

Abstract
Synapses connect discrete neurons into vast networks that send, receive, and encode diverse forms of information. Synaptic function and plasticity—the neuronal process of adapting to diverse and variable inputs—depend on the dynamic nature of synaptic molecular components, which is mediated in part by cell adhesion signaling pathways. Here, we found that the enzyme biliverdin reductase (BVR) physically links together key focal adhesion signaling molecules at the synapse. BVR-null (BVR−/−) mice exhibited substantial deficits in learning and memory on neurocognitive tests, and hippocampal slices in which BVR was postsynaptically depleted showed deficits in electrophysiological responses to stimuli. RNA-sequencing, biochemistry, and pathway analyses suggested that these deficits were mediated through the loss of focal adhesion signaling at both the transcriptional and biochemical level in the hippocampus. Independently of its catalytic function, BVR acted as a bridge between the primary focal adhesion signaling kinases FAK and Pyk2 and the effector kinase Src. Without BVR, FAK and Pyk2 did not bind to and stimulate Src, which then did not phosphorylate the N-methyl-D-aspartate (NMDA) receptor, a critical posttranslational modification for synaptic plasticity. Src itself is a molecular hub on which many signaling pathways converge to stimulate NMDAR-mediated neurotransmission, thus positioning BVR at a prominent intersection of synaptic signaling.
Introduction
The molecules that lie within synapses are the substrates for some of the most essential functions of neurons. Receptors at synapses receive and transduce signals from adjacent neurons, channels influence the excitability of the pre- and post-synaptic neuron, and adhesion molecules make and break new connections. Together, these molecules build complex bridges that connect discrete neurons into vast networks that send, receive, and encode diverse forms of information.
Synapses are plastic structures that are dynamically regulated at multiple levels to adapt to new information. Synapses can strengthen or weaken over time, and such activity-dependent changes in synaptic signaling play an important role in the development and maintenance of neural circuits (1). Depending on the inputs into a synapse, the underlying actin cytoskeleton can expand or shrink (2), channels may be inserted or endocytosed (3), or new proteins can be translated (4). This plasticity is thought to drive learning and memory, encoding them by changing the vastly interconnected neural circuits in the brain (5). Long-term potentiation (LTP) is one phenomenon in which certain synaptic inputs, such as high-frequency stimulation, result in a persistent, enduring increase in the strength of synaptic transmission (6). It is thought that the molecular processes that underlie LTP are principal components of synaptic plasticity during learning (7–9).
LTP was first discovered in the hippocampus (10) but is now known to occur throughout the brain, including the cortex, amygdala, and cerebellum (11). The exact mechanics of LTP can vary among these regions and can manifest differentially depending on a number of factors, but LTP is classically mediated by the N-methyl-D-aspartate receptor (NMDAR) (6). The NMDAR is a ligand-gated ion channel activated by the excitatory neurotransmitter glutamate. It is unique among ionotropic glutamate receptors in that it is permeable to calcium as well as sodium and potassium, whereas the others only gate sodium and potassium. Calcium influx through NMDARs is central to LTP and initiates a series of biochemical signals, including activation of calcium-calmodulin and protein kinase C (PKC) (12). NMDARs also mediate redox signaling by triggering synthesis of nitric oxide and superoxide (13, 14). Alongside calcium, nitric oxide and superoxide are key to inducing LTP in the hippocampus (15, 16).
Previously, we discovered that the heme metabolite bilirubin influences NMDAR redox signaling by scavenging superoxide (17). Mice lacking the enzyme that synthesizes bilirubin, biliverdin reductase (BVR), exhibit dysregulated NMDAR redox signaling. BVR null mice (BVR−/−) exhibit exaggerated locomotor activity when injected with NMDA or MK-801 (an NMDAR agonist and antagonist, respectively), further suggesting that BVR−/− mice exhibit irregular NMDAR signaling. Here, we considered whether losing BVR impacts NMDAR-dependent synaptic signaling. Using behavioral, computational, and biochemical approaches, we found that BVR−/− mice exhibit learning and memory deficits and that BVR physically interacts with key molecules in focal adhesion signaling to mediate synaptic signaling in the hippocampus.
Results
BVR−/− mice exhibit impaired learning and memory, but no anxiety or despair
To determine whether eliminating BVR impacts learning and memory, we evaluated the performance of BVR−/− mice against their wild-type littermates in several neurobehavioral assays. We first assessed whether BVR−/− mice exhibit spatial learning deficits in the Morris water maze. The Morris water maze reliably correlates with hippocampal synaptic plasticity (18), and animals with lesions in the dentate gyrus or subiculum perform poorly (19, 20). In this assay, mice are trained over four days to locate a submerged platform in a pool of cloudy water and are tested thereafter. Mice with deficits in spatial learning require more time to find the platform throughout training, and those with deficits in spatial learning and memory will take more time to find it during the test. Here, during the training period, WT mice quickly learned by days 3 and 4 where the platform was, whereas BVR−/− mice showed no improvement in finding the platform (Fig. 1A). On average, BVR−/− mice also spent twice as long locating the platform than did WT mice after four days of training, with some BVR−/− mice never finding the platform (Fig. 1B). BVR−/− mice were not weaker swimmers; rather, BVR−/− mice swam faster and covered a greater distance than did WT mice (Fig. 1, C and D).
Figure 1. BVR−/− mice exhibit impaired learning and memory, but no anxiety or despair.
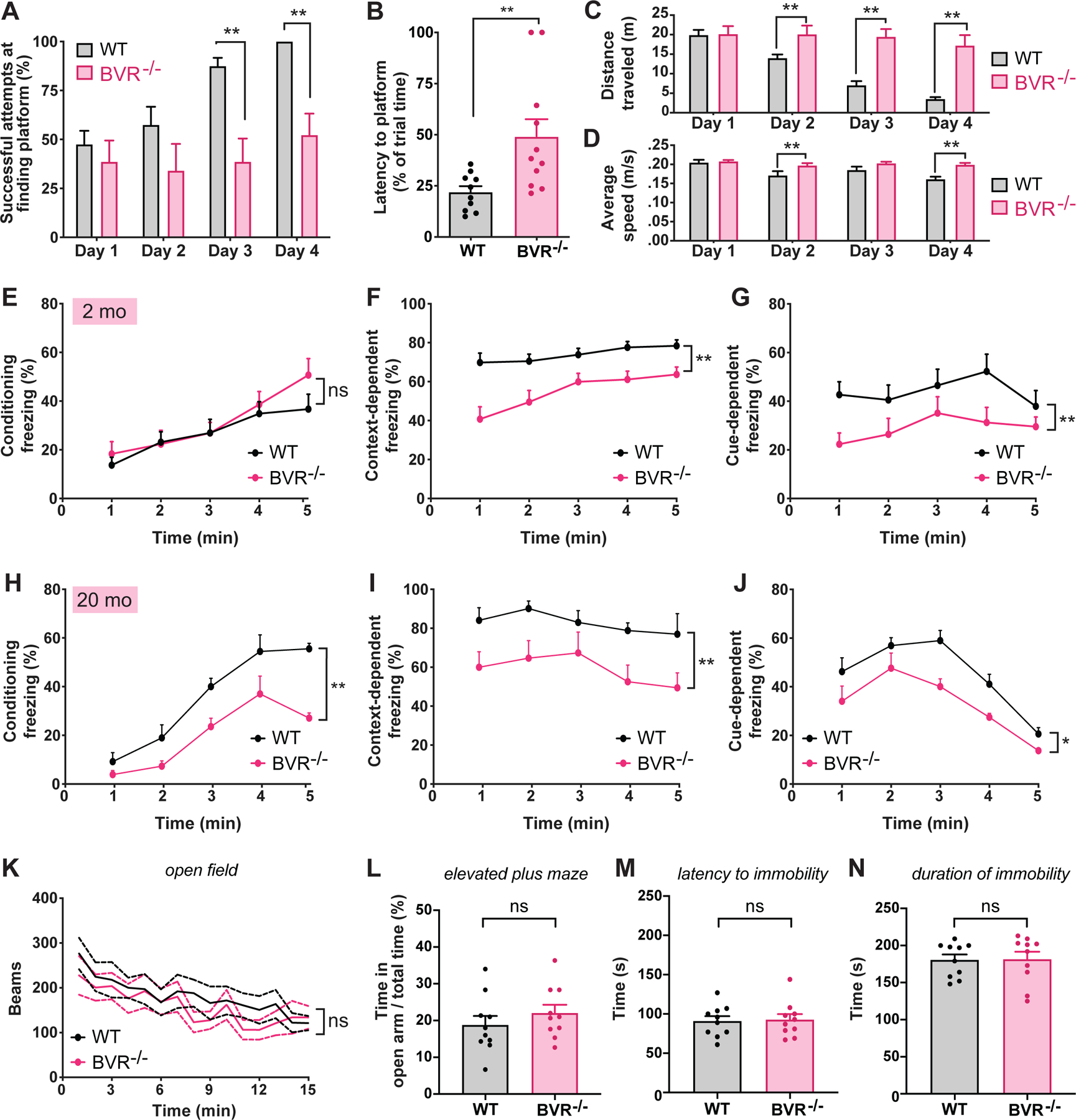
(A to D) Analysis of WT and BVR−/− mice performance in Morris water maze: (A) Percent of successful attempts to arrive at platform during trials on days 1 to 4; (B) latency to arrive at platform on test day 5; (C and D) distance traveled (m) and average swim speed (m/s) during trials on days 1 to 4. Points represent individual mice; n = 10 WT and 11 BVR−/− mice. (E to J) WT and BVR−/− mice performance in context- and cue- dependent fear conditioning at 2 months (E to G) and 20 months old (H to J), calculated as (E and H) percent of time that mice remain frozen during training trial in which a series of tone-shock pairs; (F and I) percent of time that mice remain frozen when re-exposed to training chamber the following day without delivering any shocks; and (G and J) percent of time that mice remain frozen when re-exposed to training tone in a new chamber the following day without delivering any shocks. n = 14 WT and 10 BVR−/− mice. Points represent mean of data. (K) Open field locomotor activity of WT and BVR−/− mouse over time. n = 5 WT and 5 BVR−/− mice. (L) Percent of time WT and BVR−/− mice spent in the open arms of the elevated plus maze. n = 10 WT and 10 BVR−/− mice. Points represent individual mice. (M and N) WT and BVR−/− mice performance in forced swim test. Latency to (M) and duration of (N) immobility, defined as the absence of movement or paddling with only a hind leg to keep head above water. n = 10 WT and 10 BVR−/− mice. Points represent individual mice. In (A to J, and L to N) Mean ± SEM depicted. In (K), mean ± 95% CI is depicted. * P < 0.05, ** P < 0.01, and ns = P > 0.05, by two-tailed unpaired Student’s t-test.
Fear conditioning is an additional form of learning, distinct from spatial learning, in which organisms learn to predict aversive events. Fear conditioning is a behavioral paradigm in which an offending stimulus becomes associated with an otherwise neutral context or cue. Notably, fear conditioning is thought to be learned, stored, and expressed in both the amygdala and hippocampus through the same principal synaptic signaling that encodes spatial learning (21). Electrophysiological studies have demonstrated that neurons in the amygdala undergo LTP (22, 23), and pharmacological studies that perturb synaptic signaling impede learning of conditioned responses (24). To probe whether BVR also influences fear learning, we first compared how well 2-month-old WT and BVR−/− mice associated a context or cue with an aversive sensory stimulus. In these tests, we initially trained the mice to freeze in response to a sound by mildly shocking them in the foot shortly afterwards. With each subsequent tone-shock pair, more mice will freeze after the tone in anticipation of being shocked. The following day, the mice are returned to the same chamber as the day before. Mice which associated the chamber with being shocked will freeze, whereas those that do not are more likely to move about. We then moved the mice to a new chamber and replayed the same sound from the day before. Mice which remembered that the sound precedes a shock will freeze in fear, whereas those which do not are again likely to continue moving about. Unlike in the Morris water maze in which BVR−/− mice struggled to learn the location of the hidden platform, BVR−/− mice initially learned to associate the tone with a shock and were just as likely to freeze as WT mice during training (Fig. 1E). However, BVR−/− mice did not retain either context- or cue-dependent fear learning the day after. When exposed to either the training chamber context or sound cue, BVR−/− mice froze less often than did WT mice (Fig. 1, F and G). We re-challenged the mice at 20 months old to assess whether their deficits worsened with age. We found that these older BVR−/− mice were notably worse than the older WT mice at learning the cues and context during training (Fig. 1H) and exhibited similar, if not greater, learning deficits compared to age-matched WT mice than did their younger counterparts (Fig. 1, I and J). These data suggest that younger BVR−/− mice learned the cues and context as well as WT mice but only recalled them less effectively, whereas older BVR−/− mice were also less effective at learning them to begin with. Both older WT and BVR−/− mice performed worse than their younger counterparts as well, which may reflect overall cognitive decline.
Though BVR−/− mice performed comparatively worse in both the Morris water maze and fear conditioning tests, both tests could be confounded if BVR−/− mice were more hyperactive and mobile than WT mice. If BVR−/− mice are hyperactive, they may be more preoccupied by swimming in the Morris water maze than by finding the platform. They may also freeze less in fear conditioning tests even if they remember the chamber or cue. To evaluate whether BVR−/− mice were hyperactive, we monitored their mobility in the open field assay, in which we measured the total distance traveled by mice placed in an open chamber. Whereas hyperactive mice would have covered a greater distance, we found that BVR−/− mice were only as mobile and active as their WT counterparts (Fig. 1K). We also considered that the Morris water maze and fear conditioning tests could be confounded if BVR−/− mice were more anxious or depressed. However, WT and BVR−/− mice scored similarly in the respectively associated elevated plus maze test (25, 26) and the forced swim test (Fig. 1, L to N). Furthermore, to ensure that the learning deficits we observed in BVR−/− mice were not due to gross neuroanatomical or neuroskeletal abnormalities, we measured their gross brain and skull dimensions and histologically assessed their hippocampi. All of these parameters in BVR−/− mice were similar to those of their WT counterparts (Fig. 2, A to O).
Figure 2. BVR−/− mice do not exhibit global or hippocampal gross neuroanatomical abnormalities.
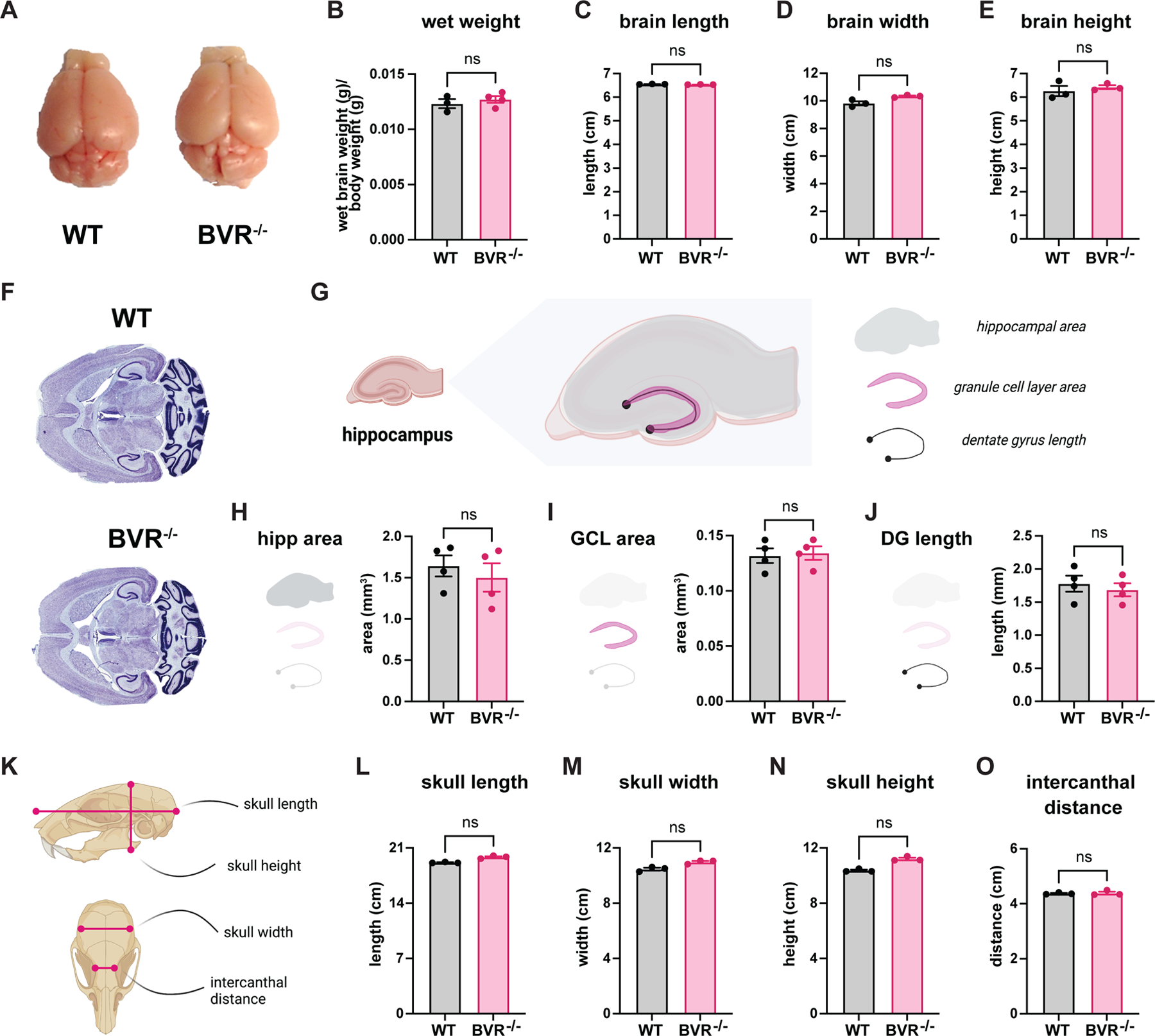
(A to E) Gross analyses of WT and BVR−/− brains from 2-month-old mice assessed for brain organ appearance (A), (B) wet weight [brain weight (g)/body weight (g)], (C) brain length measured rostrocaudally from the anterior-most frontal lobe to posterior-most occipital lobe, (D) brain width measured from widest points of the temporal lobes, and (E) brain height measured from widest points along the dorsal-ventral axis. (F) Nissl-stained transverse sections from WT and BVR−/− mice. (G to J) Depiction (G) and measurements (H to J) of hippocampal regions, measuring hippocampus area, granule cell layer (GCL) area, and dentate gyrus (DG) length. (K to O) Depiction (Kk) and measurements (L to O) of gross skull anatomy, measuring skull length, width, height, and intercanthal distance. All data are mean ± SEM, n = 3 or 4 mice each group; ns P > 0.05 by two-tailed unpaired Student’s t-test.
RNA-sequencing reveals downregulated focal adhesion signaling in BVR−/− hippocampi
Though BVR−/− mice performed comparatively worse in two neurobehavioral assays of learning and memory, neither test immediately reveals the underlying molecular pathology. However, deficits in both the Morris water maze and fear conditioning tests crudely localize to the hippocampus. To identify where in the hippocampus BVR may be expressed, we re-analyzed our previously acquired and posted single-cell RNA-sequencing data from hippocampi of WT mice (27). We identified 13 distinct cell types within the hippocampus, including populations of proliferative cells, astrocytes, radial glia, intermediate progenitors, oligodendrocyte precursor cells, and inhibitory subpopulations of somatostatin (SST)- and vasoactive intestinal peptide (VIP)-positive interneurons. Hippocampal pyramidal neurons further clustered into distinct genetic populations rooted in basic hippocampal circuitry, including the dentate gyrus, CA1, CA2/CA3, subiculum, and entorhinal cortex (Fig. 3A). BVR was ubiquitously expressed across all hippocampal cell populations, though it was perhaps slightly enriched in CA1, CA2, and CA3 pyramidal neurons compared to other cell types (Fig. 3, B and C).
Figure 3. RNA-sequencing reveals downregulated focal adhesion signaling in BVR−/− hippocampi.
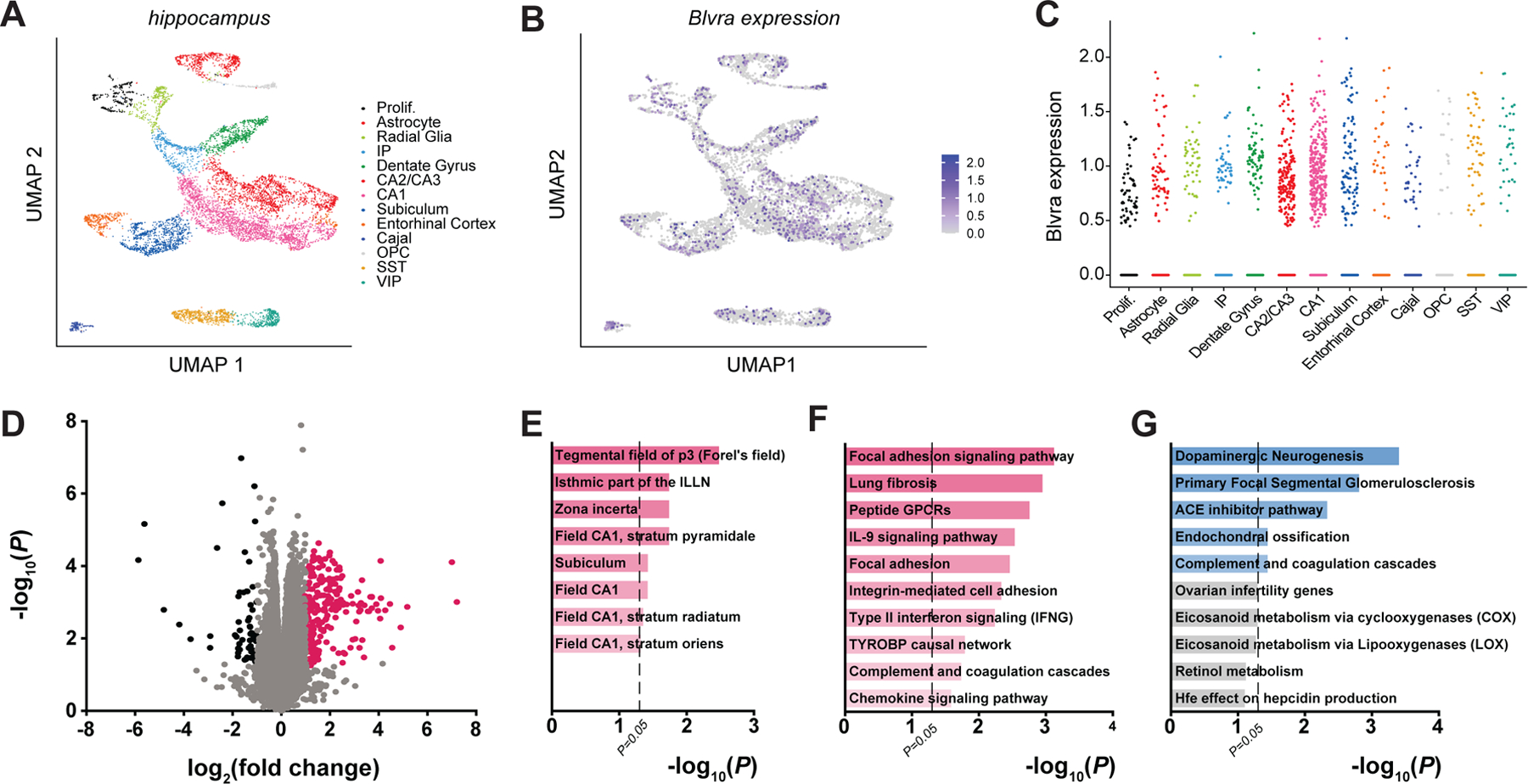
(A and B) UMAP plots of (A) all WT hippocampal cells, colored by cell cluster type (Prolif, proliferative cells; IP, intermediate progenitors; Cajal, Cajal-Retzius cells; OPC, oligodendrocyte precursor cells; SS, SST+ interneurons; VIP, VIP+ interneurons), and of the expression of Blvra (purple) (B) throughout the hippocampus and (C) in each hippocampal cell type of WT mice, analyzed from single-cell RNA-sequencing data reported elsewhere (27). Data points represent individual cells. (D) Volcano plot of transcriptome sequencing of 3 WT and 3 BVR−/− hippocampi. Points represent individual genes. Black points indicate significantly downregulated genes (FDR < 0.5, log2(fold change) ≤ −1), pink points indicate significantly upregulated genes (FDR < 0.5, log2(fold change) ≥ 1), and gray points are not differentially expressed between genotypes. Notable NRF2-target genes are labeled. (E) Genetic network analysis of neuroanatomic and neural cell-type signatures that are downregulated in BVR−/− hippocampi from transcriptome sequencing in (D). (F and G) Gene ontology analysis of molecular pathways that are (F) downregulated and (G) upregulated in the transcriptome sequencing in (D).
To determine how the loss of BVR might affect hippocampal function, we performed a whole-transcriptome RNA sequencing analysis of hippocampi from WT and BVR−/− mice (Fig. 3D and data file S1). We validated the differential expression profile with quantitative reverse-transcription PCR (qPCR) of several genes (fig. S1, A to E). Gene ontology analysis of downregulated genes via Enrichr (28) detected that loss of BVR is associated with a relative reduction in signature CA1 genes, including those expressed in the stratum pyramidale, stratum radiatum, and stratum oriens (Fig. 3E and data file S2). BVR−/− hippocampi were also depleted of transcripts linked to the Fields of Forel and zona incerta, discrete components of the subthalamus. Unbiased gene ontology surveys detected that integrin-mediated cell adhesion and downstream focal adhesion signaling were strongly downregulated in BVR−/− hippocampi (Fig. 3, F and G, and data file S3). Meanwhile, upregulated genes are more strongly enriched for ontologies associated with dopaminergic neurogenesis, though the rest involve processes with less relevance to the brain such as the nephrotic syndromic primary focal segmental glomerulosclerosis. Several pathways of genes involved in complement and chemokine signaling were also dysregulated with loss of BVR (Fig. 3, F and G, and data file S3), which is consistent with a previous study outlining how BVR regulates myeloid chemotaxis in response to complement C5a (29).
Deleting BVR disrupts synaptic focal adhesion signaling in the hippocampus
Comparison of the hippocampal transcriptomes of WT and BVR−/− mice points to the possibility that focal adhesion signaling may be disrupted in the BVR−/− hippocampus. Focal adhesions are large, dynamic structures that bridge the intracellular cytoskeleton to the extracellular matrix (30). Integrins span the membrane and anchor focal adhesions by linking extracellular proteins such as fibronectin, laminin, and collagen to actin through adapter proteins such as talin, α-actinin, and vinculin (31). Embedded among these more structural proteins is a principal signaling molecule, focal adhesion kinase (FAK). FAK is a non-receptor tyrosine kinase that relays changes in the structure of focal adhesion complexes to major signaling axes within the cell, such as phosphatidylinositol 3-kinase (PI3K) and the mitogen activated protein kinases (MAPKs) (32). As a result, FAK can rapidly and potently transduce extracellular signals into intracellular ones. Though FAK has been most extensively studied in non-neural cells, FAK is also expressed at neural synapses and similarly couples extracellular synaptic signals to long-lasting intracellular changes in synaptic strength (33–35). Integrin ligands stimulate FAK and increase synaptic NMDAR and AMPA receptor activity, whereas antibodies against specific integrin subunits prevent the induction of LTP (36–39). In neurons, focal adhesion signaling also includes the FAK ortholog proline-rich tyrosine kinase 2 (Pyk2; also known as CAKβ) (40).
FAK is activated by integrins, which recruit FAK to focal adhesions (41–43). When stimulated, FAK first autophosphorylates itself at Tyr397, generating a high-affinity ligand for the SH2 domain of the kinase Src (44, 45). Src then autophosphorylates itself at Tyr416, a crucial regulatory step that leads to a four-fold increase in Src activity (46, 47). Src then reciprocally phosphorylates FAK (42). Like FAK, Pyk2 can be recruited by integrins but is also activated directly by PKC in response to intracellular calcium (48). Canonically, active FAK and Pyk2 go on to direct cell migration by activating cytoskeletal proteins such as cofilin and paxillin (49, 50). In neurons, activated Src potently controls synaptic signaling by phosphorylating the NR2B subunit of the NMDA receptor at Tyr1472 (Fig. 4A) (38, 51, 52). Tyr1472 is basally phosphorylated in the brain but is hyperphosphorylated in CA1 hippocampal slices after tetanic stimulation (52, 53), potentiating NMDA conductance (54–57), thereby promoting synaptic plasticity and learning in various paradigms (36, 38, 58–61).
Figure 4. Deleting BVR disrupts synaptic focal adhesion signaling in the hippocampus.
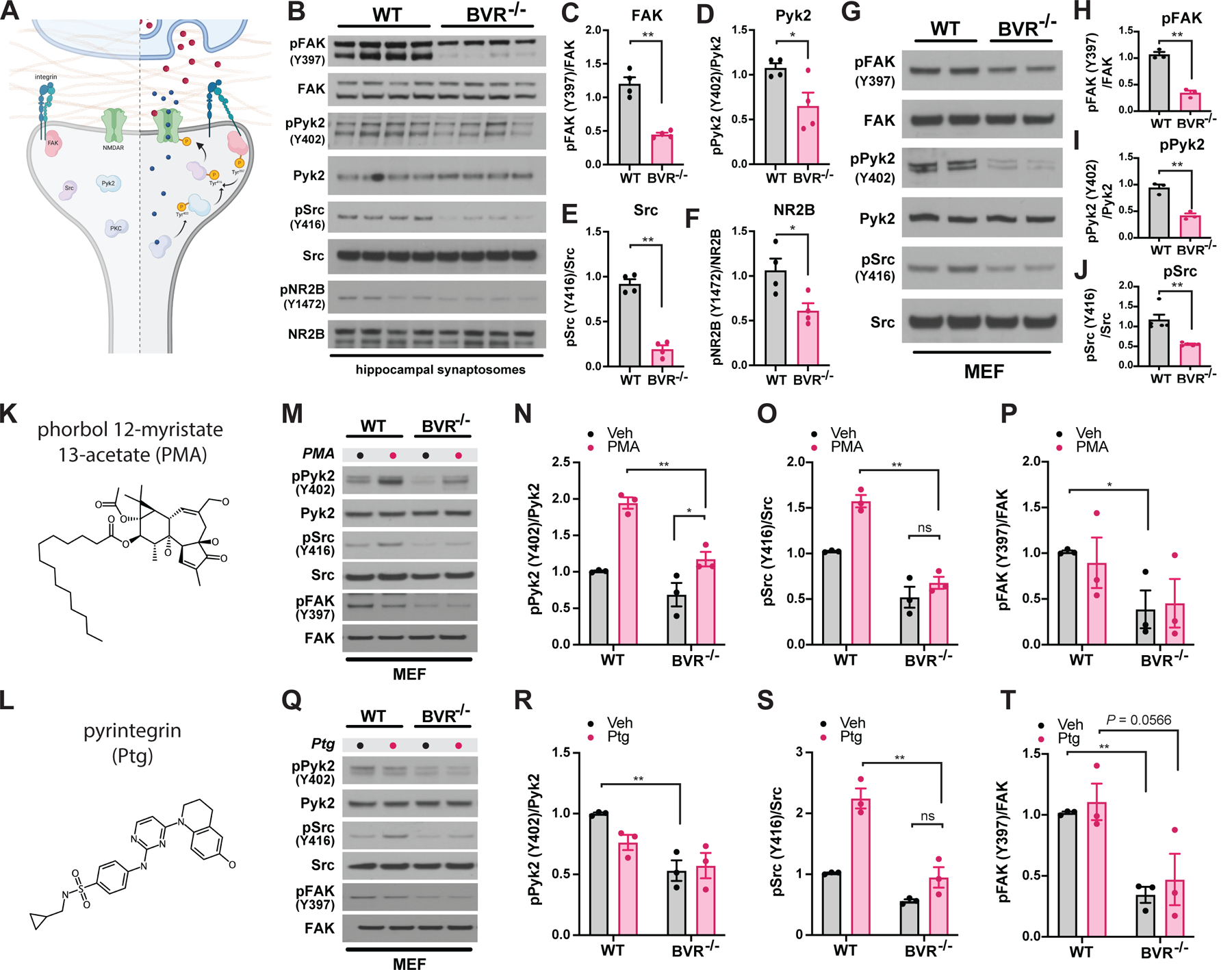
(A) Schematic depicting postsynaptic FAK-mediated signaling pathways at rest (left) and during NMDAR-mediated neurotransmission (right). Upon glutamate/co-agonist (red circles) binding and depolarization-induced removal of Mg2+ block, Ca2+ (blue circles) influx through NMDAR activates PKC. Ca2+/PKC activate Pyk2 by stimulation of autophosphorylation at Tyr402 and integrins by direct phosphorylation. The phosphorylation-induced conformational change in integrins stimulates autophosphorylation of FAK at Tyr397; autophosphorylated FAK and Pyk2 recruit and activate Src by phosphorylation at Tyr416. Src then binds and phosphorylates the NR2B subunit of the NMDAR at Tyr1472, an action which potentiates Ca2+ influx through the NMDAR. Integrins may also be activated during neurotransmission by secretion of extracellular ligands whose identities are presently unknown. (B to F) Immunoblots (B) and quantifications (C to F) of phospho-FAK (Tyr397), FAK, phospho-Pyk2 (Tyr402), Pyk2, phospho-Src (Tyr416), Src, phospho-NR2B (Tyr1472), and NR2B in crude synaptosomes isolated from hippocampi of WT and BVR−/− mice. Data are expressed as normalized ratio of phosphorylated to total protein in each mouse (n = 4 per genotype). (G to J) Immunoblots (G) and quantifications (H to J) of phospho- and total FAK, Pyk2, and Src in WT and BVR−/− MEFs (n = 3). (K to T) Molecular structures of (K) phorbol 12-myristate 13-acetate (PMA) and (L) pyrintegrin (Ptg) Corresponding respectively to immunoblots and quantitative analysis of phospho- and total Pyk2, Src, and FAK in WT and BVR−/− MEFs treated with vehicle and either (M to P) 500 nM PMA for 30 min or (Q to T) 5 µM pyrintegrin for 5 min. Data are mean ± SEM n = 3 experiments. ns P > 0.05, * P < 0.05, and ** P < 0.01, by either two-tailed unpaired Student’s t-test (C to F, H to J) or two-way ANOVA with post-hoc Šidák test (N to P, R to T).
As hinted at in our whole-transcriptome RNA sequencing analysis, we found that activation of the central components of focal adhesion signaling was profoundly downregulated in BVR−/− hippocampal synaptosomes. The initiatory phosphorylation at Tyr397 in FAK and Tyr402 in Pyk2 was considerably blunted in BVR−/− hippocampal synaptosomes, despite equivalent expression of each kinase (Fig. 4, B to D). Downstream targets of FAK and Pyk2 such as Src at Tyr416 and NR2B at Tyr1472 are hypophosphorylated as well (Fig. 4, B, E and F). When we compared whole extracts from WT and BVR−/− hippocampi however, we did not recover the same pattern we observed in isolated synaptosomes. Neither FAK, Pyk2, Src, nor NR2B were differentially phosphorylated in whole extracts (fig. S2, A to E), suggesting that losing BVR impaired synaptic focal adhesion signaling without perturbing the axis throughout the whole cell. Additionally, phosphorylation of other key molecules in synaptic signal transduction pathways—such as GluA1, CaMKII, and CREB, as well as total levels of PSD-95—were unaffected by BVR deletion (fig. S3, A to E). Signaling to canonical cytoskeletal regulators downstream of FAK apparently remained intact as well, with no change in the phosphorylation status of synaptosomal p21-activated kinase (PAK) or cofilin (fig. S4, A to C). Within synaptosomes, the loss of focal adhesion signaling did not lead to the loss of PI3K/Akt or of MAPK signaling (fig. S5, A to C).
Because focal adhesion signaling is dynamic, not static, we considered whether losing BVR disrupts acute FAK signaling beyond baseline phosphorylation events. To acutely manipulate focal adhesion signaling, we isolated and cultured mouse embryonic fibroblasts (MEFs) from WT and BVR−/− mice. Importantly, the same matrix of proteins that was hypophosphorylated in BVR−/− synaptosomes was also dysregulated in BVR−/− MEFs. Tyr397 in FAK, Tyr402 in Pyk2, and Tyr416 in Src were hypophosphorylated in BVR−/− MEFs despite equivalent expression of each kinase (Fig. 4, G to J). We have reported earlier that neither PI3K/Akt nor MAPK signaling was altered in BVR−/− MEFs (17).
To resolve where BVR falls within the matrix of proteins involved, we induced focal adhesion signaling by two orthogonal approaches (Fig. 4, K and L). Whereas phorbol esters like phorbol 12-myristate-13-acetate (PMA; Fig. 4K) stimulate PKC to activate FAK/Pyk2 (62), integrin ligands such as pyrintegrin (Fig. 4 L) act further upstream by engaging integrins (63). If neither PMA nor pyrintegrin recovered FAK/Pyk2 phosphorylation in BVR−/− MEFs, we could reason that BVR may be necessary for activating FAK/Pyk2 themselves. If PMA or pyrintegrin did stimulate FAK/Pyk2 but could not rescue Src phosphorylation, then we may reason that BVR instead somehow links active FAK/Pyk2 to Src. In preliminary experiments, we found that Src was predominantly activated by Pyk2 in MEFs and accordingly focused on Pyk2 instead of FAK here. As expected, PMA activated Pyk2 in WT MEFs with a resulting increase in Src phosphorylation (Fig. 4, M to P). PMA also increased Pyk2 phosphorylation in BVR−/− MEFs, but the effect was much milder than that in WT MEFs. However, PMA did not increase Src phosphorylation in BVR−/− MEFs, suggesting that stimulating Pyk2 alone was not sufficient to rescue Src activation without BVR. PMA did not induce much FAK phosphorylation, which may reflect that PKC is more strongly coupled to Pyk2 than FAK in MEFs. Acutely applying the integrin ligand pyrintegrin similarly increased Src activation in WT cells but was also unable to overcome the effect of losing BVR in MEFs (Fig. 4, Q to T). As a result, both acute and baseline focal adhesion signaling—particularly for activating Src—appeared to be dependent on BVR. Curiously, pyrintegrin stimulated Src but did not sustain Pyk2 or FAK phosphorylation to the same extent as PMA. This may hint that focal adhesion signaling through integrins is more labile than through PKC, but this requires further investigation in the future.
To ensure that the deficits in focal adhesion signaling in BVR−/− cells were indeed due to loss of BVR, we sought to rescue the signaling axis by re-introducing BVR into the genome of BVR−/− MEFs. We first developed a lentivirus that encodes for WT BVR and subsequently infected BVR−/− MEFs, thereby generating a genetic line that expresses BVR in an originally BVR null background (fig. S6, A and B). Upon reintroducing BVR into BVR−/− MEFs, phosphorylation of Pyk2 was rescued, as anticipated (fig. S6, C and D). As determined earlier, Pyk2 is the dominant regulator of Src signaling in MEFs.
BVR is critical for glutamatergic synaptic function in the hippocampus
To assess whether the blunted focal adhesion signaling we see in BVR−/− neurons functionally alters synaptic activity, we ballistically transfected WT organotypic hippocampal slice cultures with dsRed2 and either anti-luciferase or anti-BVR short hairpin RNA (shRNA)-coated gold particles (64). After 2 days, we collected whole-cell recordings from fluorescently-labeled CA1 area hippocampal neurons after inducing LTP at CA3–CA1 synapses (Fig. 5A). We found that silencing BVR in CA1 pyramidal neurons resulted in a significant decrease in LTP, suggesting that BVR has a critical role in glutamatergic synaptic function in the hippocampus (Fig. 5, B and C).
Figure 5. BVR is critical for LTP induction in CA1 hippocampal neurons.
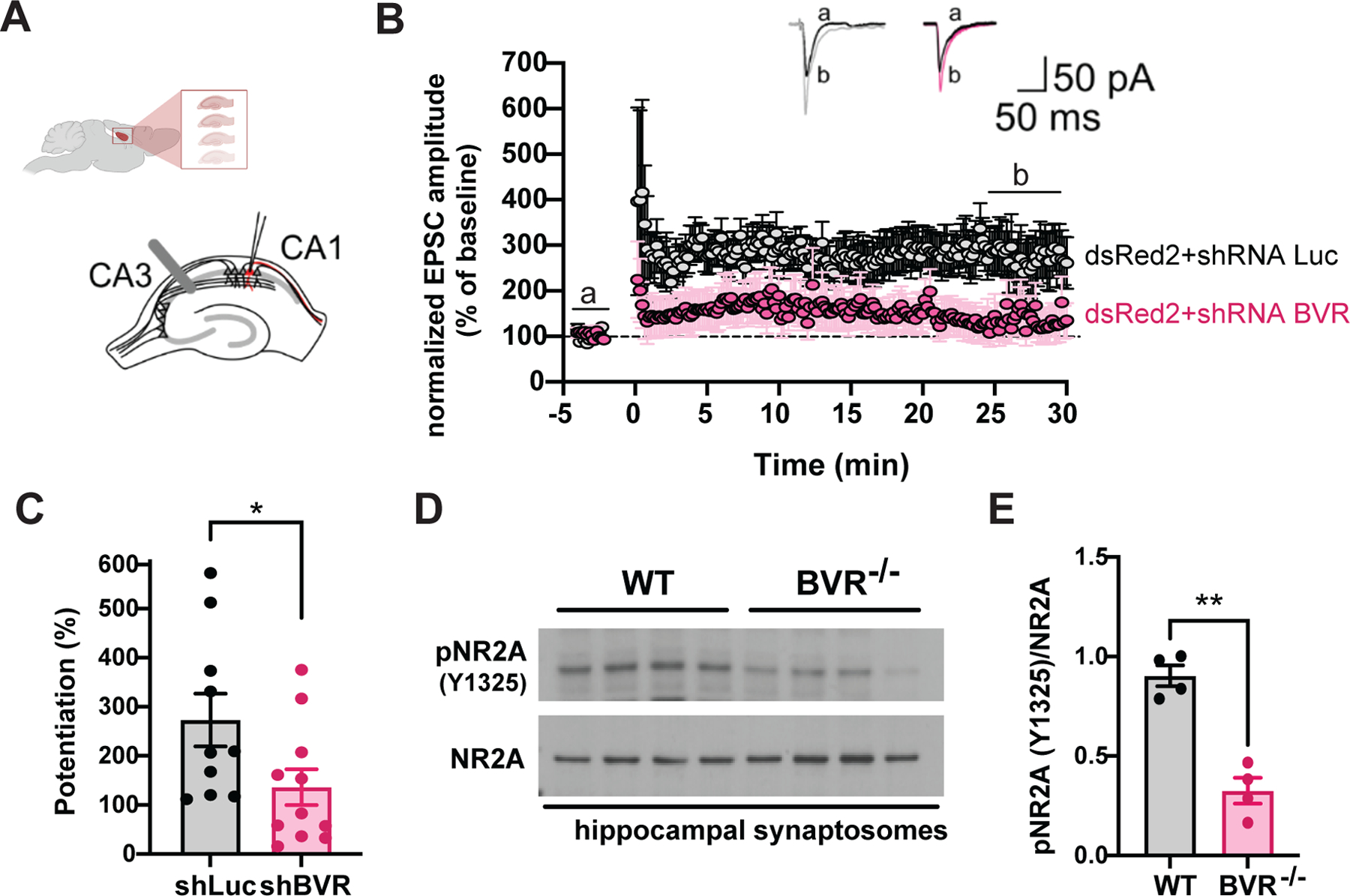
(A) Schematic representation of whole-cell recording from transfected CA1 pyramidal neuron in organotypic hippocampal slice cultures. A stimulating electrode was placed in the stratum radiatum to activate Schaffer collaterals. (B and C) Time course of LTP at CA3–CA1 synapses in hippocampal organotypic slices transfected with shRNA against luciferase (shLuc, grey) and against BVR (shBVR, pink). Results are expressed as percentages of baseline of excitatory post-synaptic current (EPSC) amplitude (= 100%). Representative traces of EPSC recorded in neurons transfected with shLuc and shBVR before (black lines) and after (grey and pink lines for shLuc and shBVR, respectively). Mean LTP values during the last 5 min were plotted (C). Data are from n = 10 (shLuc) or 11 (shBVR) independent experiments. (D and E) Immunoblots and quantitative analysis of phospho-NR2A (Tyr1325) and total NR2A in synaptosomes isolated from hippocampi of WT and BVR−/− mice. Data are expressed as normalized ratio of phosphorylated to total protein in each mouse; points represent individual mice. In (C and E), data are mean ± SEM; * P < 0.05 and ** P < 0.01 by two-tailed unpaired Student’s t-test.
Notably, these data were obtained from neurons in which BVR was depleted only post-synaptically, revealing that BVR most likely influences LTP in postsynaptic neurons. Because FAK/Pyk2 and Src also modulate LTP post-synaptically, these data are consistent with a model in which BVR helps coordinate these various molecules during NMDA neurotransmission. Intriguingly, these data also reveal that even transient (two- to three-day) loss of BVR blunts LTP, suggesting that the learning deficits in BVR null mice are less likely to be from aberrant networks during development.
We next explored the biochemical status of Tyr1325 of the NMDAR subunit NR2A in WT and BVR−/− mice. Electrophysiological studies have demonstrated that tyrosine phosphorylation of NR2A and NR2B potentiates NMDAR currents by reducing tonic inhibition; more specifically, mutating NR2A residue Tyr1325 to alanine prevents Src-mediated potentiation of NMDAR currents (53, 65). The phosphorylation of Tyr1325 of NR2A was markedly less in BVR−/− hippocampal synaptosomes (Fig. 5, D and E).
BVR physically interacts with FAK/Pyk2 and Src in a reductase-independent manner
With both acute and baseline focal adhesion signaling dependent on BVR, we considered whether BVR may mediate focal adhesion signaling directly. Because FAK/Pyk2 was unable to activate Src without BVR (Fig. 4, K to T), we wondered whether BVR may bridge FAK/Pyk2 to Src. If so, BVR might physically associate with FAK, Pyk2, and Src. Consistent with such a model, we find that glutathione-S-transferase (GST)-tagged BVR pulled down FAK, Pyk2, and Src when independently co-expressed in HEK293 cells (Fig. 6, A to C). GST by itself did not interact with any of the three kinases. We explored whether BVR also interacted with the NMDAR subunits NR2A or NR2B as well, but we found that it did not associate with either (Fig. 6, D and E).
Figure 6. BVR physically interacts with FAK/Pyk2 and Src in a reductase-independent manner.
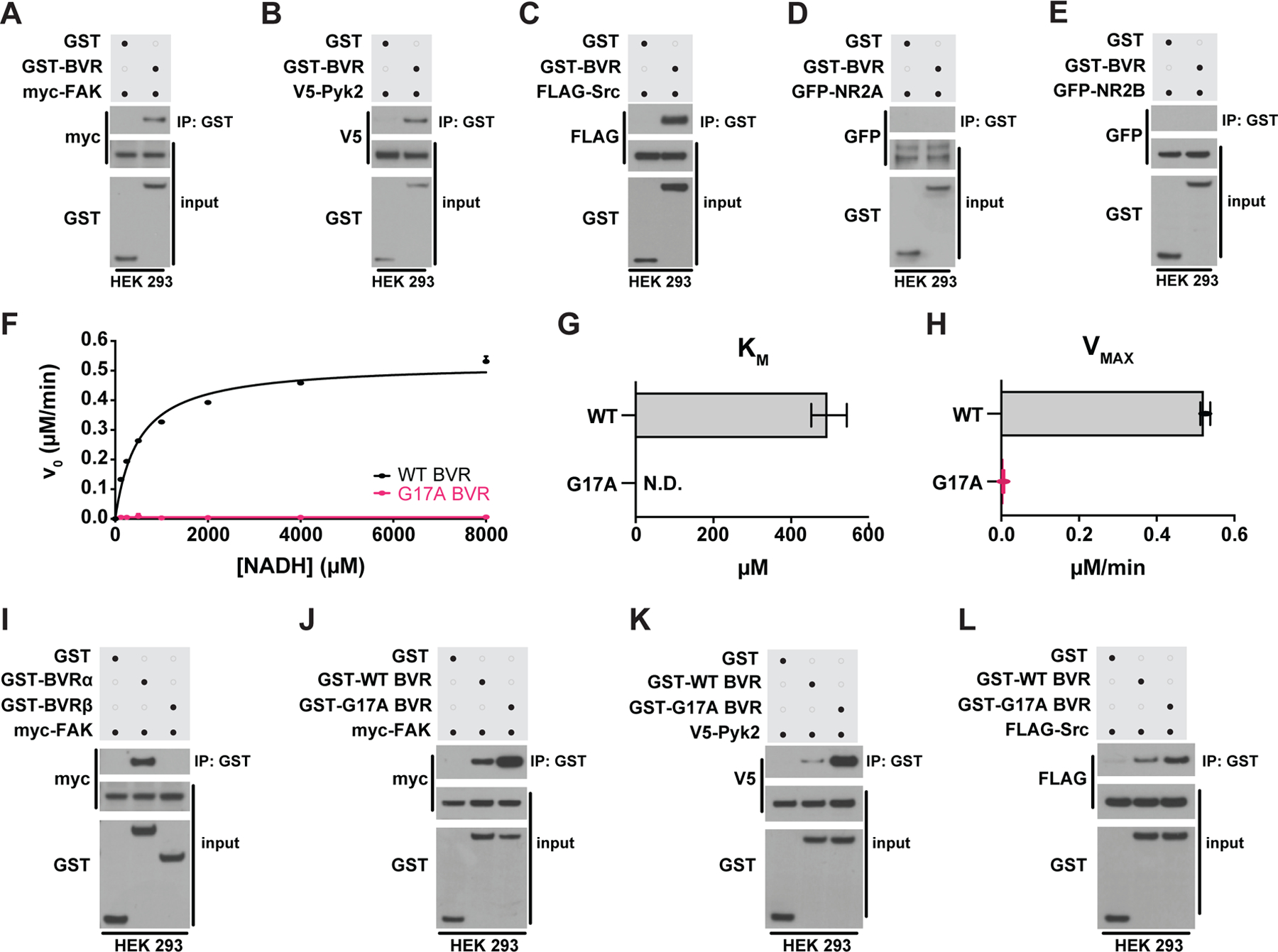
(A to E) Immunoblots of lysates (input) and GST immunoprecipitates (IP) from HEK 293 cells overexpressing GST or GST-BVR and either (A) myc-FAK, (B) V5-Pyk2, (C) FLAG-Src, (D) GFP-NR2A, or (E) GFP-NRF2B. (F to H) Michaelis-Mention kinetics of bilirubin production by WT and G17A mutant BVR with varying concentrations of NADH (F), with each enzyme’s calculated KM (G) and VMAX (H). (I) Immunoblots of lysates (input) and GST IP from HEK 293 cells overexpressing myc-FAK and either GST, GST-BVRα, or GST-BVRβ. (J to L) Immunoblots of lysates (input) and GST IP from HEK 293 cells overexpressing GST, GST-WT BVR, or GST-G17A BVR and either (J) myc-FAK, (K) V5-Pyk2, or (L) FLAG-Src. (Data are either representative of or quantified (mean ± SEM) from n = 3 independent experiments.
To judge the specificity of the interaction between BVR and FAK, we tested whether a related but structurally dissimilar enzyme also associates with FAK, Pyk2, or Src. Like BVR, the enzyme biliverdin reductase-β (BVRβ) also reduces biliverdin to bilirubin. However, classical BVR (also known as BVRα) and BVRβ reduce biliverdin to entirely different bilirubin isomers and share little-to-no sequence similarity (66). BVRβ did not interact with any of the three kinases (Fig. 6I and fig. S7, A and B), suggesting to us that the interactions between FAK, Pyk2, Src, and BVRα are unlikely to be nonspecific. BVR also did not interact with an endogenous variant of FAK (FAK-related non kinase or FRNK), further indicating that the interactions between BVR and FAK/Pyk2 are isoform-specific (fig. S7c).
To identify which residues within BVR contribute to its interactions with FAK/Pyk2 and Src, we dissected BVR into several domains based on the published crystal structures (67). We divided BVR by two complementary but orthogonal approaches: The first, focusing on BVR’s catalytically-essential Rossmann fold where nicotinamide binds (located near the amino-terminus) and its structurally notable 5-strand β-pleated sheet (located closer to the carboxy-terminus); and the second, dividing BVR in half with the β-pleated sheet as the transition point between amino- and carboxy-termini (depicted in fig. S8A). From these, we predict that BVR interacts with FAK/Pyk2 more so at the Rossman fold than with other residues, whereas Src associates best with BVR’s more complete amino terminus. Neither FAK/Pyk2 nor Src appear to interact well with BVR’s β-pleated sheet (fig. S8, B to D). As the Rossman fold is closer to the amino terminus than carboxy terminus, BVR may then bridge FAK/Pyk2 to Src by bringing the two closer in proximity through its amino terminus. Future studies that seek to further dissect how interactions between FAK/Pyk2 and Src with BVR can influence focal adhesion signaling might, therefore, first hone in on BVR’s amino terminus.
To isolate whether BVR’s canonical oxidoreductase activity is central to its interactions with FAK, Pyk2, or Src, we compared the relative affinity of fully functional wild-type BVR (WT BVR) for the kinases against a previously characterized catalytically-dead BVR (G17A BVR) (68). Crystallographic analyses reveal that Gly17 is key to binding either NADPH or NADH cofactors, (69, 70). Upon measuring both WT and G17A BVR enzyme kinetics, we found that mutating Gly17 to alanine prevents BVR from reducing biliverdin to bilirubin. Whereas WT BVR exhibited a KM of 497 μM for NADH and a Vmax of 0.5 μM bilirubin/min, G17A BVR exhibited no interaction with NADH (KM of 0 μM) with a near-zero Vmax of 0.005 μM/min bilirubin production (Fig. 6, F to H). Despite lacking enzymatic activity, G17A BVR still pulled down FAK, Pyk2, and Src to similar or greater degrees than did WT BVR (Fig. 6, J to L), suggesting that the interactions between BVR and FAK/Pyk2 or Src are not dependent on BVR’s enzymatic activity.
Though BVR still interacts with FAK, Pyk2, and Src independent of its enzymatic activity, we considered whether circumventing the enzyme with biliverdin or bilirubin directly would rescue focal adhesion signaling. However, we found that neither metabolite rescued signaling in BVR−/− cells (fig. S9, A to D). Because BVR’s substrate and product both potently influence redox homeostasis (17, 71, 72), we further investigated the contribution of oxidative stress to focal adhesion signaling by treating WT and BVR null cells with either N-acetylcysteine or 5,5-dimethyl-1-pyrroline N-oxide (DMPO), two distinct antioxidants that scavenge different classes of redox molecules. We similarly found that neither N-acetylcysteine nor DMPO rescues focal adhesion signaling (fig. S9, E to H), suggesting that focal adhesion signaling was more greatly affected by loss of BVR itself than by biliverdin, bilirubin, or oxidative stress. We lastly measured levels of the heme-catabolizing protein HO-1, given that BVR is thought to regulate its expression (73), but we did not find any differences in HO-1 levels between WT and BVR−/− synaptosomes (fig. S10, A and B).
BVR bridges FAK to Src to mediate synaptic focal adhesion signaling
As we sought to confirm these interactions in vivo, we found that the tools available to study BVR in the brain were unfortunately inadequate or unreliable. To circumvent these limitations, we generated a mouse line in which BVR is N-terminally tagged with a triplet tandem FLAG octapeptide epitope (BlvraFLAG mice). We introduced the FLAG epitope into the mouse germline just 5’ to the Blvra gene using CRISPR/Cas9-directed cleavage and homology-dependent repair (Fig. 7A) (74). We verified successful insertion of the FLAG epitope into the germline Blvra locus by Sanger sequencing, after which we backcrossed the mice over 8 generations to eliminate the possibility of additional, off-target insertions. Western blots from naive BlvraFLAG mice exhibit a single FLAG-immunopositive band between 35 and 40 kDa, consistent with the predicted molecular weight of 3xFLAG-tagged BVR (Fig. 7B).
Figure 7. BVR bridges FAK to Src to mediate synaptic focal adhesion signaling.
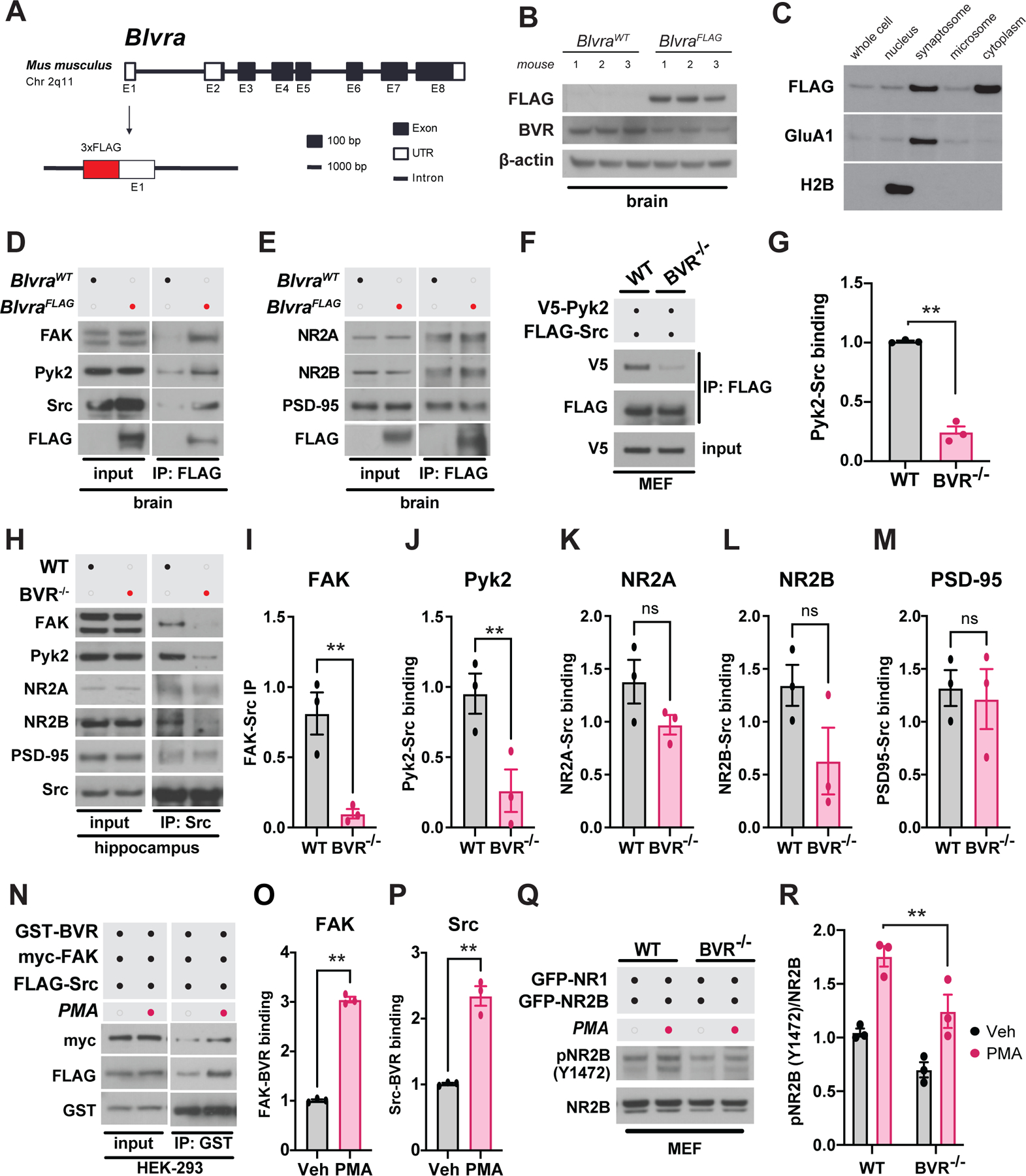
(A) Schematic depicting strategy for inserting 3xFLAG tag into the endogenous mouse Blvra locus. (B) Immunoblots of brain lysates from BlvraFLAG and 3 BlvraWT mice for FLAG and BVR. β-actin serves as loading control. (C) Immunoblot of 3xFLAG-BVR in various subcellular fractions of brain tissue from BlvraFLAG mice. H2B and GluA1 immunoblots were blotted to serve as markers of nuclear and synaptosomal fractions, respectively. (D and E) Immunoblots of (D) FAK, Pyk2, Src and FLAG-BVR and (E) NR2A, NR2B, PSD-95, and FLAG-BVR in lysates and anti-FLAG immunoprecipitates (IP) from BlvraWT and BlvraFLAG mouse brain tissue. (F and G) Immunoblots and quantification of lysates following anti-FLAG IP from WT and BVR−/− MEFs overexpressing V5-Pyk2 and FLAG-Src. Data are expressed as normalized ratio of V5-Pyk2 with anti-FLAG IP to FLAG-Src with anti-FLAG IP. V5 input serves as loading control. (H to M) Immunoblots and quantification of lysates following anti-Src IP from WT and BVR−/− hippocampi. Data are expressed as the ratios of (I) FAK, (J) Pyk2, (K) NR2A, (L) NR2B, and (m) PSD-95 that immunoprecipitated with Src, normalized to their respective expression in the input. (N to P) Immunoblots and quantification of lysates and GST IP from HEK 293 overexpressing GST-BVR, myc-FAK, and FLAG-Src treated with vehicle or 500 nM PMA for 30 min. Data are expressed as the ratios of (O) myc-FAK or (P) FLAG-Src that immunoprecipitated with GST-BVR, normalized to their respective expression in the input. (Q and R) Immunoblots and quantification of phospho- and total ectopically expressed NR2B in WT and BVR−/− MEFs treated with vehicle or 500 nM PMA for 30 min. All data are mean ± SEM depicted from independent experiments. ns P > 0.05 and ** P < 0.01 by two-tailed unpaired Student’s t-test.
With BVR successfully tagged, we first leveraged this molecular handle to isolate where BVR may reside within the brain. In fractioning hippocampi from BlvraFLAG mice into nuclear, synaptosomal, microsomal, and cytoplasmic compartments, we found that BVR is present throughout the cell. However, BVR is enriched in synaptosomes (Fig. 7C), consistent with a prominent role in synaptic signaling. We considered whether levels of BVR change with learning but found that expression of FLAG-BVR in hippocampal synaptosomes did not significantly change between naïve mice and those challenged with cue- and context- dependent learning trials (fig. S11, A and B). We then used the FLAG epitope to test whether BVR interacts with FAK, Pyk2, and Src in vivo in the brain as it does in HEK293 cells (Fig. 6, A to C). Through a series of experiments in which we immunoprecipitated 3xFLAG-BVR, we found that BVR physically interacted with all three kinases: FAK, Pyk2, and Src (Fig. 7, D and E). As in HEK293 cells, BVR did not co-immunoprecipitate with NR2A or NR2B in the brain (Fig. 7E). Lastly, we further explored whether BVR associates with NMDARs by evaluating whether BVR co-immunoprecipitated with PSD-95, a key scaffolding protein localized to the postsynaptic density of excitatory synapses alongside NMDARs. We do not find that PSD-95 associates with BVR (Fig. 7E).
Because BVR co-immunoprecipitated with both FAK and Src from the brain, we wondered whether BVR may ultimately mediate focal adhesion signaling by physically bridging FAK/Pyk2 to Src. If BVR connects FAK/Pyk2 to Src, FAK/Pyk2 may not be able to bind and stimulate Src without BVR. To explore whether losing BVR leaves FAK/Pyk2 unable to bind Src, we expressed tagged Pyk2 with Src in WT and BVR−/− MEFs and monitored co-immunoprecipitation of Src with Pyk2 in BVR−/− MEFs. Less Src co-immunoprecipitated with Pyk2 from BVR−/− MEFs than from WT MEFs, suggesting that BVR may indeed bridge FAK/Pyk2 to Src (Fig. 7, F and G). Likewise, in a series of experiments in which we immunoprecipitated Src, we found that less FAK/Pyk2 co-immunoprecipitated with Src from BVR−/− hippocampi than from WT hippocampi (Fig. 7, H to J). As a result, Src may interact less with NR2B in BVR−/− hippocampi than in WT hippocampi, though we only observed this in 2 of 3 mice and the difference fell short of statistical significance (Fig. 7L). We did not find that PSD-95 associated any differently with Src in the absence of BVR (Fig. 7M).
With FAK/Pyk2 unable to bind or activate Src without BVR, we speculated that BVR may help promote focal adhesion signaling by enhancing its interactions with FAK/Pyk2 and Src when the signaling axis is activated. We find that stimulating signaling with PMA does enhance co-immunoprecipitation of FAK/Pyk2 and Src with BVR (Fig. 7, N to P). We also probed the signaling axis one additional step downstream by reconstituting the NMDAR in MEFs by co-expressing NR1 and NR2B. The abundance of phosphorylated NR2B in response to PMA was greater in WT cells than in BVR−/− cells (Fig. 7, Q to R). Additionally, FAK, Pyk2 and Src status in the liver was unchanged between WT and BVR−/− mice (fig. S12, A to D). The results are altogether consistent with a model in which BVR links FAK/Pyk2 and Src during focal adhesion signaling in the brain to modulate NMDAR function.
Discussion
Synaptic plasticity in the hippocampus is widely considered to be the neurochemical foundation of learning and memory. The NMDA receptor (NMDAR) plays a central role by activating a cascade of kinases, enzymes, and second messengers. Following our recent discovery that bilirubin influences NMDAR redox signaling (17), we considered whether BVR itself may also impact NMDAR-dependent synaptic plasticity. In challenging BVR−/− mice with a battery of neurocognitive tasks, we first discover that BVR−/− mice exhibit profound deficits in learning and memory. We uncover that these deficits may be explained by a loss of focal adhesion signaling that is both transcriptionally and biochemically disrupted in BVR−/− hippocampi. Without BVR, FAK, Pyk2, Src, and NR2B are hypophosphorylated and thus presumably hypoactive. We found that BVR mediated focal adhesion signaling by physically bridging FAK/Pyk2 to Src and that without BVR, FAK/Pyk2 were unable to bind and stimulate Src. Src itself is a molecular hub upon which many signaling pathways converge to stimulate NMDAR (75), positioning BVR at a prominent intersection of synaptic signaling.
Outside of focal adhesions, BVR is a remarkably pleiotropic protein. Apart from reducing biliverdin to bilirubin, BVR functions as an enzyme-linked receptor that stimulates PI3K/Akt signaling in macrophages (76), hepatocytes (77), and adipocytes (78). BVR also bridges ERK to ELK1 in MAPK signaling in HEK293 cells (79). Because we do not observe any difference in the expression or phosphorylation Akt or ERK1/2 between WT and BVR−/− hippocampi, however, these alternative functions of BVR may not contribute to its activities in the brain. We have also not observed a difference in the expression, phosphorylation, or nuclear translocation of Akt or ERK1/2 between WT and BVR−/− MEFs previously (17). BVR also modulates C5aR1 expression in monocytes and thus chemotaxis in response to C5a (29), consistent with our gene ontology analyses that complement and chemokine signaling were dysregulated after deleting BVR. In the same study, Bisht and colleagues also reported that BVR null macrophages polarize towards an M1 pro-inflammatory state, thereby discovering an arm of innate immunity that is dependent on BVR. Though focal adhesion signaling was not explicitly explored in their study, it is intriguing to consider whether BVR-dependent focal adhesion signaling contributes to innate immune function. Additional gene ontology analyses in our study also hinted at an unknown role for BVR in dopaminergic neurogenesis, a potentially new avenue of future study.
Although our findings suggest that BVR interfaces with focal adhesion signaling independent of its enzymatic activity, they do not exclude some potential activities of bilirubin or biliverdin. Importantly, superoxide has been shown to be an essential mediator of LTP and contextual memory (80, 81). Thus, while our present data suggest that disruption of BVR impairs learning and memory through dysregulation of FAK- and Src-dependent modulation of the NMDA receptor, it is also possible that loss of bilirubin-mediated superoxide scavenging (17) contributes to the learning deficits observed in BVR−/− mice. Further complicating the picture is the fact that superoxide production is necessary for PKC activation during LTP (80), suggesting potential crosstalk between redox and integrin-FAK/Pyk2 signaling during synaptic plasticity. Apart from NMDA superoxide signaling, bilirubin may influence the redox state of other molecules in the synapse. Integrins interface with redox signaling by triggering oxidation and inhibition of phosphatases that dephosphorylate and inactivate FAK (82). Additionally, loss of BVR in the cortex leads to the accumulation of oxidatively-damaged proteins with impaired autophagy (83). As a result, both BVR and FAK are tightly linked to redox signaling.
Though our data suggest a prominent influence of BVR on hippocampal focal adhesion signaling, FAK and BVR are also expressed throughout the brain and other non-neural tissues (84, 85). Given that FAK/Pyk2 and Src status was unchanged in the liver between WT and BVR−/− mice, differences in focal adhesion signaling in BVR−/− cells may be cell-type and even cell compartment-specific. Aside from its activity in the hippocampus, BVR may modulate focal adhesion signaling in other brain regions important for memory formation such as the amygdala and cerebellum. It also remains to be fully determined what fraction of focal adhesion signaling BVR contributes to, and whether BVR promotes signaling moreso by enabling initial FAK/Pyk2 phosphorylation versus facilitating physical interactions between FAK/Pyk2 and Src.
Whereas LTP and the corresponding FAK/Pyk2 activation are generally regarded as NMDA receptor-dependent processes, FAK and Pyk2 can be activated by neurotransmitter receptors other than those for glutamate, including receptors for GABA, acetylcholine, and endocannabinoids (86–88). Accordingly, BVR may regulate synaptic signaling of diverse neuromodulators and neurotransmitters through a similar FAK/Pyk2-dependent Src cascade. Bilirubin also binds and activates the nuclear receptor transcription factor peroxisome proliferator-activated receptor alpha (PPAR-α) (89, 90), and may thereby also impact synaptic plasticity by altering the composition of the lipid rafts that regulate neurotransmitter exocytosis and postsynaptic receptor clustering (91, 92). Along with the mechanisms identified in the present study, these studies may inform future work that discovers how bilirubin and BVR work together to regulate synaptic signaling.
MATERIALS AND METHODS
Resources
Reagents and resources used in this study, their sources, and any associated identifiers are listed in table S1. Details of their use in this study are in the relevant sections below.
Mice
All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee at the Johns Hopkins University School of Medicine. C57BL/6J (Stock # 000664) WT mice were purchased from Jackson Labs. BVR−/− mice were generated as previously described (17, 93). Briefly, two LoxP sequences were inserted into the mouse germline flanking exon 3 of Blvra for deletion via Cre recombinase. BVR−/− mice were backcrossed to wild-type C57BL/6J mice for fourteen or more generations before these studies. WT littermates were used as controls in behavioral assays. Age-matched wild-type C57BL/6J mice were used as controls for biochemical experiments.
3x-FLAG BVR (BlvraFLAG) mice were created as follows: BlvraFLAG mice harbor a mutant Blvra allele with a triplet tandem FLAG octapeptide epitope (DYKDHD-G-DYKDHD-I-DYKDDDDK) just 5’ of BVR, generating a single 3x-FLAG BVR peptide. This FLAG epitope was inserted into the mouse germline using CRISPR/Cas9-directed cleavage and homology-directed repair (74). A CRISPR-Cas9 crRNA (5’-GAGGCCAAGATGAGTACTGAGG-3’) targeting exon 2 of Blvra was synthesized by Sigma Aldrich. A single-stranded oligo homology template (5’-ATGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGATGAGCACGGAGGTGAGCTGCCCTCAGGGGCTGTAAGGGACACCTTTGCTG-3’) was synthesized by Sigma Aldrich. The gRNA, oligo template, and Cas9 mRNA were injected into C57BL/6 embryos. Correct insertion of the FLAG epitope was confirmed by Sanger sequencing with the following primers: 5’-AACCTCTCTCTCTCTTCACGGACACTGAC-3’ (Forward) and 5’-CAGCAAAGGTGTCCCTTACAGCCCCTGA-3’ (Reverse). The BlvraWT allele returns an expected product of 93 nucleotides, whereas the BlvraFLAG results in a product of 162 nucleotides. Putative BlvraFLAG mice were then backcrossed to wild-type C57BL/6J mice for eight or more generations before these studies.
Chemicals and preparation
Pyrintegrin, phorbol 12-myristate 13-acetate (PMA), and okadaic acid were obtained from Cayman Chemical, sodium fluoride and sodium orthovanadate were obtained from Sigma-Aldrich. All compounds were prepared as 50 μl – 1,000 μl aliquots and stored at −20°C before thawing at 4°C. Freeze/thaw cycles were avoided whenever possible. Pyrintegrin and PMA were initially dissolved in 100% DMSO and then diluted to 1% DMSO in DMEM.
Plasmids/cDNA
Plasmids encoding GST-BVR (GST-BVRα) and GST-BVRβ were generated by cloning cDNA of human BLVRA and BLVRB into pCMV-GST vector (Clontech/TaKaRa). The construct encoding myc-FAK was generated as previously described (94). Plasmids encoding V5-Pyk2 (Addgene Plasmid #127233), FLAG-Src (Addgene Plasmid #118308), GFP-NR2B (Addgene Plasmid # 45447), myc-FRNK (Addgene Plasmid # 74481), and SV40T antigen (Addgene Plasmid #21826) were obtained from Addgene.
Pharmacologic treatments
Cells were treated with pyrintegrin (5 µM final, 5 min), PMA (500 nM final, 30 min) biliverdin (10 µM final, 30 min), bilirubin (10 µM final, 30 min), N-acetylcysteine (NAC; 1 mM final, 30 min), or 5,5-dimethyl-1-pyrroline N-oxide (DMPO; 4 mM final, 30 min) by removing half the volume of culture medium and replacing with an equal volume containing 2x final concentration of drug.
Neurocognitive behavior assays
Behavioral tests were performed on 2- to 4-month-old WT and BVR−/− littermate male and female mice. Behavior was assessed concomitantly or in a blocked manner with consideration for both genotype and assay.
Spatial learning was assessed with the Morris water maze. In this assay, mice were trained over four days to locate a submerged platform in a pool circular pool (120 cm diameter) filled 45 cm deep with opaque tap water. Each day, we began by revealing the platform to the mice with unique visual cues prominently displayed on the walls of the pool. We then placed the mice at semi-random locations around the pool four times and timed how long it takes them to re-locate the platform as a measure of their spatial learning. Mice were allowed 60 s to reach the platform. After four days of training, the mice are tasked once again with locating the platform but without being shown it beforehand. Each mouse’s location, speed, and latency to find the platform were recorded and automatically scored by EthoVision (Noldus Information Technology). Metrics measured in the Morris water maze were successful attempts at finding the platform, latency to reach the platform, total distance traveled, and swim speed.
Fear conditioning was assessed with cue- and context-dependent conditioning tests. The day before training, the mice were first were placed individually in the shock boxes and allowed to acclimatize to the environment for at least 30 min. The following day, mice were placed back in the shock box for training and allowed to reacclimatize for 2 min. A 20 s tone at 80 db and 2000 Hz was delivered, followed by a 2 s 0.5 mA shock to the foot shortly afterwards. Tone–shock pairs were delivered a total of four times over training. The day after, the mice are first returned the same shock chamber. They were monitored for 5 min for freezing behavior. Mice which associate the chamber with being shocked will freeze, whereas those that do not are more likely to move about. We then moved the mice to a new chamber and replay the same 20 s tone from the day before. Mice which remember that the sound precedes a shock will freeze in fear, whereas those which do not are again likely to continue moving about. Freezing was automatically recorded and scored using Cleversys Freezescan (Cleversys).
Hyperactivity was assessed with the open field test. Baseline activity in the open field was evaluated using activity chambers equipped with infrared beams (San Diego Instruments). Behavioral assays were initiated by placing mice were placed in the center of an enclosed acrylic chamber. Total locomotor activity was automatically recorded and analyzed via photobeams in the x, y, and z axes. Changes in locomotion were considered relative to WT littermate controls.
Anxiety was assessed with the elevate plus maze test. In this assay, mice were placed in a plus-shaped apparatus 1 m above the floor, built of two open and two enclosed runways pointing in orthogonal directions (San Diego Instruments). Mice were monitored for how long they spend in the open versus closed runways. Tests were initiated by placing mice in the center of the maze, which is neither in the open arms nor the closed arms. Each challenge lasted 5 min.
Despair was assessed with the forced swim test. In this assay, mice are challenged with two brief trials during which they are forced to swim in a 15 × 30 cm glass cylinder filled with room temperature water from which they cannot escape. During the second trial, mice are monitored over the course of 6 min for how long they attempt to escape before they relent and remain immobile. Immobility is defined as the absence of movement or paddling with only a hind leg to keep their heads above water.
Western Blotting
Western blotting was performed as previously described (17). Briefly, cells and tissues were homogenized at 4°C in lysis buffer (pH 7.4 solution of 50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X-100) supplemented with protease inhibitors (Sigma), 50 mM sodium fluoride, 200 µM sodium orthovanadate, and 125 nM okadaic acid (Cayman). Lysates were then pulse sonicated and centrifuged at 16,000g for 10 min at 4°C. Fifteen micrograms of cleared lysate were run on a 4–12% polyacrylamide Bis-Tris gradient gel in running buffer (pH 7.3 solution of 50 mM MES, 50 mM Tris Base, 0.1% SDS, 1 mM EDTA) and then transferred to a PVDF membrane. Membranes were blocked with 5% bovine serum albumin (BSA) (w/v) in TBS-T (pH 7.6 solution of 16 mM Tris-HCl, 140 mM NaCl, 0.1% Tween-20) for 1 hour at 25°C and then incubated with primary antibodies in 5% bovine serum albumin (BSA) (w/v) in TBS-T overnight at 4°C. The following day, membranes were washed with TBS-T, and then incubated with secondary antibodies in 5% BSA (w/v) in TBS-T for 1 hour at 25°C. Protein bands were visualized using SuperSignal West Pico Plus Chemiluminescent Substrate (ThermoFisher). Unless otherwise indicated, blots shown in figures are representative of at least three independent experiments.
The following primary antibodies were used: rabbit anti-phospho-FAK (Y397) (Cell Signaling Technology 8556, 1:1000), rabbit anti-FAK (Cell Signaling Technology 3285, 1:1000), rabbit anti-phospho-Pyk2 (Y402) (Cell Signaling Technology 3291, 1:1000), mouse anti-Pyk2 (Cell Signaling Technology 3480, 1:2000), rabbit anti-phospho-Src (Y416) (Cell Signaling Technology 6943, 1:1000), rabbit anti-Src (Cell Signaling Technology, 2109 1:2000), mouse anti-phospho-Akt (S473) (Cell Signaling Technology 4051, 1:1000), rabbit anti-Akt (Cell Signaling 9272, 1:1000), rabbit anti-phospho-ERK1/2 (T202/Y204) (Cell Signaling Technology 4370, 1:2000), rabbit anti-ERK1/2 (Cell Signaling Technology 4695, 1:2000), rabbit anti-phospho-PAK1(S199/S204)/PAK2(S192/S197) (Cell Signaling Technology 2605, 1:1000), rabbit anti-PAK1/2/3 (Cell Signaling Technology 2604, 1:1000), rabbit anti-phospho-Cofilin (S3) (Cell Signaling Technology 3313, 1:1000), rabbit anti-Cofilin (Cell Signaling Technology 5175, 1:1000), rabbit anti-phospho-NR2B (PhosphoSolutions P1516–1472, 1:1000), mouse anti-NR2B (NeuroMab 75–101, 1:1000), rabbit anti-GluA1 (Cell Signaling Technology 13185, 1:1000), mouse anti-histone H2B (Cell Signaling Technology 2934, 1:1000), rabbit anti-V5 (Cell Signaling Technology 13202, 1:1000), mouse anti-FLAG (Millipore Sigma F1804, 1:2000), mouse anti-myc (Millipore Sigma M4439, 1:4000), mouse anti-GST (Millipore Sigma 16–209, 1:10,000), mouse anti-β-actin (Santa Cruz Biotechnology sc-47778 HRP; 1:10,000), rabbit anti-BVR (Abcam ab180208, 1:1000), and rabbit anti-GFP (Cell Signaling Technology 2956, 1:1000).
The following HRP-linked secondary antibodies were used: goat anti-rabbit IgG (Cell Signaling Technology 7074; 1:5000) and horse anti-mouse IgG (Cell Signaling Technology 7076; 1:5000).
Subcellular Fractionation
Hippocampi were fractionated into nuclear, synaptosomal, microsomal, mitochondrial, and cytoplasmic compartments as previously described (95, 96). Briefly, hippocampi were first lysed in 20 volumes of homogenizing buffer (pH 7.3 solution of 0.32 M sucrose and 0.005 M Na3PO4) at 4°C supplemented with protease inhibitors (Sigma), 50 mM sodium fluoride, 200 µM sodium orthovanadate, and 125 nM okadaic acid (Cayman). Homogenates were then centrifuged at 1,000g for 10 min at 4°C, resulting in pellet P1. The supernatant was recentrifuged at 17,500g for 20 min at 4°C. The resulting pellet contains a mixture of myelin, synaptosomes, and mitochondria (pellet P2). The supernatant was centrifuged once more at 100,000g for 120 min at 4°C to pellet microsomes. The resulting supernatant was considered the cytoplasmic compartment. Pellet P2 was resuspended in homogenizing buffer and subfractioned on a discontinuous sucrose gradient of 0.4, 0.6, 0.8, 1.0, and 1.2 M by centrifugation at 61,000g for 120 min at 4°C. Fractions between 0.32–0.8 M sucrose contain myelin and synaptosomal ghosts as evidenced by electron microscopic examination of these fractions. Fractions between 0.8–1.2 M sucrose contain synaptosomes as evidenced by high [3H]naloxone and [3H]]dihydromorphine binding in previous studies (97, 98). Fractions between at 1.2 M sucrose contain mitochondria. Nuclei were purified from pellet P1 by resuspending P1 in a pH 7.4 solution of 1.5 M sucrose, 50 mM Tris-HCl, 5 mM MgCl2, and 25 mM KCl, layering it upon on a solution of 1.8 M, and centrifuging at 40,000g for 60 min at 4°C.
Immunoprecipitation
Cells or tissue were harvested on ice in lysis buffer (pH 7.6 solution of 50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, and 5% glycerol (v/v)) supplemented with protease inhibitor cocktail, passed 10 times through a 27G syringe, and centrifuged at 16,000g for 10 min at 4°C. Glutathione Sepharose 4B (GE Healthcare) or anti-FLAG M2 (Sigma) beads were added to cleared lysates and rotated overnight at 4 °C. 5% of each sample was set aside as input prior to addition of beads. The following day, beads were pelleted by centrifugation at 1,000g for 1 min at 4 °C. The supernatant was aspirated and beads were washed 5 times in lysis buffer by centrifugation, aspiration, and resuspension. Proteins were eluted from beads by boiling for 5 min in LDS sample buffer (ThermoFisher) and the eluates analyzed by Western blot as described above.
Cell Culture and Transfection
WT and BVR−/− MEFs were isolated as previously described (17). Briefly, embryonic day 14.5 (E14.5) embryos were obtained from timed BVR+/− matings. The pups were decapitated and eviscerated, after which the remaining portion was trypsinized and sheared. Isolated MEFs were plated in 2 wells of a 6-well plate and cultured overnight in DMEM (Gibco) supplemented with 10% FBS, 100 U/mL penicillin and streptomycin, and 2 mM glutamine at 37°C with 5% CO2. MEFs were then expanded to 6-well plates and transiently transfected with SV40T antigen (Addgene) and maintained until stably proliferative. DNA was isolated from the heads of the pups and genotyped to confirm the genotype of the corresponding MEF cell line.
HEK293 cells were obtained from the American Type Culture Collection. HEK293 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM), 10% fetal bovine serum, penicillin/streptomycin (100 U/ml), and glutamine (2 mM) in an atmosphere of 5% CO2 at 37°C.
HEK293 cells and MEFs were transiently transfected with PolyFect (Qiagen) and Lipofectamine 3000 (Thermo Fisher) transfection reagents, respectively, according to manufacturer’s instructions. Cells were harvested 48–72 hours after transfection.
Single-cell RNA-sequencing
Single-cell RNA-sequencing was performed as described elsewhere (27). Briefly, hippocampi from postnatal day 0 pups and dissected and dissociated for sequencing. Single cell RNA-seq libraries were constructed using the 10X Chromium Single Cell 3′ Reagent Kits v2 according to manufacturer descriptions and subsequently sequenced on a NovaSeq 6000. Reads were aligned to the mm10 genome using the 10X CellRanger pipeline with default parameters to generate the feature-barcode matrix.
Downstream quality control and analyses on feature-barcode matricies were performed with Seurat v3. Genes that were not detected in at least 4 cells were excluded. Cells with fewer than 1,000 genes or more than 5,000 genes were also excluded from analysis. We also excluded cells in which greater than 15% of reads mapped to mitochondrial genes. The filtered matrices were log-normalized and scaled to 10,000 transcripts per cell.
Using the variance-stabilizing transformation in the FindVariableFeatures function, we identified the top 2,000 most variable genes per sample. We then harmonized gene expression across datasets prior to clustering by identifying anchors between samples in each dataset using the FindIntegrationAnchors function and then computing an integrated expression matrix from these anchors as input to the IntegrateData function. We then linearly regressed the number of UMIs per cell and percentage of mitochondrial reads using the ScaleData function and performed dimensionality reduction using PCA. For each dataset, we selected the top 30 dimensions to compute a cellular distance matrix, which was used to generate a K-nearest neighbor graph. The KNN was used as input to the Louvain Clustering algorithm implemented in the FindClusters function. We chose a resolution parameter of 0.8 for clustering via Louvain. To annotate and merge clusters, we performed differential gene expression analysis on the integrated expression values between each cluster using the default parameters in the FindMarkers function.
Bulk RNA-sequencing
We performed bulk RNA-sequencing to profile the differential transcriptional signatures of WT and BVR−/− hippocampi. Total RNA was isolated from three WT and three BVR−/− hippocampi from four-month-old male mice using TRizol and RNeasy Plus Universal Kit (Qiagen) as per the manufacturer’s instructions. DNA was digested with DNase treatment. mRNA was enriched by poly-A selection and prepped for paired-end reads using an Illumina TruSeq mRNA sample preparation kit with 100-base pair adapters. Samples were sequenced by Illumina Novaseq.
Differential gene expression analysis was performed using BioJupies (99). We leveraged BioJupies to perform Pathway Enrichment Analyses through Enrichr (100) to identify the biological processes that are over-represented in the gene sets up-regulated and down-regulated by in BVR−/− hippocampi.
DNA/RNA isolation, PCR, and quantitative-PCR (q-PCR)
Total cellular or tissue DNA/RNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) for DNA or RNeasy Plus Universal Kit (Qiagen) for RNA per the manufacturer’s instructions. PCR was performed with the Platinum Taq DNA Polymerase High Fidelity Kit (Invitrogen).
Electrophysiological recordings (long-term potentiation)
Organotypic hippocampal slice cultures were prepared from P4–8 C57BL/6 mouse brains and transfected with sparse biolistic transfections as previously described (64). Briefly, shRNA- and DNA-coated gold particles were delivered with a Helios Gene Gun (BioRad). After 2–3 days of transfection, whole-cell recordings in area CA1 were done by recording responses from fluorescent transfected neurons. EPSCs were recorded in an extracellular solution bubbled with 95% O2/ 5% CO2 consisting of (in mM) 119 NaCl, 2.5 KCl, 4 CaCl2, 4 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, 11 Glucose. Intracellular solution contained (in mM) 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.3 EGTA, 5 QX314-Cl, 4 MgATP, 0.3 Na3GTP, 0.1 spermine. A bipolar stimulation electrode (FHC) was placed in stratum radiatum, and responses were evoked at 0.2 Hz. Peak EPSCs were recorded at −70 mV. To minimize runup of baseline responses during LTP, cells were held cell-attached for 1 to 2 min before breaking into the cell. To avoid “wash-out” of LTP, the induction protocol was applied within 5 min of achieving whole-cell configuration. LTP was induced by pairing 200 stimuli at 2 Hz with a holding potential of 0 mV. The magnitude of LTP was calculated on basis of the averaged EPSC values during the last 5 min of recordings (from min 25 to min 30). LTP magnitude was expressed as the percentage change in the mean EPSC peak amplitude normalized to baseline values 100% (i.e., mean values of recording before LTP induction).
Lentivirus
Lentiviruses were generated as previously described with a few modifications (101). Briefly, cDNA encoding BVR and GFP separated by a self-cleaving 2A peptide was cloned into pLenti6 and transfected HEK293 cells with psPAX2 and pMD2.G. Virus was harvested from the media and collected by centrifugation at 100,000g for 2 h. BVR−/− MEFs were infected with lentivirus in the presence of 8 μg/ml Polybrene overnight. After 48 h, cells were sorted by GFP fluorescence using fluorescence-activated cell sorting (FACS).
Histology
Histology and quantification were performed as previously described (17). Briefly, WT and BVR−/− mice were anesthetized with 50 mg/kg pentobarbital and transcardially-perfused with 20 mL cold 0.1 M heparin/PBS (pH 7.4), followed by 25 mL of cold fixative (4% formaldehyde (v/v)). Tissues were post-fixed in 4% paraformaldehyde overnight at 4°C and then cryoprotected through a series of 10%, 20%, and 30% sucrose (w/v) gradients for 48 hours at 4°C. Tissues were then embedded in Optimal Cutting Temperature compound (OCT) and sectioned in 30 μm intervals with a cryostat, after which the sections were dried on slides for at least 1 hour at 25°C. Dry sections were then stained with Nissl stain.
Hippocampal Measurements
Hippocampal area, granule cell layer area, and dentate gyrus lengths of WT and BVR−/− mice were measured from Nissl stain brain sections as previously described (102).
Gross Anatomical Measurements
For neuroanatomical neasurements, distances between landmarks were measured using calipers. Brain length was measured rostrocaudally from the anterior-most frontal lobe to posterior-most occipital lobe, brain width measured from widest points of the temporal lobes, and brain height measured from the widest points along the dorsal-ventral axis.
For neuroskeletal measurements, distances between landmarks were measured using calipers. Skull length was measured rostrocaudally from the anterior-most nasal bone to the occiput, skull width measured from widest points of the zygomatic processes, skull height measured from the dorsal-most portion of the frontal bone to the ventral-most portion of the mandible, and intercanthal distance measured from the medial canthi bilaterally.
WT and G17A BVR protein purification
WT and G17A were cloned into in the pGEX-6P2–6xHIS vector and then transformed into BL21(DE3) cells. Starter cultures were grown to saturation overnight in Luria Broth (LB) at 37°C and then diluted 10-fold in LB and grown to an OD600 of 0.3 at 37°C, after which cultures were moved to 18°C. At OD600 of 0.6, BVR expression was induced by the addition of 400 μM isopropyl β-D-1-thiogalactopyranoside for 16 hours at 18°C. Cells were harvested by centrifugation, resuspended in 15 mL resuspension buffer (30 mM Tris-HCl, 300 mM NaCl, 1 mM TCEP, 10% glycerol, 1% TritonX-100, 1 mM PMSF, 2.34 μM leupeptin, 1.45 μM pepstatin at pH 7.6) per liter of culture, and flash-frozen in liquid nitrogen. Pellets were thawed on ice and sonicated to lyse. Lysate was clarified by centrifugation at 26,000g at 4°C for 30 min and subsequently loaded onto a glutathione column. The columns were first washed in resuspension buffer and then equilibrated into protease buffer (50 mM Tris, 150 mM NaCl, 0.5 mM TCEP, 10% glycerol, 0.01% TritonX-100 at pH 7.4). The proteins were then cleaved off the columns by the addition of Prescission Protease (GE Healthcare) for 16 h at 4°C. WT and G17A BVR were further purified via gel-filtration as previously described, with minior modifications (103). Briefly, proteins were run over a S-200 gel-filtration column in 50 mM HEPES, 300 mM NaCl, 0.5 mM TCEP, 10% glycerol at pH 7.4, concentrated, and flash frozen in gel filtration buffer at 30% glycerol for storage at −80°C.
BVR Michaelis-Menten kinetics
BVR activity was assayed in 50 mM Tris at pH 6.5 at 37°C with 100 nM purified recombinant BVR, with 10 µM biliverdin and varying concentrations of NADH. Reaction rates were determined by monitoring the change in absorbance at 442 nm over time.
Quantification and statistical analysis
All data were plotted and expressed as the median and range, mean ± SEM, or mean ± 95% CI, as noted. Statistical comparisons were performed using two-tailed unpaired Student’s t-tests, Fisher’s exact tests, or ANOVA analyses, as noted. Differences were considered significant at P < 0.05. All in vivo experiments were performed concomitantly or in a blocked manner (meaning WT and KO were interspersed and rotated in the analysis) with consideration for both genotype and treatment.
Supplementary Material
Acknowledgements:
We thank Dr. Paul Jenkins and Dr. Qianwen Zhu for thoughtful feedback and discussion. Illustrations were created with BioRender.
Funding:
This work was supported by NIH NIDA grant P50 DA044123 (to S.H.S.), T32 GM136577 (to C.V.), NIH 1R21AG073684–01, (to B.D.P), the American Heart Association (AHA)-Allen Initiative in Brain Health and Cognitive Impairment (to B.D.P. and associates), a Research Scholar Award from the Neurosurgery Pain Research Institute at Johns Hopkins Medicine (to C.V.), and Funding: Conrad N. Hilton Foundation (17316, to M.D.K.). We also acknowledge the contribution of the Core Facility G-STeP “Electrophysiology” and “Ricerca Corrente” Funds (C.G.) from Fondazione Policlinico Universitario A. Gemelli IRCCS.
Footnotes
Competing interests: C.V. and R.S.D are paid consultants and stockholders of AstraZeneca. M.D.K. has received consulting fees from Biogen Idec, Janssen, OptumRx, Novartis, and TG Therapeutics. All other authors declare that they have no competing financial interests.
Data and materials availability:
RNA-sequencing of hippocampi from WT and BVR−/− mice is deposited to NIH Sequence Read Archive (SRA), accession ID: PRJNA826563 (BioSample accessions: SAMN27572251, SAMN27572252, SAMN27572253, SAMN27572254, SAMN27572255, and SAMN27572256). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Solomon Snyder (ssnyder@jhmi.edu) and Dr. Bindu Paul (bpaul8@jhmi.edu). Requests for mice will require a non-restrictive material transfer agreement from the Johns Hopkins University School of Medicine.
REFERENCES AND NOTES
- 1.Sheng M, Kim MJ, Postsynaptic Signaling and Plasticity Mechanisms. Science 298, 776–780 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Cingolani LA, Goda Y, Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9, 344–356 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Huganir RL, Nicoll RA, AMPARs and Synaptic Plasticity: The Last 25 Years. Neuron 80, 704–717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutton MA, Schuman EM, Dendritic Protein Synthesis, Synaptic Plasticity, and Memory. Cell 127, 49–58 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Mayford M, Siegelbaum SA, Kandel ER, Synapses and Memory Storage. Csh Perspect Biol 4, a005751 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lüscher C, Malenka RC, NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Csh Perspect Biol 4, a005710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kandel ER, Dudai Y, Mayford MR, The Molecular and Systems Biology of Memory. Cell 157, 163–186 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Whitlock JR, Heynen AJ, Shuler MG, Bear MF, Learning Induces Long-Term Potentiation in the Hippocampus. Science 313, 1093–1097 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R, Engineering a memory with LTD and LTP. Nature (2014), doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bliss TV, Lomo T, Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. The Journal of Physiology 232, 331–356 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malenka RC, Bear MF, LTP and LTD An Embarrassment of Riches. Neuron 44, 5–21 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Wang J-H, Kelly PT, Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 15, 443–452 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Bredt DS, Snyder SH, Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc National Acad Sci 86, 9030–9033 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lafon-Cazal M, PIETRI S, CULCASI M, BOCKAERT J, NMDA-dependent superoxide production and neurotoxicity. Nature 364, 535–537 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Schuman E, Madison D, A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506 (1991). [DOI] [PubMed] [Google Scholar]
- 16.Klann E, Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. Journal of Neurophysiology 80, 452–457 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Vasavda C, Kothari R, Malla AP, Tokhunts R, Lin A, Ji M, Ricco C, Xu R, Saavedra HG, Sbodio JI, Snowman AM, Albacarys L, Hester L, Sedlak TW, Paul BD, Snyder SH, Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem Biol 26, 1450–1460.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris RGM, Anderson E, Lynch GS, Baudry M, Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319, 774–776 (1986). [DOI] [PubMed] [Google Scholar]
- 19.Sutherland RJ, Whishaw IQ, Kolb B, A behavioural analysis of spatial localization following electrolytic, kainate- or colchicine-induced damage to the hippocampal formation in the rat. Behav Brain Res 7, 133–153 (1983). [DOI] [PubMed] [Google Scholar]
- 20.Morris RGM, Garrud P, Rawlins JNP, O’Keefe J, Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683 (1982). [DOI] [PubMed] [Google Scholar]
- 21.Maren S, Quirk GJ, Neuronal signalling of fear memory. Nat Rev Neurosci 5, 844–852 (2004). [DOI] [PubMed] [Google Scholar]
- 22.McKernan MG, Shinnick-Gallagher P, Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Rogan MT, Stäubli UV, LeDoux JE, Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390, 604–607 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Bauer EP, Schafe GE, LeDoux JE, NMDA Receptors and L-Type Voltage-Gated Calcium Channels Contribute to Long-Term Potentiation and Different Components of Fear Memory Formation in the Lateral Amygdala. J Neurosci 22, 5239–5249 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walf AA, Frye CA, The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2, 322–328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez LE, File SE, A Five Minute Experience in the Elevated Plus-Maze Alters the State of the Benzodiazepine Receptor in the Dorsal Raphe Nucleus. J Neurosci 17, 1505–1511 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dugger SA, Dhindsa RS, Sampaio GDA, Rafikian EE, Petri S, Letts VA, Teoh J, Ye J, Colombo S, Yang M, Boland MJ, Frankel WN, Goldstein DB, Neurodevelopmental deficits and cell-type-specific transcriptomic perturbations in a mouse model of HNRNPU haploinsufficiency. Biorxiv (2020), doi: 10.1101/2020.05.01.072512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. Bmc Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bisht K, Canesin G, Cheytan T, Li M, Nemeth Z, Csizmadia E, Woodruff TM, Stec DE, Bulmer AC, Otterbein LE, Wegiel B, Deletion of Biliverdin Reductase A in Myeloid Cells Promotes Chemokine Expression and Chemotaxis in Part via a Complement C5a--C5aR1 Pathway. Journal of immunology (Baltimore, Md. : 1950) 202, 2982–2990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitra SK, Hanson DA, Schlaepfer DD, Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Bio 6, 56–68 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Park YK, Goda Y, Integrins in synapse regulation. Nat Rev Neurosci 17, 745–756 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P, Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 372, 786–791 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SGN, Proteomic analysis of NMDA receptor–adhesion protein signaling complexes. Nat Neurosci 3, 661–669 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Yang YC, Ma YL, Chen SK, Wang CW, Lee EHY, Focal Adhesion Kinase Is Required, But Not Sufficient, for the Induction of Long-Term Potentiation in Dentate Gyrus Neurons In Vivo. J Neurosci 23, 4072–4080 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babayan AH, Kramár EA, Barrett RM, Jafari M, Häettig J, Chen LY, Rex CS, Lauterborn JC, Wood MA, Gall CM, Lynch G, Integrin Dynamics Produce a Delayed Stage of Long-Term Potentiation and Memory Consolidation. J Neurosci 32, 12854–12861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernard‐Trifilo JA, Kramár EA, Torp R, Lin C, Pineda EA, Lynch G, Gall CM, Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J Neurochem 93, 834–849 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Juhász G, Vass G, Bozsó Z, Budai D, Penke B, Szegedi V, Integrin activation modulates NMDA and AMPA receptor function of CA1 cells in a dose-related fashion in vivo. Brain Res 1233, 20–26 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Lin B, Arai AC, Lynch G, Gall CM, Integrins Regulate NMDA Receptor-Mediated Synaptic Currents. J Neurophysiol 89, 2874–2878 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Kramár EA, Lin B, Rex CS, Gall CM, Lynch G, Integrin-driven actin polymerization consolidates long-term potentiation. Proc National Acad Sci 103, 5579–5584 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y-Q, Lu W-Y, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF, CAKβ/Pyk2 Kinase Is a Signaling Link for Induction of Long-Term Potentiation in CA1 Hippocampus. Neuron 29, 485–496 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Hanks SK, Calalb MB, Harper MC, Patel SK, Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc National Acad Sci 89, 8487–8491 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calalb MB, Polte TR, Hanks SK, Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 15, 954–963 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL, Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biological Chem 267, 23439–42 (1992). [PubMed] [Google Scholar]
- 44.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT, Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol 14, 1680–1688 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas JW, Ellis B, Boerner RJ, Knight WB, White GC, Schaller MD, SH2- and SH3-mediated Interactions between Focal Adhesion Kinase and Src. J Biol Chem 273, 577–583 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Cooper JA, MacAuley A, Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proc National Acad Sci 85, 4232–4236 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boczek EE, Luo Q, Dehling M, Röpke M, Mader SL, Seidl A, Kaila VRI, Buchner J, Autophosphorylation activates c-Src kinase through global structural rearrangements. J Biol Chem 294, 13186–13197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J, Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376, 737–745 (1995). [DOI] [PubMed] [Google Scholar]
- 49.Schaller MD, Parsons JT, pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol 15, 2635–2645 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM, Focal Adhesion Kinase Acts Downstream of EphB Receptors to Maintain Mature Dendritic Spines by Regulating Cofilin Activity. J Neurosci 29, 8129–8142 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheung HH, Gurd JW, Tyrosine phosphorylation of the N‐methyl‐d‐aspartate receptor by exogenous and postsynaptic density‐associated Src‐family kinases. J Neurochem 78, 524–534 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T, Characterization of Fyn-mediated Tyrosine Phosphorylation Sites on GluRε2 (NR2B) Subunit of theN-Methyl-d-aspartate Receptor. J Biol Chem 276, 693–699 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Yang M, Leonard JP, Identification of mouse NMDA receptor subunit NR2A C‐terminal tyrosine sites phosphorylated by coexpression with v‐Src. J Neurochem 77, 580–588 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Yu X-M, Askalan R, Keil GJ, Salter MW, NMDA Channel Regulation by Channel-Associated Protein Tyrosine Kinase Src. Science 275, 674–678 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Yu X-M, Salter MW, Src, a molecular switch governing gain control of synaptic transmission mediated by N-methyl-d-aspartate receptors. Proc National Acad Sci 96, 7697–7704 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takasu MA, Dalva MB, Zigmond RE, Greenberg ME, Modulation of NMDA Receptor- Dependent Calcium Influx and Gene Expression Through EphB Receptors. Science 295, 491–495 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Chen S, Leonard JP, Protein Tyrosine Kinase‐Mediated Potentiation of Currents from Cloned NMDA Receptors. J Neurochem 67, 194–200 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima‐Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T, NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. Embo J 25, 2867–2877 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinai L, Duffy S, Roder JC, Src inhibition reduces NR2B surface expression and synaptic plasticity in the amygdala. Learn Memory 17, 364–371 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Zalewska T, Ziemka-Nałęcz M, Domańska-Janik K, Transient forebrain ischemia effects interaction of Src, FAK, and PYK2 with the NR2B subunit of N-methyl-d-aspartate receptor in gerbil hippocampus. Brain Res 1042, 214–223 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Li H, Jackson MF, Yang K, Trepanier C, Salter MW, Orser BA, MacDonald JF, Plasticity of synaptic glun receptors is required for the Src‐dependent induction of long‐term potentiation at CA3‐CA1 synapses. Hippocampus 21, 1053–1061 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y, Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biological Chem 257, 7847–51 (1982). [PubMed] [Google Scholar]
- 63.Xu Y, Zhu X, Hahm HS, Wei W, Hao E, Hayek A, Ding S, Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc National Acad Sci 107, 8129–8134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barone E, Tramutola A, Triani F, Calcagnini S, Domenico FD, Ripoli C, Gaetani S, Grassi C, Butterfield DA, Cassano T, Perluigi M, Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol Neurobiol 56, 2922–2943 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Zheng F, Gingrich MB, Traynelis SF, Conn PJ, Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1, 185–191 (1998). [DOI] [PubMed] [Google Scholar]
- 66.Yamaguchi T, Komuro A, Nakano Y, Tomita M, Nakajima H, Complete Amino Acid Sequence of Biliverdin-IXβ Reductase from Human Liver. Biochem Bioph Res Co 197, 1518–1523 (1993). [DOI] [PubMed] [Google Scholar]
- 67.Takao H, Hirabayashi K, Nishigaya Y, Kouriki H, Nakaniwa T, Hagiwara Y, Harada J, Sato H, Yamazaki T, Sakakibara Y, Suiko M, Asada Y, Takahashi Y, Yamamoto K, Fukuyama K, Sugishima M, Wada K, A substrate-bound structure of cyanobacterial biliverdin reductase identifies stacked substrates as critical for activity. Nat Commun 8, 14397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lerner‐Marmarosh N, Miralem T, Gibbs PEM, Maines MD, Regulation of TNF‐α‐activated PKC‐ζ signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. Faseb J 21, 3949–3962 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Kikuchi A, Park S-Y, Miyatake H, Sun D, Sato M, Yoshida T, Shiro Y, Crystal structure of rat biliverdin reductase. Nature Structural Biology 8, 221–225 (2001). [DOI] [PubMed] [Google Scholar]
- 70.Whitby FG, Phillips JD, Hill CP, McCoubrey W, Maines MD, Crystal Structure of a Biliverdin IXα Reductase Enzyme–Cofactor Complex. J Mol Biol 319, 1199–1210 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN, Bilirubin Is an Antioxidant of Possible Physiological Importance. Science 235, 1043–1046 (1987). [DOI] [PubMed] [Google Scholar]
- 72.Barañano DE, Rao M, Ferris CD, Snyder SH, Biliverdin reductase: A major physiologic cytoprotectant. Proc National Acad Sci 99, 16093–16098 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kravets A, Hu Z, Miralem T, Torno MD, Maines MD, Biliverdin Reductase, a Novel Regulator for Induction of Activating Transcription Factor-2 and Heme Oxygenase-1*. J Biol Chem 279, 19916–19923 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (New York, NY) 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salter MW, Kalia LV, Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5, 317–328 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, Chin BY, Kaczmarek E, Alam J, Bach FH, Zuckerbraun BS, Otterbein LE, Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. The Journal of biological chemistry 284, 21369–21378 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hinds Terry DJ, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Heuvel JPV, Stec DE, Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. The Journal of biological chemistry (2016), doi: 10.1074/jbc.m116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stec DE, Gordon DM, Nestor-Kalinoski AL, Donald MC, Mitchell ZL, Creeden JF, Hinds TD, Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomol 10, 387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lerner-Marmarosh N, Miralem T, Gibbs PEM, Maines MD, Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proceedings of the National Academy of Sciences of the United States of America 105, 6870–6875 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klann E, Roberson ED, Knapp LT, Sweatt JD, A Role for Superoxide in Protein Kinase C Activation and Induction of Long-term Potentiation*. J Biol Chem 273, 4516–4522 (1998). [DOI] [PubMed] [Google Scholar]
- 81.Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E, Impairment of Long-term Potentiation and Associative Memory in Mice That Overexpress Extracellular Superoxide Dismutase. J Neurosci 20, 7631–7639 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G, Reactive oxygen species as essential mediators of cell adhesion. J Cell Biology 161, 933–944 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lanzillotta C, Zuliani I, Vasavda C, Snyder SH, Paul BD, Perluigi M, Domenico FD, Barone E, BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain—Implications for Neurodegeneration. Antioxidants 9, 671 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ewing JF, Maines MD, Immunohistochemical localization of biliverdin reductase in rat brain: age related expression of protein and transcript. Brain Res 672, 29–41 (1995). [DOI] [PubMed] [Google Scholar]
- 85.Burgaya F, Menegon A, Menegoz M, Valtorta F, Girault J, Focal Adhesion Kinase in Rat Central Nervous System. Eur J Neurosci 7, 1810–1821 (1995). [DOI] [PubMed] [Google Scholar]
- 86.Dahmani S, Tesnière A, Rouelle D, Toutant M, Desmonts J-M, Mantz J, Effects of Anesthetic Agents on Focal Adhesion Kinase (pp125FAK) Tyrosine Phosphorylation in Rat Hippocampal Slices. Anesthesiology 101, 344–353 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Felsch JS, Cachero TG, Peralta EG, Activation of protein tyrosine kinase PYK2 by the m1 muscarinic acetylcholine receptor. Proc National Acad Sci 95, 5051–5056 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Derkinderen P, Toutant M, Burgaya F, Bert ML, Siciliano JC, de Franciscis V, Gelman M, Girault J-A, Regulation of a Neuronal Form of Focal Adhesion Kinase by Anandamide. Science 273, 1719–1722 (1996). [DOI] [PubMed] [Google Scholar]
- 89.Gordon DM, Neifer KL, Hamoud A-RA, Hawk CF, Nestor-Kalinoski AL, Miruzzi SA, Morran MP, Adeosun SO, Sarver JG, Erhardt PW, McCullumsmith RE, Stec DE, Hinds TD, Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator–activated receptor α. J Biol Chem 295, 9804–9822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, Hinds TD, Bilirubin Binding to PPARα Inhibits Lipid Accumulation. Plos One 11, e0153427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korade Z, Kenworthy AK, Lipid rafts, cholesterol, and the brain. Neuropharmacology 55, 1265–1273 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM, Lipid rafts in neuronal signaling and function. Trends Neurosci 25, 412–417 (2002). [DOI] [PubMed] [Google Scholar]
- 93.Meixiong J, Vasavda C, Green D, Zheng Q, Qi L, Kwatra SG, Hamilton JP, Snyder SH, Dong X, Identification of a bilirubin receptor that may mediate a component of cholestatic itch. Elife 8 (2019), doi: 10.7554/elife.44116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fu C, Xu J, Cheng W, Rojas T, Chin AC, Snowman AM, Harraz MM, Snyder SH, Neuronal migration is mediated by inositol hexakisphosphate kinase 1 via α-actinin and focal adhesion kinase. Proc National Acad Sci 114, 2036–2041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pert CB, Snowman AM, Snyder SH, Localization of opiate receptor binding in synaptic membranes of rat brain. Brain Res 70, 184–188 (1974). [DOI] [PubMed] [Google Scholar]
- 96.Whittaker V, Michaelson I, Kirkland R, The separation of synaptic vesicles from nerve-ending particles (‘synaptosomes’). Biochem J 90, 293–303 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pert CB, Snyder SH, Opiate Receptor: Demonstration in Nervous Tissue. Science (New York, NY) 179, 1011–1014 (1973). [DOI] [PubMed] [Google Scholar]
- 98.Pert CB, Snyder SH, Properties of Opiate-Receptor Binding in Rat Brain. Proc National Acad Sci 70, 2243–2247 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Torre D, Lachmann A, Ma’ayan A, BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst 7, 556–561.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jenkins PM, Vasavda C, Hostettler J, Davis JQ, Abdi K, Bennett V, E-cadherin Polarity Is Determined by a Multifunction Motif Mediating Lateral Membrane Retention through Ankyrin-G and Apical-lateral Transcytosis through Clathrin. J Biol Chem 288, 14018–14031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ahmad SAI, Anam MB, Istiaq A, Ito N, Ohta K, Tsukushi is essential for proper maintenance and terminal differentiation of mouse hippocampal neural stem cells. Dev Growth Differ 62, 108–117 (2020). [DOI] [PubMed] [Google Scholar]
- 103.Lee JH, Mosher EP, Lee Y-S, Bumpus NN, Berger JM, Control of topoisomerase II activity and chemotherapeutic inhibition by TCA cycle metabolites. Cell Chem Biol 29, 476–489.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsai RYL, Reed RR, Using a Eukaryotic GST Fusion Vector for Proteins Difficult to Express in E. coli. Biotechniques 23, 794–800 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing of hippocampi from WT and BVR−/− mice is deposited to NIH Sequence Read Archive (SRA), accession ID: PRJNA826563 (BioSample accessions: SAMN27572251, SAMN27572252, SAMN27572253, SAMN27572254, SAMN27572255, and SAMN27572256). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Solomon Snyder (ssnyder@jhmi.edu) and Dr. Bindu Paul (bpaul8@jhmi.edu). Requests for mice will require a non-restrictive material transfer agreement from the Johns Hopkins University School of Medicine.