Liposome were 80.1% and 79.6%, which indicated that the lipophilic RA-V and BMS were mostly entrapped inside the liposome.
Figure 1.

BMS/RA@CC-Liposome physicochemical characterization. (A) Size intensity curves of BMS/RA@Liposome (blue), BMS/RA@CC-Liposome (red) and cancer cell membrane vesicles (green) measured by Malvern Zetasizer nano instrument at 25 °C. (B) Hydrodynamic size of BMS/RA@Liposome, BMS/RA@CC-Liposome and cancer cell membrane vesicles. Data are given as mean ± SD (n = 3). (C) Zeta potential of BMS/RA@Liposome, BMS/RA@CC-Liposome and cancer cell membrane vesicles. Data are given as mean ± SD (n = 3). (D) Transmission electron micrographs of (I) a BMS/RA@Liposome; (II) a cancer cell membrane vesicle; (III) a BMS/RA@CC-Liposome; and (IV) multiple BMS/RA@CC-Liposome. All scale bars = 200 nm.
3.2. Validation of cancer cell membrane antigen functionalization
To further investigate the complete cancer cell membrane coating of BMS/RA@CC-Liposome in the cellular environment, the cancer cells membrane were fluorescently labeled with DiO (green), and the pH-sensitive liposome was loaded with Cy5.5 (red), respectively. CT26 cancer cells were then incubated with the dual-labeled BMS/RA@CC-Liposome for 1 and 2 h, and imaged by confocal laser scanning microscopy (Fig. 2A, Supporting Information Fig. S2). As the results show, DiO and Cy5.5 both distributed in CT26 cells and co-localized with each other, demonstrating that BMS/RA@CC-Liposome was uptake by CT26 cells with the help of cell membrane (Supporting Information Fig. S2). Interestingly, DiO and Cy5.5 did not match well after cellular endocytosis (the colocalization efficient was 0.33 ± 0.02), which suggested that cell membranes were mostly detached from the surface of liposome when the BMS/RA@CC-Liposome was subjected to the slightly acidic tumor microenvironment40. This feature is advantageous for targeting cancer cells but does not affect drugs release.
Figure 2.

Validation of cancer cell membrane antigen functionalization and investigation of long-time stability and pH-responsive property of BMS/RA@CC-Liposome. (A) Colocalization of liposome and cancer cell membrane upon cellular uptake; BMS/RA@CC-Liposome was synthesized with liposome loaded with Cy5.5 (red channel) and cancer cell membrane labeled with Dio (green channel). Scale bar = 5 μm. (B) SDS-PAGE protein analysis of raw cell lysate, cytoplasm and cancer cell membrane vesicles. E1: marker, E2: raw cell lysate, E3: cytoplasm, E4: cancer cell membrane vesicles. Samples were loaded at equal protein concentration. (C) Western blotting analysis for membrane-specific and intracellular protein markers, including GAPDH, Lamin B, COXIV, Na+/K+-ATPase, E-cadherin and CD47. (D) Hydrodynamic size as measured by Malvern Zetasizer nano instrument at 25 °C at varying membrane protein to liposome volume ratios right after synthesis, after adjusting to PBS for 7 and 21 days. Data are given as mean ± SD (n = 3). Insert: Western blotting analysis of Na+/K+-ATPase for nanoparticles at varying membrane protein to liposome ratios (3:1, 1.5:1, 1:1, 0.5:1, 0.25:1, 0:1; v/v). (E) In vitro release profiles of RA-V from BMS/RA@Liposome and BMS/RA@CC-Liposome at pH 5.0, 6.5 and 7.4. Data are given as mean ± SD (n = 3). ns, not significant.
Next, we analyzed the protein content of cancer cell membrane vesicles for confirming successful purification from cancer cells. Gel electrophoresis experiment showed the types and expression quantity of proteins in cancer cell membrane vesicles are different from to raw cell lysate or cytoplasm (Fig. 2B), which was later confirmed by Western blotting analysis of a series of membrane and intracellular protein markers (Fig. 2C). GAPDH as a protein marker of cytosol, lamin B as the nucleus marker, and COXIV as the mitochondria marker36 were all lower present on cell membrane vesicles compared to raw cell lysate or cytoplasm, which suggested the successful prefractionation of cell membrane vesicles from cancer cells. Moreover, a significant enrichment of Na+K+-ATPase in cell membrane vesicles, as an indispensable marker of membrane protein, further validated the successful separation of cell membranes through the fabrication process. CD47 has been identified as a crucial protein expressed on the surface of cells in many cancers, allowing them to evade phagocytic activity of macrophages. As shown in Western blot results of CD47, the contents of E4 are more than that of E2 or E3, which suggested that CT26 cell membrane protein retained CD47 after purification from cancer cells. Moreover, E-cadherin41, a widely reported protein that is related to homotypic cell adhesion, also still remained in the cancer cell membrane vesicles, which demonstrates preferential retention of membrane antigens after the extraction process. These results illustrate cell membrane vesicles could be successfully prepared from cancer cells, which still remained adhesion of homotypic tumor cells mediated by membrane proteins such as E-cadherin. Collectively, it is believed that the cell membrane vesicles possess the potential to target homologous tumor.
3.3. Long-time stability and pH-responsive property of BMS/RA@CC-Liposome
In order to investigate the effect of cell membrane coverage on liposome nanoparticles, BMS/RA@CC-Liposome was synthesized at different membrane-to-liposome volume ratios ranging from 6:1 to 0.25:1 (Fig. 2D). The BMS/RA@CC-Liposome at different membrane-to-liposome volume ratios retained the coverage of cell membrane as the observed membrane maker (Na+/K+-ATPase). Moreover, there is slight decreased changes but still near to 200 nm (suitable for passive targeting of EPR effect) in the hydrodynamic diameter when the particles at membrane-to-liposome volume ratio (1.5:1) were adjusted to 1 × PBS for 7 and 21 days. In addition, the stability of the BMS/RA@CC-Liposome was also evaluated at 37 °C. As Supporting Information Fig. S3 shows, the average hydrodynamic diameter of BMS/RA@CC-Liposome did not change obviously during 3 days, suggesting the nanoparticles were stable for practical application conditions of 37 °C. These results demonstrated that membrane coverage on liposome at 1.5:1 has a good stability and could be used for further research. Next, the release of RA-V from BMS/RA@CC-Liposome or BMS/RA@Liposome was performed to characterize the effects of membrane coverage on liposome (Fig. 2E). At pH 7.4, both BMS/RA@CC-Liposome and BMS/RA@Liposome showed no more than 20% amounts of RA-V released even over 25-h incubation, reflecting good stability at normal physiological environment. However, the release of RA-V was dramatically accelerated at pH 5.0, demonstrating an obvious pH-dependent gradient in vitro. After about 25-h incubation, 82.03 ± 3.26% and 74.53 ± 2.40% of RA-V were released from BMS/RA@Liposome and BMS/RA@CC-Liposome, respectively. The exposure to acidic pH could result in the molecular shape of DOPE phospholipids accumulating in inverse hexagonal phases, which in turn induced the drug release42. Due to outer shell membrane acting as a diffusion barrier41, the release of RA-V from BMS/RA@CC-Liposome is less than that of BMS/RA@Liposome, but with no statistic difference, indicating that the cell membrane coating had negligible effects on the drug release. Similar results were observed from the release profiles of BMS (Supporting Information Fig. S4). As a result, the biomimetic pH-sensitive liposome realized precisely controlled drug release in the intracellular acidic environments.
3.4. Investigation of anti-phagocytosis capability and homologous adhesion property of BMS/RA@CC-Liposome
Biocompatibility investigation is an important toxicity assessment for biomedical applications. Therefore, sulphorhodamine B (SRB) assay was firstly used to assess cytotoxicity of empty cancer cell membrane-coating liposome (CC-Liposome). The cytotoxicity of CC-Liposome in different concentrations on various colon cancer cells and normal cells were negligible (Supporting Information Fig. S5). Moreover, hemolytic test in vitro experiment (Supporting Information Fig. S6) demonstrated that the hemolytic rate of CC-Liposome was lower than 1%, which was safe in the biological function test of biomaterial. In addition, considering the specialty of CC-Liposome as drug delivery carriers from endogenous substances, colony forming assay was used to examine the capability of a single cell to grow into a large colony in vitro after CC-Liposome incubation43. As shown in Supporting Information Fig. S7, there was no obvious increase or decrease in the numbers of colonies compared to control group, suggesting both CC-Liposome and liposome had no obvious influence on colony formation activity in various colon cancer cells, as well as normal cells. Collectively, the above results demonstrated a good biocompatibility of CC-Liposome as drug delivery carrier for the application in biology.
Next, the anti-phagocytosis capability of BMS/RA@CC-Liposome was investigated. Biomimetic drug delivery carrier has been demonstrated to selectively accumulate in tumor site, utilizing the biological properties of membrane such as anti-phagocytosis and homologous binding capabilities44. To investigate the anti-phagocytosis capability of BMS/RA@CC-Liposome, RAW 264.7 cells as murine macrophage-like cell, were incubated with three kinds of nanoparticles (BMS/RA@Liposome, BMS/RA@CC-Liposome and BMS/RA@RBC-Liposome), and then imaged using confocal laser scanning microscope (CLSM). The RAW264.7 cells treated with BMS/RA@Liposome (without membrane-coating) exhibited bright red fluorescence along with the incubation time, while BMS/RA@CC-Liposome-treated cells showed much weaker intensity of the red fluorescence (Fig. 3A and B), indicating that the cell membrane on the outer shell of the liposome remarkably decreased the cellular uptake of BMS/RA@CC-Liposome. This result suggested that BMS/RA@CC-Liposome could be disguised as endogenous substances and reduced reticuloendothelial system uptake, which might be attributed to anti-phagocytosis capability of membrane protein. To further determine the crucial role of the membrane proteins in escape from uptake in RAW264.7 cells, red blood cells (RBCs), the recognized natural long-circulating delivery vehicles that have anti-phagocytosis ability45 were used to prepare RBC-membrane-coated liposome (BMS/RA@RBC-Liposome) as positive control. The BMS/RA@RBC-Liposome exhibited a lower uptake rate (Fig. 3C), which is comparable with that of BMS/RA@CC-Liposome, confirmed that BMS/RA@CC-Liposome possessed anti-phagocytosis ability. Quantification of flow cytometry analysis also showed the similar results (Supporting Information Fig. S8A). These results demonstrate that BMS/RA@CC-Liposome was hardly taken up by macrophage cells in reticuloendothelial system (RES) due to certain surface proteins of cancer cell membrane similar to red blood cells membrane (such as CD47, serves as an anti-phagocytic or “don't eat me” signal46), contributing to a long circulation time, which was comparable to RBC membrane-coated nanoparticles (BMS/RA@RBC-Liposome).
Figure 3.

Investigation of anti-phagocytosis capability and homologous adhesion property of BMS/RA@CC-Liposome. (A) Intracellular uptake of BMS/RA@Liposome in RAW264.7 cells after 1–2 h incubation. The nucleus was stained with Hoechst 33342 (blue). The BMS/RA@Liposome was labeled with Cy5.5 (red). Scale bar = 10 μm. (B) Intracellular uptake of BMS/RA@CC-Liposome in RAW264.7 cells after 1–2 h incubation. The nucleus was stained with Hoechst 33342 (blue). The BMS/RA@CC-Liposome was labeled with Cy5.5 (red). Scale bar = 10 μm. (C) Intracellular uptake of BMS/RA@RBC-Liposome in RAW264.7 cells after 1–2 h incubation. The nucleus was stained with Hoechst 33342 (blue). The BMS/RA@RBC-Liposome was labeled with Cy5.5 (red). Scale bar = 10 μm. (D) Intracellular uptake of BMS/RA@CC-Liposome in CT26, HCT116, SW620, RKO and A549 cells after 3 h incubation. The nucleus was stained with Hoechst 33342 (blue). The BMS/RA@CC-Liposome was labeled with Cy5.5 (red). Scale bar = 10 μm. (E) Quantitative analysis of BMS/RA@CC-Liposome uptake with CT26, HCT116, SW620, RKO and A549 cells. Data are given as mean ± SD (n = 3). ∗∗∗P < 0.001.
Then, the homologous adhesion property of BMS/RA@CC-Liposome was investigated. The retaining membrane antigens such as TF-antigen and E-cadherin, endowing the homologous targeting ability, have been utilized for the targeted drug delivery in some researches41,47. As Supporting Information Fig. S9 displays, after 2-h incubation, BMS/RA@CC-Liposome exhibited relative stronger fluorescence intensity in CT26 cells than HCT116, SW620 and RKO cells, which indicated an initial homologous targeting ability though the differences were not significant. However, along with the incubation time to 3 h, BMS/RA@CC-Liposome exhibited remarkable fluorescence in CT26 cells, while showed much weaker fluorescence in HCT116, SW620 and RKO cells (Fig. 3D and E), indicating the superior selectivity of BMS/RA@CC-Liposome to CT26 cells, which could be attributed to the homotypic adhesive interactions mediated by the adhesion molecules on CT26-cancer cell membrane. In contrast, BMS/RA@CC-Liposome incubated in the human lung adenocarcinoma A549 cells exhibited negligible fluorescence, suggesting that the enhanced internalization of BMS/RA@CC-Liposome in colon cancer cells, especially in CT26 cells, was probably caused by the excellent affinity of BMS/RA@CC-Liposome to homotypic cell lines. Moreover, the obvious differences in uptake of CT26, HCT116, SW620, RKO and A549 cells were also observed in Fig. S8B. The results imply that when cell membrane of BMS/RA@CC-Liposome matched homogeneous cancer cells, such as CT26 and HCT116 cells, remarkable binding property was observed. Moreover, to investigate the cellular uptake mechanism of BMS/RA@CC-Liposome, the CT26 cells were incubated with BMS/RA@CC-Liposome, BMS/RA@Liposome and BMS/RA@CC-Liposome (pre-treated with TF-antigen antibody and E-cadherin antibody). As shown in Supporting Information Fig. S10, the CT26 cells displayed strong fluorescence after incubation of BMS/RA@CC-Liposome, while the cells pre-treated with TF-antigen antibody and E-cadherin antibody, and then incubated with BMS/RA@CC-Liposome showed much weaker fluorescence. Moreover, the CT26 cells incubated with BMS/RA@Liposome also were observed relative weaker fluorescence, compared with the cells incubated with BMS/RA@CC-Liposome. These results suggest that TF-antigen and E-cadherin remaining on the cell membrane of BMS/RA@CC-Liposome were engaged in the internalization process, contributing to the homologous targeting ability. Therefore, the BMS/RA@CC-Liposome possessing homologous binding ability and immune evasion ability is broadly applicable to the same kind of cancer cells.
3.5. Investigation of RA-V-induced dual reduction of HIF-1α and PD-L1 expression
Many studies have demonstrated the important contribution of hypoxia to tumor immune escape in cancer treatment. Especially in adaptive immunity, hypoxia increased the expression of PD-L1, leading to immunosuppression through PD-1/PD-L1-mediated interaction between tumor cells and T cells27. So there are some intrinsic relations between hypoxia and PD-L1 signaling in colon cancer cells. Hypoxia-inducible factor 1α (HIF-1α) as an important marker in hypoxia environment characterizes the degree of hypoxia cancer cells. Considering RA-V can overcome hypoxia by down-regulating the expression of HIF-1α in tumor cells24, we further investigated the effect of RA-V on PD-L1 expression driving immune escape. Western blotting assays revealed that RA-V down-regulated PD-L1 expression remarkably in a HIF-1α-dependent manner (Fig. 4A and B). Moreover, immunofluorescence assay showed that RA-V effectively reduced the fluorescence intensity of FITC-labelled PD-L1 (Fig. 4C), further confirming the inhibition of PD-L1 expression mediated by RA-V.
Figure 4.

Investigation of RA-V-induced dual reduction of HIF-1α and PD-L1 expression in colon cancer cells. (A) Effects of RA-V-treatment on CT26 cells with different concentrations. Western blotting analysis for the expression of HIF-1α, PD-L1, HMGA1, p-Erk and p-Akt. (B) Quantitative analysis of the expression of HIF-1α and PD-L1 in Fig. 4A by optical densitometry using Image J. Data are given as mean ± SD (n = 3). (C) Immunofluorescence assay of PD-L1 expression in CT26 cells after different concentrations. The nucleus was stained with Hoechst 33342 (blue). The PD-L1 protein was labeled with FITC (Green). Scale bar = 10 μm. (D) Effects of RA-V-treatment on HCT116 cells with different concentrations. Western blotting analysis for the expression of HIF-1α, PD-L1, HMGA1, p-Erk and p-Akt. (E) Effects of RA-V-treatment on SW620 cells with different concentrations. Western blotting analysis for the expression of HIF-1α, PD-L1, HMGA1, p-Erk and p-Akt. All the samples from total cell lysates for Western blotting analysis ran at equal protein (20 μg). ∗P < 0.05, ∗∗∗P < 0.001.
It was reported that PD-L1 promotes colorectal cancer stem cell expansion by activating high-mobility group A1 (HMGA1)-dependent signaling pathways. And PD-L1 silencing resulted in significant down-regulation of HMGA1, p-AKT, and p-ERK expression. Thus, HMGA1, p-Akt and p-Erk as the crucial proteins play important roles in PD-L1-mediated immune response48, 49, 50. After RA-V-treatment, HMGA1 expression was inhibited significantly in CT26, HCT116 and SW620 cells (Fig. 4A, D and E). In addition, RA-V also decreased the level of p-Akt expression and p-Erk expression both in CT26, HCT116 and SW620 cells. The results above suggested that RA-V could suppress immune escape of tumor cells by inhibiting HMGA1-dependent signaling pathways, including the PI3K/Akt and MEK/ERK pathways. And the similar results were also observed in BMS/RA@CC-Liposome-treated CT26, HCT116 and SW620 cells, which demonstrated that RA-V encapsulated in nanoparticles still held the potential for inhibiting HMGA1-dependent signaling pathways (Supporting Information Fig. S11). BMS-202 as a commercial inhibitor, blocking PD-l/PD-Ll protein/protein interaction, mainly be used to treat cancers. However, the effect of BMS on HIF-1α and PD-L1 expression in cancer cells is unknown. To further explore the mechanism of the BMS/RA@CC-Liposome in blocking PD-L1/PD-1 axis, the effect of BMS on cancer cells was studied. As shown in Supporting Information Fig. S12, BMS alone did not inhibit HIF-1α and PD-L1 expression in CT26, SW620, HCT116 cells. Furthermore, quantitative analysis of the expression of HIF-1α and PD-L1 in Supporting Information Fig. S13 demonstrates that BMS/RA@CC-Liposome exhibited similar inhibitory effect on the HIF-1α and PD-L1 expression in CT26, SW620, HCT116 cells, which is comparable to RA-V treatment alone. Those results suggest that the combination mechanism of BMS/RA@CC-Liposome in blocking PD-L1/PD-1 axis could be as following: one is cutting of the source of PD-L1 expression on cancer cells under the effect of RA-V; the other is that BMS directly blocks the interaction of PD-1/PD-L1 protein/protein in case of partial PD-L1 remained on cancer cells binds to PD-1 protein for mediating immune escape. Moreover, in order to investigate whether RA-V could also induce immunogenic cell death (ICD) effect, which could stimulate the maturation of DCs and promote antitumor immune responses, we detected the characteristics of ICD18 induced by RA-V on HCT116 and CT26 cells. As shown in Supporting Information Fig. S14, RA-V failed to elicit significant pre-apoptotic CRT exposure to membrane surface and release little HMGB1 from nuclei to cytosol, as determined by immunofluorescence staining. From the above, RA-V-induced slight ICD effect and did not participate in the early antitumor immune responses stage. So, RA-V actually induced antitumor immune responses by inhibition PD-L1 expression in cancer cells for blocking PD-L1/PD-1 axis.
Moreover, to further validate the feasibility of RA-V as chemotherapeutics directly killing cancer cells for combination with immune checkpoint inhibitors, the cytotoxicity was studied using sulphorhodamine B (SRB) assay in vitro. The cytotoxicity of the BMS/RA@CC-Liposome-treatment group and RA-V@CC-Liposome group in HCT116 or SW620 cells were obviously higher than those of control group and BMS@CC-Liposome-treatment group. And, it was noted that the cytotoxicity in the BMS/RA@CC-Liposome-treatment group showed no significant difference to RA-V@CC-Liposome group despite co-loading BMS, suggesting that in vitro cytotoxicity was attributed to the RA-V-induced destruction but not subjected to the effect of the BMS, resulting from the absence of immune cells during in vitro cell experiment (Supporting Information Fig. S15). Meanwhile, the cytotoxicity of the BMS/RA@CC-Liposome-treatment group, RA-V@CC-Liposome group and BMS/RA@CC-Liposome-treatment group in CT26 cells also showed the similar results. To further investigate the cell death mechanism, flow cytometry assay and JC-1 assay were carried out with CT26 cells (Supporting Information Fig. S16). The population of apoptotic cells treated with the BMS/RA@CC-Liposome and RA-V@CC-Liposome were higher than that of the cells treated with other treatments. JC-1 assay was performed to investigate cell mitochondrial damage on CT26 cells. Increasing green fluorescence and decreasing red fluorescence were observed after BMS/RA@CC-Liposome-treatment, suggesting the structural and functional impairment of mitochondria induced by RA-V. The results demonstrated that RA-V induced the cell apoptosis through mitochondrion-mediated apoptosis pathway. Therefore, RA-V is an ideal chemotherapeutic candidate combined with PD-1/PD-L1 inhibitor to improve antitumor immunity by down-regulating PD-L1 expression.
3.6. In vivo homologous-targeting fluorescence imaging and antitumor on mice
To evaluate the in vivo tumor-targeting ability of BMS/RA@CC-Liposome, time-dependent whole body near-infrared imaging and ex vivo fluorescence analysis were performed. The liposome co-loading BMS and RA-V without cell membranes (BMS/RA@Liposome), red blood cell membrane-derived liposome (BMS/RA@RBC-Liposome) and CT26 cell membranes-coated liposome (BMS/RA@CC-Liposome) were respectively injected into CT26 tumor-bearing nude mice through the tail vein. The mice treated with BMS/RA@CC-Liposome exhibited a significant fluorescence enhancement in tumor site at 4 h post injection, and could be distinguished from the normal tissues within 24 h post injection (Fig. 5A and C), indicating the high tumor specificity of BMS/RA@CC-Liposome. As a control, BMS/RA@Liposome-treated mice showed lower fluorescence in tumor site at 24 h post injection, while the fluorescence in liver and kidney were higher than that of BMS/RA@CC-Liposome-treated mice, suggesting the essential role of the CT26 cancer cell membranes in tumor targeting. To further validate the tumor targeting was induced by the homologous adhesion property of CT26 cell membranes to cancer cells, BMS/RA@RBC-Liposome was also used for in vivo imaging. BMS/RA@RBC-Liposome showed higher accumulation and longer retained time in tumor region compared to BMS/RA@Liposome, which was attributed to unique anti-phagocytosis capability. However, BMS/RA@RBC-Liposome exhibited lower tumor targeting ability than BMS/RA@CC-Liposome, due to the mismatch between the donor cancer cell membrane and host tumors. The ex vivo analysis of three groups also confirmed the highest accumulation of BMS/RA@CC-Liposome at tumor sites at 24 h after injection (Fig. 5B and D). In addition, to further investigate the homing specificity of BMS/RA@CC-Liposome, three types of mice models bearing CT26, SW620 and A549 tumor respectively, were also in vivo imaged after receiving the same volume injection of BMS/RA@CC-Liposome. As shown in Supporting Information Fig. S17, CT26 tumor-bearing mice exhibited the brightest fluorescence in tumor region, the SW620 tumor-bearing mice came second, while A549 tumor-bearing mice showed much weaker fluorescence than SW620 tumor-bearing mice. These results indicated that CT26 cancer cell membrane-cloaked particles not only possessed the anti-phagocytosis capability similar of RBC membrane-coated liposomes, but also achieved in vivo homologous tumor targeting ability. In addition, BMS/RA@CC-Liposome significantly increased the area under the RA-V or BMS concentration–time curve (AUC0‒t) compared to BMS/RA@Liposome group and Free RA-V/BMS group, demonstrated that BMS/RA@CC-Liposome had a long circulation time, endowed by the biomimetic membrane camouflage (Supporting Information Figs. S18 and S19).
Figure 5.

In vivo homologous-targeting fluorescence imaging and antitumor treatment on mice. (A) In vivo images of mice after tail vein injection of BMS/RA@Liposome, BMS/RA@RBC-Liposome and BMS/RA@CC-Liposome at 1, 4, 8, 12, and 24 h, respectively. (B) Ex vivo images of tumor and other tissues from the mice after 24 h administration with the different formulations. 1: heart; 2: liver; 3: spleen; 4: lung; 5: kidney; 6: tumor. (C) Quantification of fluorescence intensity of (A) (n = 3). (D) Quantification of fluorescence intensity of (B) (n = 3). (E) Changes of tumor volume upon different treatments (n = 8). (F) Changes of body weight upon different treatments (n = 8). (G) Tumors weight of mice with different formulations treatment at the end point of the experiment (n = 8). (H) Representative images of the CT26 tumors after treatment with different formulations on Day 22. I: Control group; II: BMS@CC-Liposome group; III: RA-V@CC-Liposome group; IV: BMS/RA@CC-Liposome group. (I) Representative H&E stained images of tumor slices collected from different groups of mice upon different treatments. Scale bar = 50 μm. Data are given as mean ± SD. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Combinations of anti-PD therapy with conventional therapeutic strategies have shown great potential for reducing side effects and increasing effectiveness of cancer immunotherapy. However, some combinations are not designed based on the basic principles of anti-PD therapy and impair the effects of anti-PD therapies8. In this system, chemotherapeutic agent RA-V for efficient combination with BMS-mediated checkpoint blockade immunotherapy was chosen based on the biology of PD-1/PD-L1 pathway. On the basis of the excellent homologous tumor targeting of BMS/RA@CC-Liposome and the dual blockage of PD-1/PD-L1 pathway, we further investigated the combination antitumor effects of BMS/RA@CC-Liposome in CT26 tumor-bearing mice. In order to evaluate the antitumor activity in vivo, a CT26 tumor-bearing BALB/c mice model was developed by subcutaneously injecting CT26 cells into the right flank regions of mice. When the tumors reached ∼200 mm3, the mice were divided randomly into four groups: (I) control group, (II) BMS@CC-Liposome group, (III) RA-V@CC-Liposome group and (IV) BMS/RA@CC-Liposome group. The tumor-bearing mice were intravenously injected every two days for 22 days, and meanwhile the tumor sizes were monitored. All mice were sacrificed on Day 23 for further characterization. Remarkable inhibition of tumor growth was observed in the mice treated with BMS/RA@CC-Liposome (Fig. 5E, G and H), and body weights exhibited the same changes in groups during the treatments (Fig. 5F), which implied the high in vivo antitumor efficiency and the slight systemic toxicity of BMS/RA@CC-Liposome. In contrast, the mice treated with RA-V@CC-Liposome or BMS@CC-Liposome exhibited lower tumor growth inhibition rates compared with mice treated with BMS/RA@CC-Liposome, validating the enhancement in therapeutic outcomes through an attractive synergy between chemotherapy and checkpoint blockade immunotherapy. Moreover, hematoxylin and eosin (H&E) staining on tumor sections and organ slices after different treatments was investigated. The largest damaged area was observed in tumor tissue of BMS/RA@CC-Liposome group (Fig. 5I), while a few cell damage or degradation was found in other groups, suggesting that monotherapy was not enough to eradicate tumor. Moreover, the H&E staining assay of organ slices further indicated the low systematic toxicity in BMS/RA@CC-Liposome group (Supporting Information Fig. S20). In addition, BMS/RA@CC-Liposome group also remarkably prolonged the survival time of tumor-bearing mice after 32 days (Supporting Information Fig. S21), which was significantly higher than the other treatment groups. All of these results revealed a great potential of this biomimetic drug delivery system was biocompatible for more efficient combination therapy in colon cancer.
3.7. Intratumoral activation of immune response and immunologic mechanisms
Blocking PD-1/PD-L1 interactions would lead to the activation of adaptive immune, which is closely related to the recruitment of cytotoxic CD8+ T cells for directly killing tumor cells and helper CD4+ T cells for regulating of adaptive immunities7. To investigate the activation of immune response and suppression of immune resistance in mice treated with BMS/RA@CC-Liposome, tumor-infiltrating cytotoxic CD8+ T cells and helper CD4+ T cells were analyzed by immunofluorescence staining. As shown in Fig. 6, mice treated with BMS/RA@CC-Liposome exhibited the most proportion of tumor-infiltrating CD4+CD8+ T cells while lower levels in RA@CC-Liposome-treatment and BMS@CC-Liposome-treatment mice, suggesting combined utilization of PD-1/PD-L1 inhibitor (BMS) and chemotherapeutic agent (RA-V) effectively disrupted the immune blockade of PD-1/PD-L1 and reverted T cell exhaustion within the tumor microenvironment, leading to the intratumoral enhancement of immune response. Moreover, quantification of cytotoxic CD8+ T cells and helper CD4+ T cells within the tumors in different groups analyzed by the flow cytometry further confirmed reinvigoration exhausted T cells in mice treated with BMS/RA@CC-Liposome (Fig. 7A and D). In addition, IFN-γ secretion by CD8+ T cells secreting is critical to induce a specific cytotoxic response for the clearance of cancer cells. As shown in Fig. 7C and F, BMS/RA@CC-Liposome-treated mice were determined increasing IFN-γ+ CD8+ T cells compared with other treatments mice. In contrast, regulatory T cells (Tregs, CD4+CD25+Foxp3+ T cells), which have an adverse impact on the proliferation of tumor infiltrated CD8+ T cells51, were significantly decreased by determined in mice with BMS/RA@CC-Liposome administration. The results suggeste that BMS/RA@CC-Liposome succeeded in depleting Tregs to improve the response rate of immunity response (Fig. 7B and E). To further evaluate the activation of immune response, we measured the immune response-related factors in tumor tissue and serum. Certain immune factors, involving in the activation of T cells (TNF-α, IFN-γ, IL-1β)52 were determined by the enzyme linked immunosorbent assay (ELISA). It was observed that BMS/RA@CC-Liposome treatments significantly improved the levels of TNF-α, IFN-γ, IL-1β in tumor tissue and serum (Fig. 8A and B), suggesting that BMS/RA@CC-Liposome could successfully activate the adaptive immune response and cause inflammation in vivo, which was superior to BMS@CC-Liposome and RA-V@CC-Liposome. All results above demonstrated that BMS/RA@CC-Liposome had a synergistic effect on activation of systematic antitumor immunity, which was much more efficient than the treatment with RA-V or BMS alone. BMS/RA@CC-Liposome triggered an antitumor immune response during BMS-mediated blockade of PD-1/PD-L1 axis, simultaneously, RA-V in BMS/RA@CC-Liposome obviously enhanced the antitumor immune by inhibition of PD-L1 expression during the chemotherapy.
Figure 6.

Intratumoral activation of tumor-infiltrating CD4+CD8+ T cells. (A) Representative image of immunofluorescence staining of the tumor sections from all groups showing infiltrated CD4+ T cells and CD8+ T cells. The nucleus were imaged at blue channel, CD4+ T cells were imaged at red channel, CD8+ T cells were imaged at green channel. Scale bar = 20 μm. (B) Quantification of fluorescence intensity of (A). Data are given as mean ± SD (n = 3). ∗∗∗P < 0.001.
Figure 7.

Intratumoral activation of immune response and immunologic mechanisms. (A) FCM examination of tumor-infiltrating CD8+ and CD4+ T cells of tumors from mice with different treatments (Gated by CD3+ T cells). (B) FCM examination of tumor-infiltrating Tregs (CD4+CD25+Foxp3+) of tumors from mice with different treatments (Gated by CD4+ T cells). (C) FCM examination of IFN-γ+CD8+ T cells (Gated by CD3+ T cells). (D) Proportion of CD4+ and CD8+ T cells according to (A). (E) Proportion of Tregs according to (B). (F) Proportion of IFN-γ+CD8+ T cells according to (C). All error bars present as mean ± SD (n = 3). ∗∗∗P < 0.001.
Figure 8.

Intratumoral activation of immune response and immunologic mechanisms. (A) Quantitative analysis of the expression of TNF-α, IFN-γ, IL-1β in tumors from mice with different treatments by enzyme-linked immunosorbent assay (ELISA, n = 8). (B) Quantitative analysis of the expression of TNF-α, IFN-γ, IL-1β in serum collected from mice with different treatments by enzyme linked immunosorbent assay (ELISA, n = 8). (C) Representative image of immunohistochemical staining of the tumor slices. Analysis of the expression of HIF-1α, PD-L1, HMGA1, MPO and CD68 in tumor sections from all groups. Scar bar = 100 μm. Data are given as mean ± SD. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Subsequently, to illuminate the immunologic mechanisms behind the synergistic therapeutic effects of BMS/RA@CC-Liposome, immunohistochemical staining was performed (Fig. 8C). Myeloperoxidase (MPO) and CD68, present in the immune cells of tumor microenvironment (TME), contribute to triggering potent antitumor immune responses. It was observed that BMS/RA@CC-Liposome-treated mice group effectively decreased the expression of HIF-1α, PD-L1 and HMGA1, compared to the other groups treated with monotherapy. Meanwhile, the distinct increase of MPO and CD68 expression in BMS/RA@CC-Liposome-treated mice group demonstrated that BMS/RA@CC-Liposome induced a prominent inflammatory reaction, contributed to boosting antitumor immunity response against cancer cells. These results revealed that BMS/RA@CC-Liposome disrupted the immunosuppression of tumor microenvironment and inhibited tumor growth, which might be attributed to highly therapeutic efficiency in reducing PD-L1 expression in HIF-1α-dependent manner, with targeted inhibition of HMGA1-mediated PI3K-Akt and MEK-ERK pathways.
4. Conclusions
In summary, new cancer-cell-biomimetic drug delivery system (BMS/RA@CC-Liposome) was designed for efficient chemotherapy and immunotherapy. In this system, biomimetic membrane camouflage endowed BMS/RA@CC-Liposome superior anti-phagocytosis capability and homotypic binding capacities, realizing elongated blood circulation and preferential tumor accumulation. Moreover, the BMS/RA@CC-Liposome treatment could induce synergistic antitumor immunity and suppressed the tumor growth in tumor-bearing mice. The blocking of PD-L1/PD-1 axis by PD-L1/PD-1 inhibitor directly recruited robust tumor infiltrating lymphocytes (TILs) to attack tumor, which promoted antitumor T cell response. Simultaneously, the expression of PD-L1 on tumor cells was down-regulated during RA-V-mediated chemotherapy, synergistically blocking PD-L1/PD-1 axis to enhance the activation of lymphocytes. Briefly, the combination therapy not only achieved a dual disruption of the PD-1/PD-L1 axis to elicit significant immune response, but also presented cell-killing effect on cancer cells effect by RA-V-mediated chemotherapy. The BMS/RA@CC-Liposome provides new strategy for optimal combinatorial therapy based on immunology of the PD-1/PD-L1 pathway, contributing to exciting therapeutic benefits to cancer immunotherapy.
Acknowledgments
The authors wish to express sincere gratitude to National Natural Science Foundation of China (No. 32070356), the National New Drug Innovation Major Project of Ministry of Science and Technology of China (No. 2017ZX09309027), the Program of Innovative Research Team of Jiangsu Province, “Double First-Class” University Project of China Pharmaceutical University (No. CPU2018GF05, China) and the Fundamental Research Funds for the Central Universities (No. 2632018ZD08, China).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Appendix A. Supporting information
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.10.010.
Contributor Information
Huachao Chen, Email: huachao.chen@cpu.edu.cn.
Ninghua Tan, Email: nhtan@cpu.edu.cn.
Author contributions
Ninghua Tan supervised the project, administrated all participants, and wrote and revised the manuscript. Huachao Chen helped and provided advice for the experimental details, assisted in the experiments, wrote and revised the manuscript. Yongrong Yao conceived and designed the work, undertook the synthesis and characterization of the biomimetic drug delivery system, performed the cell experiments, and animal experiments, wrote the manuscript. All authors reviewed the manuscript and discussed the results.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Peng M., Mo Y.Z., Wang Y., Wu P., Zhang Y.J., Xiong F., et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg S.A. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192:5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodgers D.T., Mazagova M., Hampton E.N., Cao Y., Ramadoss N.S., Hardy I.R., et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113:E459–E468. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xing R.R., Li S.K., Zhang N., Shen G.Z., Möhwald H., Yan X.H. Self-assembled injectable peptide hydrogels capable of triggering antitumor immune response. Biomacromolecules. 2017;18:3514–3523. doi: 10.1021/acs.biomac.7b00787. [DOI] [PubMed] [Google Scholar]
- 5.Souza-Fonseca-Guimaraes F., Cursons J., Huntington N.D. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 2019;40:142–158. doi: 10.1016/j.it.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Lan G.X., Ni K.Y., Xu Z.W., Veroneau S.S., Song Y., Lin W.B. Nanoscale metal-organic framework overcomes hypoxia for photodynamic therapy primed cancer immunotherapy. J Am Chem Soc. 2018;140:5670–5673. doi: 10.1021/jacs.8b01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Constantinidou A., Alifieris C., Trafalis D.T. Targeting programmed cell death-1 (PD-1) and ligand (PD-L1): a new era in cancer active immunotherapy. Pharmacol Therapeut. 2019;194:84–106. doi: 10.1016/j.pharmthera.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Sanmamed M.F., Chen L.P. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–326. doi: 10.1016/j.cell.2018.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwak G., Kim D., Nam G.H., Wang S.Y., Kim I.S., Kim S.H., et al. Programmed cell death protein ligand-1 silencing with polyethylenimine-dermatan sulfate complex for dual inhibition of melanoma growth. ACS Nano. 2017;11:10135–10146. doi: 10.1021/acsnano.7b04717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gajewski T.F., Woo S.R., Zha Y.Y., Spaapen R., Zheng Y., Corrales L., et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Zou M.Z., Liu W.L., Li C.X., Zheng D.W., Zeng J.Y., Gao F., et al. A multifunctional biomimetic nanoplatform for relieving hypoxia to enhance chemotherapy and inhibit the PD-1/PD-L1 axis. Small. 2018;14:1801120. doi: 10.1002/smll.201801120. [DOI] [PubMed] [Google Scholar]
- 15.Yang G.B., Xu L.G., Chao Y., Xu J., Sun X.Q., Wu Y.F., et al. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat Commun. 2017;8:902. doi: 10.1038/s41467-017-01050-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathew M., Enzler T., Shu C.A., Rizvi N.A. Combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol Therapeut. 2018;186:130–137. doi: 10.1016/j.pharmthera.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Duan X.P., Chan C., Lin W.B. Nanoparticle mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem Int Ed. 2019;58:670–680. doi: 10.1002/anie.201804882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Y.C., Kuai R., Xu Y., Ochyl L.J., Irvine D.J., Moon J.J. Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett. 2017;17:7387–7393. doi: 10.1021/acs.nanolett.7b03218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan N.H., Zhou J. Plant cyclopeptides. Chem Rev. 2006;106:840–895. doi: 10.1021/cr040699h. [DOI] [PubMed] [Google Scholar]
- 20.Fan J.T., Su J., Peng Y.M., Li Y., Li J., Zhou Y.B., et al. Rubiyunnanins C-H, cytotoxic cyclic hexapeptides from Rubia yunnanensis inhibiting nitric oxide production and NF-κB activation. Bioorg Med Chem. 2010;18:8226–8234. doi: 10.1016/j.bmc.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Fang X.Y., Chen W., Fan J.T., Song R., Wang L., Gu Y.H., et al. Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction. Toxicol Appl Pharmacol. 2013;267:95–103. doi: 10.1016/j.taap.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 22.Leung H.W., Wang Z., Yue G.G.L., Zhao S.M., Lee J.K.M., Fung K.P., et al. Cyclopeptide RA-V inhibits cell adhesion and invasion in both estrogen receptor positive and negative breast cancer cells via PI3K/AKT and NF-κB signaling pathways. Biochim Biophys Acta. 2015;1853:1827–1840. doi: 10.1016/j.bbamcr.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 23.Yue G.G., Fan J.T., Lee J.K.M., Zeng G.Z., Ho T.W.F., Fung K.P., et al. Cyclopeptide RA-V inhibits angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and HMEC-1 endothelial cells. Br J Pharmacol. 2011;164:1883–1898. doi: 10.1111/j.1476-5381.2011.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao Y.R., Feng L., Wang Z., Chen H.C., Tan N.H. Programmed delivery of cyclopeptide RA-V and antisense oligonucleotide for combination therapy on hypoxic tumors and therapeutic self-monitoring. Biomater Sci. 2020;8:256–265. doi: 10.1039/c9bm00905a. [DOI] [PubMed] [Google Scholar]
- 25.Siemens D.R., Hu N.P., Sheikhi A.K., Chung E., Frederiksen L.J., Pross H., et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. 2008;68:4746–4753. doi: 10.1158/0008-5472.CAN-08-0054. [DOI] [PubMed] [Google Scholar]
- 26.Mohme M., Riethdorf S., Pantel K. Circulating and disseminated tumour cells-mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. 2017;14:155–167. doi: 10.1038/nrclinonc.2016.144. [DOI] [PubMed] [Google Scholar]
- 27.Barsoum I.B., Smallwood C.A., Siemens D.R., Graham C.H.A. Mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 28.Li S.K., Zou Q.L., Xing R.R., Govindaraju T., Fakhrullin R., Yan X.H. Peptide-modulated self-assembly as a versatile strategy for tumor supramolecular nanotheranostics. Theranostics. 2019;9:3249–3261. doi: 10.7150/thno.31814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pitchaimani A., Nguyen T.D.T., Aryal S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials. 2018;160:124–137. doi: 10.1016/j.biomaterials.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 30.Chen Z.W., Wang Z.J., Gu Z. Bioinspired and biomimetic nanomedicines. Accounts Chem Res. 2019;52:1255–1264. doi: 10.1021/acs.accounts.9b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z.W., Wen D., Gu Z. Cargo-encapsulated cells for drug delivery. Sci China Life Sci. 2020;63:599–601. doi: 10.1007/s11427-020-1653-y. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X.D., Wang J.Q., Chen Z.W., Hu Q.Y., Wang C., Yan J.J., et al. Engineering PD-1-presenting platelets for cancer immunotherapy. Nano Lett. 2018;18:5716–5725. doi: 10.1021/acs.nanolett.8b02321. [DOI] [PubMed] [Google Scholar]
- 33.Xue J.W., Zhao Z.K., Zhang L., Xue L.J., Shen S.Y., Wen Y.J., et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12:692–700. doi: 10.1038/nnano.2017.54. [DOI] [PubMed] [Google Scholar]
- 34.Yang X., Yang Y., Gao F., Wei J.J., Qian C.G., Sun M.J. Biomimetic hybrid nanozymes with self-supplied H+ and accelerated O2 generation for enhanced starvation and photodynamic therapy against hypoxic tumors. Nano Lett. 2019;19:4334–4342. doi: 10.1021/acs.nanolett.9b00934. [DOI] [PubMed] [Google Scholar]
- 35.Li S.Y., Cheng H., Xie B.R., Qiu W.X., Zeng J.Y., Li C.X., et al. Cancer cell membrane camouflaged cascade bioreactor for cancer targeted starvation and photodynamic therapy. ACS Nano. 2017;11:7006–7018. doi: 10.1021/acsnano.7b02533. [DOI] [PubMed] [Google Scholar]
- 36.Fang R.H., Hu C.M.J., Luk B.T., Gao W.W., Copp J.A., Tai Y.Y., et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Q.V., Lee J., Lee H., Shim G., Oh Y.K. Cell membrane-derived vesicles for delivery of therapeutic agents. Acta Pharm Sin B. 2021;11:2096–2113. doi: 10.1016/j.apsb.2021.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun H.P., Su J.H., Meng Q.S., Yin Q., Chen L.L., Gu W.W., et al. Cancer cell membrane-coated gold nanocages with hyperthermia-triggered drug release and homotypic target inhibit growth and metastasis of breast cancer. Adv Funct Mater. 2017;27:1604300. [Google Scholar]
- 39.Chen H.C., Tian J.W., He W.J., Guo Z.J. H2O2-activatable and O2-evolving nanoparticles for highly efficient and selective photodynamic therapy against hypoxic tumor cells. J Am Chem Soc. 2015;137:1539–1547. doi: 10.1021/ja511420n. [DOI] [PubMed] [Google Scholar]
- 40.Wang H., Wang Z.H., Tu Y.B., Li Y.K., Xu T., Yang M., et al. Homotypic targeting upconversion nano-reactor for cascade cancer starvation and deep-tissue phototherapy. Biomaterials. 2020;235:119765. doi: 10.1016/j.biomaterials.2020.119765. [DOI] [PubMed] [Google Scholar]
- 41.Sun H.P., Su J.H., Meng Q.S., Yin Q., Chen L.L., Gu W.W., et al. Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv Mater. 2016;28:9581–9588. doi: 10.1002/adma.201602173. [DOI] [PubMed] [Google Scholar]
- 42.Draffehn S., Kumke M.U. Monitoring the collapse of pH-sensitive liposomal nanocarriers and environmental pH simultaneously: a fluorescence-based approach. Mol Pharm. 2016;13:1608–1617. doi: 10.1021/acs.molpharmaceut.6b00064. [DOI] [PubMed] [Google Scholar]
- 43.Rajendran V., Jain M.V. In vitro tumorigenic assay: colony forming assay for cancer stem cells. Methods Mol Biol. 2018;1692:89–95. doi: 10.1007/978-1-4939-7401-6_8. [DOI] [PubMed] [Google Scholar]
- 44.Cheng H., Jiang X.Y., Zheng R.R., Zuo S.J., Zhao L.P., Fan G.L., et al. A biomimetic cascade nanoreactor for tumor targeted starvation therapy-amplified chemotherapy. Biomaterials. 2019;195:75–85. doi: 10.1016/j.biomaterials.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Luk B.T., Zhang L.F. Cell membrane-camouflaged nanoparticles for drug delivery. J Contr Release. 2015;220:600–607. doi: 10.1016/j.jconrel.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh E., Lee E.J., Nam G.H., Hong Y., Cho E., Yang S., et al. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials. 2017;121:121–129. doi: 10.1016/j.biomaterials.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z., Zhao P.F., Luo Z.Y., Zheng M.B., Tian H., Gong P., et al. Cancer cell membrane-biomimetic nanoparticles for homologous-targeting dual-modal imaging and photothermal therapy. ACS Nano. 2016;10:10049–10057. doi: 10.1021/acsnano.6b04695. [DOI] [PubMed] [Google Scholar]
- 48.Wei F., Zhang T., Deng S.C., Wei J.C., Yang P., Wang Q., et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019;450:1–13. doi: 10.1016/j.canlet.2019.02.022. [DOI] [PubMed] [Google Scholar]
- 49.Xian L.L., Georgess D., Huso T., Cope L., Belton A., Chang Y.T., et al. HMGA1 amplifies Wnt signalling and expands the intestinal stem cell compartment and paneth cell niche. Nat Commun. 2017;8:15008. doi: 10.1038/ncomms15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng X.L., Liu Y., Zhu S., Peng X., Li H., Jiao W.H., et al. Co-targeting PI3K/Akt and MAPK/ERK pathways leads to an enhanced antitumor effect on human hypopharyngeal squamous cell carcinoma. J Cancer Res Clin Oncol. 2019;145:2921–2936. doi: 10.1007/s00432-019-03047-2. [DOI] [PubMed] [Google Scholar]
- 51.Luo L.H., Zhu C.Q., Yin H., Jiang M.S., Zhang J.L., Qin B., et al. Laser immunotherapy in combination with perdurable PD-1 blocking for the treatment of metastatic tumors. ACS Nano. 2018;12:7647–7662. doi: 10.1021/acsnano.8b00204. [DOI] [PubMed] [Google Scholar]
- 52.Dai L.L., Li K., Li M.H., Zhao X.J., Luo Z., Lu L., et al. Size/charge changeable acidity-responsive micelleplex for photodynamic-improved PD-L1 immunotherapy with enhanced tumor penetration. Adv Funct Mater. 2018;28:1707249. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.