

Abstract
Respiratory syncytial virus (RSV) is a major health problem. A better understanding of the geographical and temporal dynamics of RSV circulation will assist in tracking resistance against therapeutics currently under development. Since 2015, the field of RSV molecular epidemiology has evolved rapidly with around 20–30 published articles per year. The objective of this systematic review is to identify knowledge gaps in recent RSV genetic literature to guide global molecular epidemiology research. We included 78 studies published between 2015 and 2020 describing 12,998 RSV sequences of which 8,233 (63%) have been uploaded to GenBank. Seventeen (22%) studies were performed in low‐ and middle‐income countries (LMICs), and seven (9%) studies sequenced whole‐genomes. Although most reported polymorphisms for monoclonal antibodies in clinical development (nirsevimab, MK‐1654) have not been tested for resistance in neutralisation essays, known resistance was detected at low levels for the nirsevimab and palivizumab binding site. High resistance was found for the suptavumab binding site. We present the first literature review of an enormous amount of RSV genetic data. The need for global monitoring of RSV molecular epidemiology becomes increasingly important in evaluating the effectiveness of monoclonal antibody candidates as they reach their final stages of clinical development. We have identified the following three knowledge gaps: whole‐genome data to study global RSV evolution, data from LMICs and data from global surveillance programs.
Keywords: molecular epidemiology, monoclonal antibodies, respiratory syncytial virus, RSV, systematic review
List of abbreviations
- F protein
fusion protein
- LMICS
low‐ and middle‐income countries
- RSV
respiratory syncytial virus
- mAb
monoclonal antibody
- WG
whole genome
1. INTRODUCTION
Respiratory syncytial virus (RSV) is a global cause of morbidity and mortality in children under the age of five, predominantly in low‐ and middle‐income countries (LMICs). 1 , 2 Although RSV has been recognised as a major health problem, there are no licensed vaccines available. 3 Prevention of severe RSV illness is limited to an approved monoclonal antibody (mAb) administration of palivizumab. 4 Although effective in high‐risk populations, there is a critical need for affordable mAbs for healthy infants to reduce severe disease. More than 20 vaccine candidates and mAbs are currently in clinical development. 5 The main target for vaccine and mAb development, the fusion (F) protein, is characterised by a high genetic and antigenic stability. 6 The F protein has a pre‐ and post‐fusion conformation and contains six antigenic sites (Ø ‐V) that are key epitopes for prophylactic neutralising of mAbs. With promising F targeting mAbs on the horizon, it is likely that they will replace palivizumab in the near future. Two next‐generation candidate mAbs with prolonged half‐life are nirsevimab (targeting site Ø) and MK‐1654 (targeting site IV), which are in phase 3 and phase 2b development, respectively. 7 , 8 With both mAbs in the final stages of clinical development, global monitoring of RSV resistance development grows more important. 9 Although the binding epitopes are known to be conserved, mutations may emerge and spread. 10 The emergence of escape variants may result in prophylaxis resistance, as seen in the suptavumab trial. This phase 3 trial was discontinued due to a lack of efficacy caused by two amino acid substitutions (L172Q and S173L) in the binding epitope of all circulating RSV B strains. These escape mutants had emerged globally over three RSV seasons and were identified during F sequences analyses. 11
Previously, only Zhu et al. had reviewed viral neutralisation susceptibility for nirsevimab‐related amino acid substitutions in strains collected from 1956 to 2014. 12 , 13 In addition, Mas et al. evaluated the variability of F sequences until 2017, but this review lacks an analysis of mAb binding epitopes. 14 Despite these efforts, potential escape mutants have not been reviewed systematically and gaps in the recent knowledge remain with respect to recent polymorphisms. In addition to detecting resistance against therapeutics, understanding the molecular epidemiology of RSV is fundamental for elucidating temporal and spatial dynamics, which can help inform surveillance efforts and guide interventions in future epidemics.
As the molecular epidemiology of RSV is a rapidly evolving research field, we provide a comprehensive overview of literature to date. This systematic review aimed to identify knowledge gaps in recent RSV genomic literature required to study global RSV evolution and transmission patterns and, at the same time provide guidance for monitoring next generation mAbs before and after licensure.
2. METHODS
2.1. Search strategy and selection criteria
This review was registered at PROSPERO (International Prospective Register of Systematic Reviews) under registration number CRD42021237337. We have followed the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) reporting guidelines and the Cochrane Handbook. 15 , 16
We searched for studies of any design and in any setting that included sequencing of human RSV samples. We searched MEDLINE and Embase for studies published from 1 January 2015 until 1 December 2020. The search included the terms ‘RSV’ and ‘sequences’ (see Table S1 for full search). The search strategy was not limited by language. Case reports were excluded. Reference lists of included studies were assessed for additional relevant studies. We classified LMICs based on the World Bank list of economies from 2020. 17
We selected studies describing the RSV genotype distribution and/or the amino acid polymorphisms in the F protein as we felt that articles with these criteria describe complete and high‐quality data. Genotype distribution was defined as the number of individually reported genotypes for RSV A and/or RSV B. Amino acid polymorphisms in the F protein were reported as the amino acid change, neutralising capacity, number and percentage, country and year. Studies without genotype data or amino acid polymorphisms in the F protein were excluded. Antibody binding sites of RSV F were defined using the mAb binding epitopes Ø, II, IV and V described in the original publications: aa 62‐69, 196‐212 for nirsevimab, 12 , 13 254‐277 for palivizumab, 11 426‐447 for MK‐1654 7 and 161‐182 for suptavumab. 11
2.2. Data collection
Titles and abstracts were screened and full texts were reviewed by three authors (ACL, ERH and BK) independently, using the web app Rayyan. 18 Endnote X9 (Thomson Reuters) was used to upload full text articles. 19 Disagreements between reviewers were resolved by consensus. Data were extracted by two authors (ACL, ERH) and checked by the third author (BK). We extracted data for: first author, year of publication, study period, country, study design, study population, age, main finding, sequenced gene(s), sequencing platform, reference strains, availability of data in public domain and quantitative outcomes on subtyping, genotyping and clinic. The overall quality of the studies included was independently assessed by two authors (ACL, ERH) using the Critical Appraisal Skills Program (CASP) checklist for cohort studies.
2.3. Data analysis
We performed a descriptive analysis to describe the number of RSV positives, generated sequences, genes, genotypes, and availability of data in the public domain. Maps and Gantt charts were made in R 20 3D structures of the prefusion and postfusion RSV F protein were generated with BioRender.com using 4MMT 6 and 3RRR. 21
3. RESULTS
We identified 78 studies from 43 countries across six continents in 89 articles for our systematic review (Figures 1 and 2, Tables S2 and S3). Seventy‐seven (99%) studies were observational in nature; one (1%) interventional trial was identified. 11 , 22 No studies were excluded based on lack of quality.
FIGURE 1.
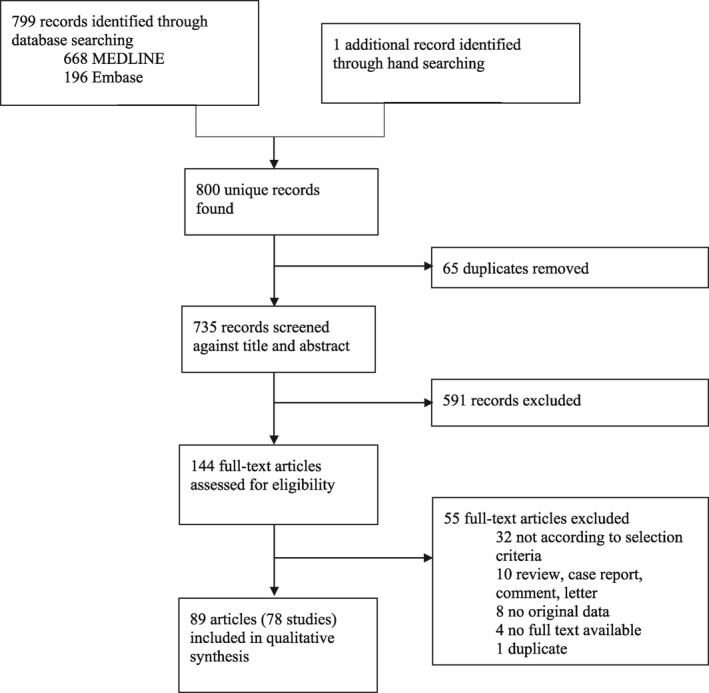
Study selection
FIGURE 2.
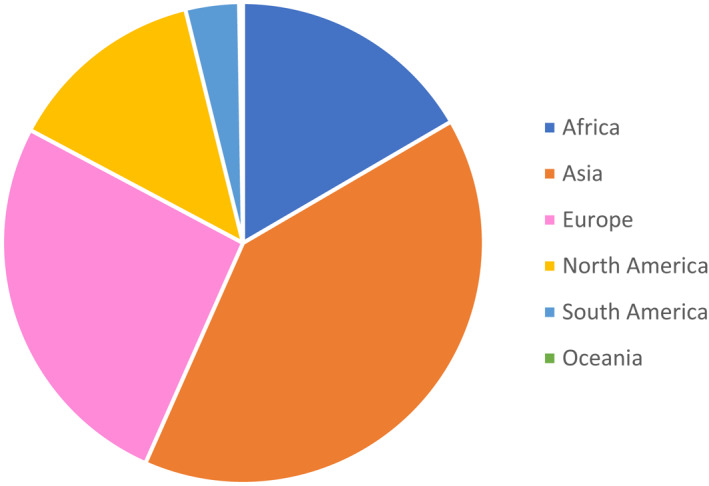
Continental distribution of sequences (n = 6 continents)
A total of 223,857 samples were identified of which 59,721 (32%) were RSV positive (Table S4). 12,994 sequences were successfully generated of which 8,233 (63%) were uploaded into GenBank. Overall, seven studies (9%) sequenced full genomes accounting for 593 whole genome (WG) sequences and 71 studies (91%) sequenced individual genes. Of these individual genes, 65 studies (92%) sequenced the attachment (G) gene, 22 (31%) the F gene, and 2 (3%) the SH gene. 75 (96%) studies involved children, mainly under the age of five years. Only a few studies (n = 3, 4%) focused on adults. The time from sample collection to publication ranged from 1 to 9 years with an average of 3 years.
3.1. International studies
From 78 studies included, 75 (96%) were performed in a single country. One study had analysed sequences from two countries (the UK, Spain). 23 Only two studies with patients from ≥5 continents were included in this systematic review. 11 , 24 Country of origin from the sequences (n = 47) from the suptavumab trial could not be analysed as this information was not provided in the manuscript. 11 Tabor et al. have published data (n = 476 RSV positives; n = 410 successful sequences) from 2017 to 2018 from Spain, the UK, the Netherlands, Finland, Japan, South Africa, Brazil and Australia. 24
3.2. LMICs
LMICs have contributed to global data with 17 studies (21.8%), including 7,084 RSV positive subjects (15%) (Table S4). In total, 266 WG sequences (4%) derived from LMICs: Philippines (n = 13), Kenya (n = 184) and Vietnam (n = 69). A large proportion of LMIC sequence data (43%) were published by Kenyan researchers who conducted a longitudinal surveillance study and a community outpatient study. 25 , 26 , 27 , 28 , 29 , 30
3.3. Natural polymorphisms in the F protein
Out of 22 studies sequencing the F gene, WG sequences included, 12 studies (55%) described the amino acid diversity in the binding epitopes of RSV A F (n = 2,474) and RSV B F (n = 3,238) for mAbs in clinical development (Figure 3). F sequences originated from Africa (Kenya, South Africa), Asia (China, Japan, Philippines, Iran, Lebanon), Europe (The Netherlands, The UK, Spain, Finland), South America (Brazil), North America (the USA), and Oceania (Australia), and were collected from 2005 to 2018.
FIGURE 3.
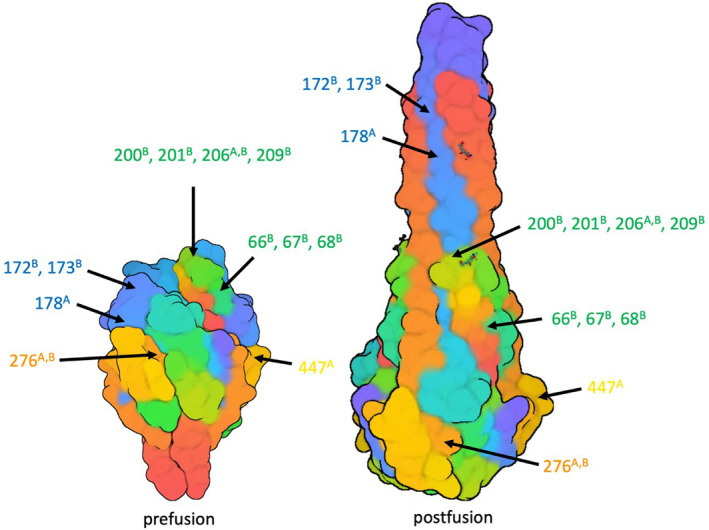
Amino acid polymorphisms detected at ≥10% frequency (Table S4) are highlighted with arrows. Previously defined mAb binding sites are delineated in colour (green = nirsevimab, orange = palivuzumab, blue = suptavumab, yellow = MK‐1654). A and B superscripts denote subtype A and B, respectively
All amino acid changes found in literature were reported per country and study period (Figure 3, Table S4). Most of the changes identified in the antibody binding sites have not been reported in literature before. Known detected resistance‐associated mutations include L172Q (967/1,401; South Africa 2015–2017, the USA, 2015–2017, global 2015–2018, Korea 2009–2015, Kenya 2015–2016) and S173L (948/1,409; South Africa 2015–2017, global 2015–2018, the USA 2015–2017, Philippines 2015–2016, Korea 2009–2015, Kenya 2015–2016) for suptavumab. The number of suptavumab escape mutants (L172Q and S173L) increased to almost 100% in 2017–2018 (Table S4). Changes associated with partial resistance for nirsevimab were E66K (27/27; Iran 2015–2016), K68N (210/787; Kenya, the USA 2016–2017), N201S (8/376; South Africa 2015/2017, The Netherlands 2017–2018, Korea 2009–2015), Q209K (183/1,007; China 2014–2016, South Africa 2015, The Netherlands 2017–2018, Korea 2009–2015, Philippines 2014–2016, the USA 2015–2017) and Q209L (88/856; Brazil 2017–2018, the USA 2015–2017). For palivizumab, S276N (377/535; South Africa 2017–2018, Korea 2009–2015, China 2014–2016, Iran 2015–2016, Lebanon 2016–2017) was found in RSV A strains. S276N for RSV B samples in the palivizumab binding site was not associated with resistance. No changes associated with resistance were detected for MK‐1654.
3.4. Geographical and temporal distribution
While sequences were obtained in all continents, most sequences were derived from China (n = 1,514; 11.6%) followed by Kenya (n = 1,360; 10.5%) and the USA (n = 1,153; 8.9%) (Figure 4). Temporal distribution is shown in Figure 5. Although the INFORM study 24 has contributed to samples in the 2017‐2018 season, data from 2015 to 2019 were lacking for multiple countries. Especially in Africa and South America recent data were rare.
FIGURE 4.
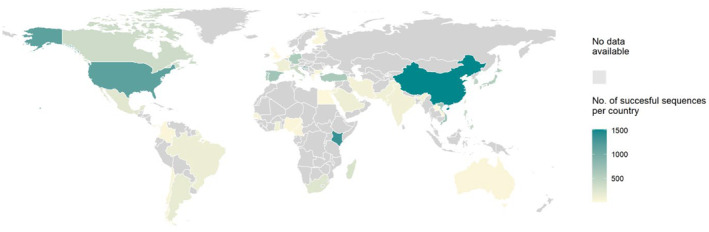
Geographic distribution of all sequences (n = 43 countries)
FIGURE 5.

Temporal distribution of recent sequences. The bars show the time period of sample collection. Countries included in this review are shown on the y‐axis. The time line from 2010–2020 is shown on the x‐axis. The continents are coloured according to the legend
3.5. Genotype distribution
Individually reported genotypes per study were summarised to give an overall impression of the genotype distribution in literature per continent from 2015–2020 (Figure 6). Ontario (ON) 1 and Buenos Aires (BA) 9 were the most reported genotypes for RSV A and B, respectively.
FIGURE 6.
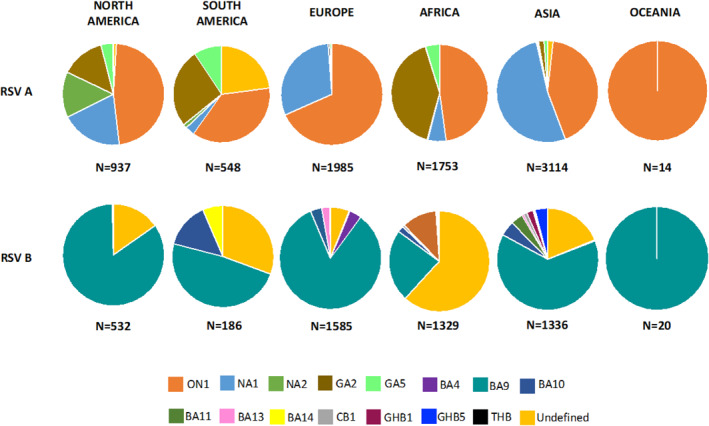
Reported RSV A and RSV B genotype distribution (2015–2020). RSV, respiratory syncytial virus
4. DISCUSSION
This systematic review aimed at identifying knowledge gaps about RSV molecular epidemiology in order to guide future research. While most of the reported amino acid changes have not been tested in neutralization assays, changes associated with partial resistance were only reported in a limited amount for nirsevimab and palivizumab. The suptavumab binding site was associated with high levels of complete resistance for RSV B strains. We have identified a time lag between data collection and publication, as it took the authors of the articles included on average 3 years to publish their sequencing data. This indicates a delay of recent sequences.
This review of available data has identified three knowledge gaps in the field of molecular epidemiology of RSV. First, only a few studies performed WG sequencing. The availability of whole‐genome sequences is important for both mAb as well as vaccine development as sequencing data increase the opportunity of the identification of novel vaccine candidates. 31 In more detail, full genomes are needed for two reasons. The first reason is the higher chance of detecting variation in WG compared to individual genes which makes WG sequences essential for reconstructing relatedness. The second reason for the added value of WG sequences is the possibility of amino acid changes outside G, and even outside F, that could have an impact on replication rate and capacity. Currently, data are lacking for genome regions other than the G gene. 32 While the F gene is the most important target for mAb development, only a few studies have sequenced it. The G gene is considered to have the highest genetic diversity. As this confers higher resolution for phylogeny‐based reconstructions compared to other genes, the G gene has been the preferred target for sequencing. This is reflected in the fact that nearly all of the included studies (65/78) focused on the G gene. Although F gene sequences were derived from five continents, non‐wild type amino acid polymorphisms in F were found in only 14 countries indicating that many countries remain without data. Mas et al. analysed amino acid variability within the F protein from sequences uploaded in GenBank. F sequences originated from 19 countries. Although a high number of changes were identified, the authors showed that a certain level of variation is natural within evolution. They suggest prospective sample collection to identify potential evolutionary changes driven by passive immunisation programs. 14
Second, most patients came from China, Kenya and the USA (n = 21,589; 45%). Interestingly, only 7,084 patients (14.8%) were recruited in LMICs, while the burden of RSV disease is disproportionally high in these countries. More than 90% of ALRIs and 99% of RSV related childhood mortality occur in these settings. Furthermore, 58% of deaths in LMICs occur in children aged younger than six months. 1 , 33 Global numbers on RSV positive children in LMICs are underestimated due to limited access to care. The number of RSV positive children in LMICs reported in this systematic review is not representative for all LMICs as most of the samples derived from a large study in Kenya. LMICs should be considered for RSV research, and included in global studies. Ideally, sampling efforts should focus on places with high burden. 1 Because we lack the data on the global burden of RSV, we used a high population number (>50,000,000) as a proxy for a high burden. LMICs without sequencing data fulfilling this criterion include Indonesia, Nigeria, Bangladesh, Ethiopia, Congo, and Thailand should be prioritised for RSV studies. Sampling was not only uneven in space, but also in time. The temporal distribution showed a lack of recent sampling efforts. In addition to the lack of LMIC data, our review identified specific geographical gaps of sampling. While sequencing data from countries with large populations (China, India, the USA) were available, data from other large populations, for example, Russia with a current population over 145,000,000, would be valuable in a global context.
Third, ongoing molecular surveillance is crucial with regard to the introduction of next generation mAbs. We should learn from past failures like the suptavumab trial. 11 Current programs include the WHO Global RSV Surveillance program 34 (Mozambique, Sierra Leone, South Africa, Argentina, Brazil, Canada, Chile, Egypt, Russia, the UK, India, Thailand, Australia, Mongolia) and the OUTSMART program (the USA). 35 We demonstrated that almost all global RSV B strains collected between 2015 and 2018 reported amino acid mutations in the suptavumab target epitope sequence. In order to learn from past failures continued monitoring should take place on a global scale.
To the best of our knowledge, this is the first systematic review evaluating the molecular epidemiology of RSV worldwide. There are limitations to this systematic review. Although we attempted to correct for overlapping data in articles that described the same study population, some overlap may remain. Another limitation was that clinical data including age, sex, comorbidity, and disease severity, were lacking in a considerable number of papers. In addition, we only included articles when authors described polymorphisms in the F protein or when genotype data were shown. A further limitation is that the genotype classification as reported by individual papers is error‐prone. Ramaekers et al. described the challenges in identifying RSV genotypes from sequencing data. Neither the hypervariable region (HVR) 2 fragment nor the G gene contain sufficient phylogenetic signal to define genotypes. Therefore, a new classification procedure has been proposed based on full genomes. 36 We recommend that future studies perform WG sequencing and apply the new genotype criteria to determine genotypes. Next to the limitation of the genotype classification, we did not correct for studies that focused on a specific RSV subtype or genotype. We only showed an overall coverage of genotypes as reported in literature. Because reporting bias may have occurred, the genotype distribution could be different on population level. An additional limitation was that this review focused on genetic data in published articles and not on the total number of RSV sequences uploaded in GenBank. A final limitation is that confirmation bias could have impacted our review, as the amino acid changes that have been reported could be an overestimation. More papers could have investigated these changes, but did not find any and therefore did not report on them.
In conclusion, with this systematic review we would like to provide guidance to the scientific community for future RSV research. Sampling effort is crucial in LMICs and other countries with high burden and/or high population to timely detect the development of resistance to RSV therapeutics. Global WG surveillance studies are required to ultimately understand the mechanisms underlying RSV global circulation patterns.
CONFLICTS OF INTEREST
Annefleur C. Langedijk, Eline R. Harding, Burak Konya, Bram Vrancken, Robert Jan Lebbink, Anouk Evers, Joukje Willemsen and Philippe Lemey do not report conflicts of interest. Louis J. Bont has not received personal fees or other personal benefits. UMCU has received funding from Abbvie, AstraZeneca, Janssen, the Bill and Melinda Gates Foundation, Nutricia (Danone) and MeMed Diagnostics. UMCU has received major cash or in kind funding as part of the public private partnership IMI‐funded RESCEU project from GSK, Novavax, Janssen, AstraZeneca, Pfizer and Sanofi. UMCU has received major funding by Julius Clinical for participating in the INFORM study sponsored by AstraZeneca. UMCU has received minor funding for participation in trials by Regeneron and Janssen from 2015 to 2017. UMCU received minor funding for consultation and invited lectures by AbbVie, AstraZeneca, Ablynx, Bavaria Nordic, MabXience, Novavax, Pfizer, Janssen. LJB is the founding chairman of the ReSViNET Foundation. Nirsevimab development is jointly funded by AstraZeneca and Sanofi Pasteur.
AUTHOR CONTRIBUTIONS
The study team conceived the project. ACL, ERH and BK were the review authors. ACL drafted the first version of the manuscript. All authors contributed and endorsed the final version of the manuscript.
Supporting information
Supporting Information S1
ACKNOWLEDGEMENTS
No sources of funding.
Langedijk AC, Harding ER, Konya B, et al. A systematic review on global RSV genetic data: identification of knowledge gaps. Rev Med Virol. 2022;32(3):e2284. 10.1002/rmv.2284
REFERENCES
- 1. Shi T, McAllister DA, O'Brien KL, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. 2017;390(10098):946‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hall CB, Weinberg GA, Iwane MK, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360(6):588‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mazur NI, Higgins D, Nunes MC, et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect Dis. 2018;18(10):e295‐e311. [DOI] [PubMed] [Google Scholar]
- 4. American Academy of Pediatrics Committee on Infectious D, American Academy of Pediatrics Bronchiolitis Guidelines C . Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014;134(2):415‐420. [DOI] [PubMed] [Google Scholar]
- 5. PATH. RSV Vaccine and mAb Snapshot . 2020. Accessed March 1, 2021 https://www.path.org/resources/rsv‐vaccine‐and‐mab‐snapshot/ [Google Scholar]
- 6. McLellan JS, Chen M, Joyce MG, et al. Structure‐based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013;342(6158):592‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang A, Chen Z, Cox KS, et al. A potent broadly neutralizing human RSV antibody targets conserved site IV of the fusion glycoprotein. Nat Commun. 2019;10(1):4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffin MP, Yuan Y, Takas T, et al. Single‐dose Nirsevimab for prevention of RSV in preterm infants. N Engl J Med. 2020;383(5):415‐425. [DOI] [PubMed] [Google Scholar]
- 9. Langedijk AC, Bont LJ. How viral sequence analysis may guide development of RSV monoclonal antibodies. Clin Infect Dis. 2020;ciaa944. doi: 10.1093/cid/ciaa944. PMID: 32640025 [DOI] [PubMed] [Google Scholar]
- 10. Bose ME, He J, Shrivastava S, et al. Sequencing and analysis of globally obtained human respiratory syncytial virus A and B genomes. PLoS One. 2015;10(3):e0120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simoes EAF, Forleo‐Neto E, Geba GP, et al. Suptavumab for the prevention of medically attended respiratory syncytial virus infection in preterm infants. Clin Infect Dis. 2020;ciaa951. doi: 10.1093/cid/ciaa951. PMID: 32897368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Q, McLellan JS, Kallewaard NL, et al. A highly potent extended half‐life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med 2017;9(388):eaaj1928. doi: 10.1126/scitranslmed.aaj1928 [DOI] [PubMed] [Google Scholar]
- 13. Zhu Q, Lu B, McTamney P, et al. Prevalence and significance of substitutions in the fusion protein of respiratory syncytial virus resulting in neutralization escape from antibody MEDI8897. J Infect Dis. 2018;218(4):572‐580. [DOI] [PubMed] [Google Scholar]
- 14. Mas V, Nair H, Campbell H, Melero JA, Williams TC. Antigenic and sequence variability of the human respiratory syncytial virus F glycoprotein compared to related viruses in a comprehensive dataset. Vaccine. 2018;36(45):6660‐6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higgins JPTTJ, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.2. 2021. Accessed March 1, 2021 www.training.cochrane.org/handbook [Google Scholar]
- 16. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. J Clin Epidemiol. 2009;62(10):1006‐1012. [DOI] [PubMed] [Google Scholar]
- 17. Lower Middle Income Countries. 2020. Accessed on March 1, 2021 https://data.worldbank.org/country/XN [Google Scholar]
- 18. Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan‐a web and mobile app for systematic reviews. Syst Rev. 2016;5(1):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Team TE . EndNote. EndNote . X9 ed. Philadelphia, PA: Clarivate; 2013. [Google Scholar]
- 20. Team RC. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2017. [Google Scholar]
- 21. McLellan JS, Yang Y, Graham BS, Kwong PD. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol. 2011;85(15):7788‐7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Avadhanula V, Chemaly RF, Shah DP, et al. Infection with novel respiratory syncytial virus genotype Ontario (ON1) in adult hematopoietic cell transplant recipients, Texas, 2011‐2013. J Infect Dis. 2015;211(4):582‐589. [DOI] [PubMed] [Google Scholar]
- 23. Lin G‐L, Golubchik T, Drysdale S, et al. Simultaneous viral whole‐genome sequencing and differential expression profiling in respiratory syncytial virus infection of infants. J Infect Dis. 2020;222(7):S666‐S71. [DOI] [PubMed] [Google Scholar]
- 24. Tabor DE, Fernandes F, Langedijk AC, et al. Global molecular epidemiology of respiratory syncytial virus from the 2017‐2018 INFORM‐RSV study. J Clin Microbiol. 2020;59(1):e01828‐20. doi: 10.1128/JCM.01828-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Otieno JR, Kamau EM, Oketch JW, et al. Whole genome analysis of local Kenyan and global sequences unravels the epidemiological and molecular evolutionary dynamics of RSV genotype ON1 strains. Virus Evol. 2018;4(2):vey027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otieno JR, Kamau EM, Agoti CN, et al. Spread and Evolution of Respiratory Syncytial Virus A Genotype ON1, Coastal Kenya, 2010‐ 2010‐2015. Emerg Infect Dis. 2017;23(2):264‐271. doi: 10.3201/eid2302.161149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Otieno JR, Agoti CN, Gitahi CW, et al. Molecular evolutionary dynamics of respiratory syncytial virus group A in recurrent epidemics in coastal Kenya. J Virol. 2016;90(10):4990‐5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamau E, Otieno JR, Murunga N, et al. Genomic epidemiology and evolutionary dynamics of respiratory syncytial virus group B in Kilifi, Kenya, 2015‐17. Virus Evol. 2020;6(2):veaa050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Agoti CN, Otieno JR, Munywoki PK, et al. Local evolutionary patterns of human respiratory syncytial virus derived from whole‐genome sequencing. J Virol 2015;89(7):3444‐3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Agoti CN, Otieno JR, Ngama M, et al. Successive respiratory syncytial virus epidemics in local populations arise from multiple variant introductions, providing insights into virus persistence. J Virol. 2015;89(22):11630‐11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bambini S, Muzzi A, Olcen P, Rappuoli R, Pizza M, Comanducci M. Distribution and genetic variability of three vaccine components in a panel of strains representative of the diversity of serogroup B meningococcus. Vaccine. 2009;27(21):2794‐2803. [DOI] [PubMed] [Google Scholar]
- 32. Schobel SA, Stucker KM, Moore ML, et al. Respiratory Syncytial Virus whole‐genome sequencing identifies convergent evolution of sequence duplication in the C‐terminus of the G gene. Sci Rep 2016;6:26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scheltema NM, Gentile A, Lucion F, et al. Global respiratory syncytial virus‐associated mortality in young children (RSV GOLD): a retrospective case series. Lancet Glob Health. 2017;5(10):e984‐e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jackson S, Peret TCT, Ziegler TT, et al. Results from the WHO external quality assessment for the respiratory syncytial virus pilot, 2016‐17. Influenza Other Respir Viruses. 2020;14(6):671‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruzin A, Pastula ST, Levin‐Sparenberg E, et al. Characterization of circulating RSV strains among subjects in the OUTSMART‐RSV surveillance program during the 2016‐17 winter viral season in the United States. PLoS One. 2018;13(7):e0200319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramaekers K, Rector A, Cuypers L, Lemey P, Keyaerts E, Van Ranst M. Towards a unified classification for human respiratory syncytial virus genotypes. Virus Evol. 2020;6(2):veaa052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1