

Abstract
Background
Metastatic epithelioid sarcoma (EPS) remains a largely unmet clinical need in children, adolescents and young adults despite the advent of EZH2 inhibitor tazemetostat.
Methods
In order to realise consistently effective drug therapies, a functional genomics approach was used to identify key signalling pathway vulnerabilities in a spectrum of EPS patient samples. EPS biopsies/surgical resections and cell lines were studied by next‐generation DNA exome and RNA deep sequencing, then EPS cell cultures were tested against a panel of chemical probes to discover signalling pathway targets with the most significant contributions to EPS tumour cell maintenance.
Results
Other biologically inspired functional interrogations of EPS cultures using gene knockdown or chemical probes demonstrated only limited to modest efficacy in vitro. However, our molecular studies uncovered distinguishing features (including retained dysfunctional SMARCB1 expression and elevated GLI3, FYN and CXCL12 expression) of distal, paediatric/young adult‐associated EPS versus proximal, adult‐associated EPS.
Conclusions
Overall results highlight the complexity of the disease and a limited chemical space for therapeutic advancement. However, subtle differences between the two EPS subtypes highlight the biological disparities between younger and older EPS patients and emphasise the need to approach the two subtypes as molecularly and clinically distinct diseases.
Keywords: distal, epithelioid sarcoma, functional genomics, proximal, SMARCB1
Metastatic Epithelioid Sarcoma has a largely unmet clinical need in pediatrics and young adults. To identify potential targets for inhibition we conducted next generation DNA exome and RNA deep sequencing and chemical screens in EPS patient samples and cell lines. We uncovered distinguishing features of the two subtypes of EPS, distal and proximal.

1. BACKGROUND
Epithelioid sarcoma (EPS) is a rare soft tissue sarcoma of children and young adults, which often presents as a seemingly benign growth, yet is very aggressive with metastatic spread occurring in up to 50% of cases with 1‐ and 5‐year survival rates of 46% and 0%, respectively. 1 , 2 EPS was first described by F.M. Enzinger in 1970 as a mesenchymal tumour with epithelioid‐like features. 3 EPS is generally segmented into two distinct clinicopathological subgroups: the more prevalent distal (or classical) EPS which occurs in younger individuals (20–40 years of age), and proximal EPS which occurs predominantly in older populations (20–65 years of age). 4 , 5 Late relapse with metastases results in a significant unmet clinical need as surgery is not always possible and effective targeted therapies have yet to be discovered. Preclinical and translational research is needed to improve outcomes for patients with EPS, yet progress in EPS‐focused therapeutic development has been slow in part due to the paucity of EPS study models. Specifically, while several immortalised cell lines 6 and patient samples exist globally, one centralised EPS biobank has not historically existed. The genomic and transcriptomic landscape are defined in only a limited number of biopsies, 7 but even those data sets are not shared publicly. Furthermore, to our knowledge, few patient‐derived xenograft (PDX) mouse models exist for testing promising therapeutics, 8 , 9 highlighting multiple opportunities for this challenging disease. Recent clinical trials have identified tazemetostat, an EZH2 inhibitor, as promising monotherapy treatment for proximal and distal EPS, with 9 of 62 epithelioid sarcoma patients (∼15%) demonstrating objective response to tazemetostat, although all responders were classified as partial responders. Median progression‐free survival and overall survival was 5.5 months and 19 months, respectively. 10 Tazemetostat is effective clinically for a portion of patients and provides an actionable therapeutic option for patients with significant unmet clinical need, yet the biological basis for variable responses remains unknown and the rationale for drug combinations is undefined.
Previous studies have shown that loss of tumour suppressor gene SMARCB1 is the most common mutation in EPS, occurring in up to 93% of EPS cases. 6 , 11 While loss of SMARCB1 is believed to play a crucial role in the pathogenesis of EPS, restoration of this protein‐coding gene does not solely stop the progression of disease. 12 Thus, other signalling pathway mutations likely contribute to the disease and pathogenesis is a result of a complex genetic landscape. 6 Targeting a single signalling pathway has conventionally proven insufficient, conceivably due to crosstalk between diverse biological pathways, 12 , 13 suggesting that therapies, which simultaneously target multiple biological signalling pathways are likely needed to improve patient outcomes. In this study, we have performed drug screening in combination with next‐generation sequencing (i.e. functional genomics) to define patient specific and disease‐wide therapeutic vulnerabilities, that is, ideally tazemetostat combination therapies. While this comprehensive approach did not yield a pan‐EPS drug combination for immediate clinical investigation, we have elucidated the molecular and functional characteristics of the two distinct EPS subtypes as a foundation for broader study.
2. METHODS AND MATERIALS
2.1. Cell lines
HS‐ES‐1, HS‐ES‐2R and HS‐ES‐2M were purchased from Riken (RCB2364, RCB2361, RCB2360, respectively, Riken, Japan) and maintained in DMEM supplemented with 10% foetal bovine serum (FBS) and 1% penicillin‐streptomycin (PS). VA‐ES‐BJ was purchased from ATCC (CRL‐2138, ATCC, Manassas, VA, USA) and also maintained in DMEM supplemented with 10% FBS and 1% PS. ESX, developed and provided by T. Tsukahara (Japan), 23 was maintained in IMDM supplemented with 10% heat‐inactivated FBS and 1% PS. YCUS‐5 was developed and provided by co‐author Hiroaki Goto (Japan) 24 and maintained in RPMI 1640 with 10% FBS and 1% PS. Epi‐544 was developed and provided by co‐author Keila E. Torres (Houston, TX, USA) and maintained in DMEM supplemented with 10% FBS and 1% PS. The BT‐12 cell line was provided by the Children's Oncology Group (COG) and maintained in Iscove's Modified Dulbecco's Medium (IMDM) plus 20% FBS, 4mM L‐Glutamine, 5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml selenous acid and 1% PS. The G‐401 cell line was purchased from ATCC (CRL‐1441, ATCC) and cultured in McCoy's 5A medium supplemented with 10% FBS and 1% PS. HEK293 cells were also purchased from ATCC (CRL‐1573, ATCC) and cultured in RPMI with 10% FBS and 1% PS. All cell lines were grown at 37°C and 5% CO2. All cell cultures were tested to ensure mycoplasma negativity and authenticated by short tandem repeat analysis (University of Arizona Genetics Core, Table S4) using the Promega PowerPlex16HS Assay (Madison, WI, USA).
2.2. Primary cell culture generation
The EPS primary cell culture CF‐00442‐2 was received through the CuReFAST tumour bank program at the Children's Cancer Therapy Development Institute (cc‐TDI.org). Tumour tissue was minced and processed using a gentleMacs tissue dissociator and associated tumour dissociation kit (130‐093‐235 and 130‐095‐929, respectively, Miltenyi Biotec, Germany) per manufacturer's instructions. The resultant culture was grown in DMEM supplemented with 20% FBS and 1% (PS). The EPS primary cell culture PCB490‐5 was generated as described previously 6 and maintained in RPMI 1640 with 10% FBS and 1% PS. All tissue samples for primary cell culture development were collected with patient consent (Advarra, protocol # cc‐TDI‐IRB‐1).
2.3. EPS tissue samples
Eps samples were gathered using the CuReFAST Biobank with informed consent obtained from patients and families. Co‐author Paul Huang and Robin L. Jones from the Royal Marsden Institute and Institute of Cancer Research supplied the tissue for CF‐01427 through CF‐01439. Co‐author Thomas G. P. Grünewald supplied the data for Eps_1 through Eps_11.
2.4. Therapeutic compound screen
The following compounds were purchased from (MedChemExpress Monmouth Junction, NJ, USA): CCT251545, MTX‐211, CUDC305, CUDC427, AMG232, ML329 and fasudil. Both 666‐15 and GSK‐J4 were purchased from R&D Systems (Minneapolis, MN, USA). Larotrectinib was purchased from Abmole (Houston, TX, USA). All other agents were purchased from Selleckchem (Houston, TX, USA). All agents were received as a dried powder and were reconstituted in DMSO (D8418, Sigma Aldrich, St. Louis, MO, USA) to a concentration of 10 mM or lower based on solubility specifications per manufacturer. Agents were plated at four concentrations (2, 0.2, 0.02 and 0.002 μM) in triplicate in a 384‐well format using the HP Tecan D300e and Perkin Elmer SciClone G3 digital dispensers. Cells were grown in T‐75 flasks until 80% confluent, trypsinised and plated at a density of 2000–2500 cells per well and incubated at 37°C with humidified 5% CO2 for 72 h. Cell viability was measured using CellTiter‐Glo 2.0® (G9243, Promega, Madison, WI, USA) per manufacturer's instructions. Luminescence was measured using a BioTek Synergy HT plate reader (BioTek, Winooski, VT). IC50 values were determined using a nonlinear best fit method.
2.5. PABPC1 RNA interference
PABPC1 was silenced using shRNA kit from Dharmacon (L‐019598‐00‐0005, Lafayette, CO, USA) according to manufacturer instructions. PCB‐00490‐5 was cultured and seeded into a 384‐well dish and incubated for 24 h. The cells were then transfected and incubated for an additional 24 h. The 384‐well plate was then dosed using the Tecan D300e with Selinexor and HCQ and incubated for 72 h. The plate was then imaged using CellTiter‐Glo 2.0 according to manufacturer instructions.
2.6. Tazemetostat pretreatment combination therapy
VA‐ES‐BJ was cultured in growth media with tazemetostat at 300 nM for 144 h. The pretreated cells were seeded into half of a 384 multi‐well plate with growth media dosed with tazemetostat and untreated cells were seeded into the other half in growth media and incubated for 24 h. Plates was dosed with (+)‐JQ1 and UNC0642 at a concentration range of 0.01‐10 μM or with caffeine and theophylline at a concentration range of 0.005–100 μM and incubated for 72 h and imaged using CellTiter‐Glo 2.0.
2.7. BRD7/9 inhibition
VZ185 degrader and its control were obtained from Boehringer Ingelheim onpME Portal (Ingelheim, Germany). BRD7/9 protein levels were monitored in cells treated with VZ185 and its non‐degrading control cis‐VZ185 at 10 and 1000 nM. There was significant degradation in BRD9 but not in BRD7 as shown in Figure S11B. Inhibition curves were done using VZ185 on cell lines VA‐ES‐BJ and PCB‐490‐5; however, the IC50s were found to be in the tens of micromolars and the degrader molecule was not further pursued.
2.8. DNA and RNA extraction and sequencing
Material for the generation of whole exome and RNA sequencing data was isolated from all EPS cell lines as well as all FFPE tissue specimens. Each cell line was grown to 80% confluency, trypsinised and snap frozen. RNA and DNA were extracted and sequenced by Beijing Genomics Institute (BGI, San Jose, CA). The quality of DNA prior to extraction was adequate for each cell line (DNA fragment ≥ 250 bp), as well as the quality of RNA (DV < 200%). HiSeq 4000 was used for paired‐end sequencing with 40 million reads for RNA and 100× coverage for tumour DNA.
2.9. Whole exome and whole transcriptome sequencing analysis
Raw FASTQ sequencing files were run through our in‐house computational pipeline. Somatic mutations, variations and indels were called using Genome Analysis Toolkit (GATK) Version 4.0 and the GRCh38 human reference genome. Gene copy number variations were quantified as a log ratio of tumour copy to normal copy using Samtools and VarScan2. RNA sequencing data was analysed for gene expression and gene fusion events. Transcriptome data were aligned to STAR‐derived human transcriptome from GRCh38 human reference genome. Normalised gene expression was quantified using STAR aligner with RSEM.
2.10. Eigengene analysis of proximal versus distal epithelioid sarcoma
The top 5000 most variable genes in terms of expression in the first n samples were determined using standard variance and then clustered into co‐expression modules using the WGCNA package in R 3.4.1. 25 , 26 After clustering, sets of co‐expressed genes were annotated for functional overrepresentation using the DAVID web service, version 6.8. 27 Functional overrepresentation was determined using the false discovery rate. 28 For visualisation, the eigengene or central tendency of each module was plotted in heatmap form and used to cluster samples. After the original clustering, additional samples were clustered using the module members determined in the initial analysis, but the modules were not recalculated based on these new members.
2.11. Supervised learning analysis of proximal versus distal epithelioid sarcoma
The supervised machine learning approach to identify differential gene features is adapted from the PTM‐Biomarker analysis platform. 29 In brief, post‐analysis whole exome and whole transcriptome sequencing data were ingested into the learning framework, which generates Boolean gene feature relationships differentiating proximal versus distal EPS. Top prioritised gene features were merged to create a joint heatmap of differential gene features linked by common biological roles as defined by interaction network and gene ontology analyses.
2.12. Western blot analysis
All cell samples were extracted in RIPA lysis buffer supplemented with complete protease inhibitor and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA; #89901 and #78441, respectively). After incubation on ice for 30 min, samples were centrifuged at 12 000 × g for 5 min (at 4˚C), and supernatant was collected. Protein was quantified using the Pierce BCA assay kit (Thermo Fisher Scientific, Waltham, MA; #23224). Fifty micrograms of protein from each sample was loaded and separated in 7.5% SDS‐PAGE gel and transferred onto a 0.2 mm PVDF membrane using wet transfer method (90V for 90 min). The PVDF membrane was incubated with primary mouse anti‐BAF47 (SMARCB1) antibody (BD Transduction Laboratories, #612110) at a dilution of 1:500 in non‐fat powdered milk and tris buffered saline with tween (TBST), then placed on a rocker overnight at 4°C. For the BRD7/9 western blot anti‐BRD7 (Bethyl Labs, #A302‐304‐M) and anti‐BRD9 (Bethyl Labs, #A303‐781A) were used and thePABCP1 western blot used anti‐PABP (Abcam, ab153930). The membrane was then incubated with peroxidase labelled anti‐mouse IgG secondary antibody (Vector Laboratories, Burlingame, CA; #BA9200) at a dilution of 1:5000 and placed on a rocker for 1 h prior to protein visualisation. Following primary antibody visualisation, the PVDF membrane was incubated with primary rabbit anti‐GAPDH antibody (Cell Signaling Technology, Danvers, MA; #14C10) for the SMARCB1 and PABPC1 western blot, which was used as a positive control, at a dilution of 1:5000 then placed on a rocker for 1‐h prior. β‐actin (Abcam, #ab8227) was used for the BRD7/9 western blot with the same dilutions. The membrane was then incubated with peroxidase labelled anti‐rabbit IgG secondary antibody (Vector Laboratories, Burlingame, CA; #PI‐1000) diluted 1:5000 for 1 h at room temperature with gentle rocking prior to visualisation. All proteins were detected using enhanced chemiluminescence (ECL) and read on an IVIS Lumina Imaging System, with an exposure time of 5–10 s.
2.13. ICC staining
A Fisher brand premium glass cover slip (Fisher Scientific #12‐548‐BP) was placed into each well of a 6‐well plate. A total of 300 000 cells of each cell line were plated with 1.5 ml of appropriate media into each well, covering the glass cover slip. After 24 h, the cells were 50%–80% confluent. Cells were washed with PBS, then fixed with ice cold 100% MEOH for 5 min. Cells were washed three times with cold PBS, then blocked with 1% BSA in PBST (PBS+0.1% Tween20) for 30 min. Cells on the coverslip were then placed face up on parafilm and 100 μl of 1:100 BD Biosciences mouse anti‐Baf47 (SMARCB1) (612110) in blocking buffer was applied to each coverslip. Parafilm, coverslip and antibody were then covered with tinfoil to shield from light and allowed to incubate for 2 h at room temperature. After incubation, antibody was removed, and coverslips were placed into 6‐well dishes and washed three times with PBS. For secondary antibody treatment, cover slips were removed from the 6‐well plates and once again placed face up on parafilm. 150 μl of 1:1000 mouse Alexa fluor 488 (Invitrogen #A32723TR, Carlsbad, CA, USA) in blocking buffer was applied to each coverslip, then covered and allowed to incubate for 1 h at room temperature. Secondary antibody was then removed, and cover slips were placed into 6‐well dishes and washed three times with PBS. One drop of Vectashield antifade mounting medium with DAPI (Vector laboratories #H‐1200) was applied to each glass slide and the cover slip was placed on it with cells facing down. Nail polish was used to fix coverslip to slide. Slides were imaged using a LSM800 confocal inverted laser scanning microscope (Zeiss, Oberkochen, Germany).
2.14. Immunohistochemistry
In Figure 1 deparaffinised sections of each tumour were stained with anti‐INI1/SMARCB1 antibody (anti‐BAF47) mouse monoclonal antibody (Sigma‐Aldrich, Burlington MA), using heat‐induced epitope retrieval. Appropriate positive and negative controls were used throughout.
FIGURE 1.
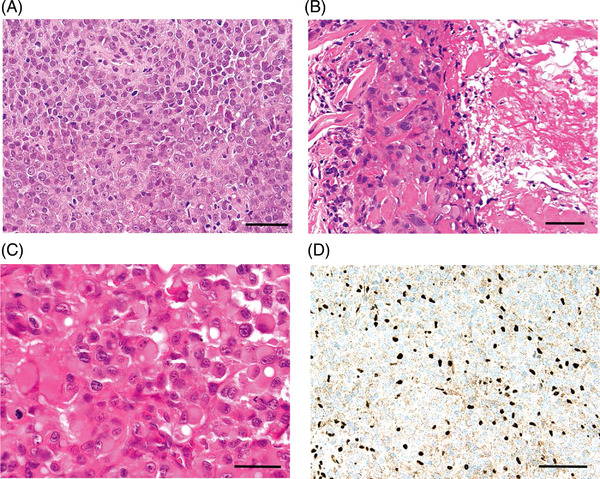
Proximal versus distal histology. (A) Conventional‐type/distal‐type epithelioid sarcoma. The tumour is composed of sheets of essentially uniform and relatively small to medium‐sized cells with rounded to ovoid vesicular nuclei, prominent nuclei and moderate amounts of eosinophilic cytoplasm. The cells have a somewhat histiocytoid‐appearance, and cellular atypia is minimal. A mild chronic inflammatory infiltrate, largely of small lymphocytes, is intermingled (haematoxylin and eosin, ×100). Scale bar, 100 μm. (B) Conventional‐type/distal‐type epithelioid sarcoma. Nests and sheets of epithelioid cells, here with abundant eosinophilic cytoplasm, are present adjacent to a large area of fibrinoid material/incipient geographic tumour necrosis (right of field) (haematoxylin and eosin, ×200). Scale bar, 50 μm. (C) Proximal‐type epithelioid sarcoma. This tumour is composed of monotonous sheets of large, epithelioid and polygonal cells with ovoid vesicular nuclei and prominent, sometimes multiple nucleoli and abundant, palely eosinophilic cytoplasm. In many cells, the combination of extensive eosinophilic cytoplasm and eccentrically oriented nuclei give the cells marked rhabdoid appearances (haematoxylin and eosin, ×400). Scale bar, 25 μm. (D) Conventional‐type/distal‐type epithelioid sarcoma. Immunohistochemistry for INI1 (SMARCB1). This protein is absent in nuclei in approximately 90% of both classic‐type and proximal‐type epithelioid sarcoma, and here, the lesional nuclei show diffuse absence of expression of INI1. This is in contrast to the surrounding smaller numbers of lymphocytes and local stromal and endothelial cells, which show strong expression of the intact protein in their nuclei (immunoperoxidase, ×200). Scale bar, 50 μm
2.15. Microsatellite instability analysis
Microsatellite instability (MSI) PCR testing was performed by LabCorp Research Services (Burlington, NC, USA) using the MSI Analysis System v1.2 (MD1641; Promega, Madison, WI, USA). Fluorescently labelled primers were used for co‐amplification of five mononucleotide repeat markers (BAT‐25, BAT‐26, NR‐21, NR‐24 and MONO‐27) to detect MSI, as well as two pentanucleotide repeat markers (Penta C and Penta D) included to confirm sample identity. The PCR products were sized with an Applied Biosystems 3500 xL Genetic Analyzer (Invitrogen) and instability was defined as heterozygosity or allele size shifts compared to the associated normal reference tissue sample. Tumour samples were categorised as MSI‐High if two or more loci demonstrated instability, MSI‐Low if one locus demonstrated instability, and stable if all loci matched between the tumour sample and its normal reference.
For tumour samples, which lacked an available normal reference sample, instability was defined as a 3 bp shift or greater from the common allele sizes of the five quasi‐monomorphic markers (NR‐21 = 98 bp, BAT‐26 = 113 bp, BAT‐25 = 120 bp, NR‐24 = 130 bp, MONO‐27 = 150 bp) as described by Bacher et al. 30 Given that instability in the form of 1 bp shifts cannot be appreciated without the direct normal comparison, samples which showed no apparent instability by this unpaired method of analysis were categorised as ‘No MSI detected’ rather than certainly stable.
Msisensor2 (available at https://github.com/niu‐lab/msisensor2), an updated version of MSIsensor, 31 was used in order to determine MSI status. The tool outputs an MSI score, which is the percentage of all valid sites – defined as sites with sequencing coverage over a user defined threshold – that were classified as MSI by a machine learning model. The developers recommend calling a sample as MSI‐H if the MSI score is above 20%. We validated the results generated by the tool by comparing them against a subset of samples where MSI was determined with support from LabCorp. Specifically, for samples where we had matched tumour‐normal pairs, MSI‐PCR was used. In cases with just tumour only samples, the pentaplex PCR panel was used and marker length compared to allele frequency tables.
2.16. Tumour mutational burden
High/modifier (HM) and low/moderate (LM) mutation VCFs were used for all samples. Non‐silent mutations were denoted as all SNPs in the HM mutation results, whereas silent were denoted as all the SNPs in the LM mutation results. Indels were denoted as all mutations that had an insertion or deletion greater than ‘2 nucleotides’ among both HM and LM mutations; however, the majority of InDels came from HM mutations. A stacked bar‐graph was then created using the silent, non‐silent and InDels in every patient using the ‘Seaborn’ python package.
2.17. Copy number variation
All biopsy and cell‐line data were subset as Distal or Proximal. An average of the exponential version of the log_ratio gain/loss data was computed across samples for the cytogenetic bands and these bands were sorted based on their position in the chromosome from 1 through 23, X and Y. A bar graph was created with the processed input using the ‘Seaborn’ python package.
2.18. Statistical analysis
All statistical analyses were performed with GraphPad Prism V9.3.1. Low, Mid and High in Tables 1 and 2 were determined using one‐sided Student's t‐test performed in Microsoft Excel, with Mid representing no significant difference between the expression in the sample and the expression in the GTExII data set, low representing statistically low expression, and high representing statistically high expression. Statistical tests used are described for each result presented.
TABLE 1.
Procured epithelioid sarcoma cell line resources
| SMARCB1 status this report | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Line/culture name | Age (years) | Sex | Location | Primary mutation; secondary mutation(s) | Year reported | Cell line/culture source | Protein status | Mutation status | CNV status | RNA status | Ref |
|
HS‐ES‐1 (CF‐00775) |
60 | M | Primary, right perineum |
SMARCB1– +vimentin, +cytokeratin, +EMA/MUC1 |
1997 | Sonobe (Riken, Japan) | Null | N/A | Hom intragenic loss (3344 bp) | Low (0×) | 32 |
|
HS‐ES‐2M (CF‐00776) |
‐ | M | Lung mets (2m) | SMARCB1– | 1993 | Sonobe (Riken, Japan) | Null | WT | Het gene loss | Low (0.14×) | 12 |
|
HS‐ES‐2R (CF‐00777) |
EPS recurrence (2r) | ||||||||||
| Primary unknown | |||||||||||
|
YCUS‐5 (CF‐00748) |
3 | F | Primary, neck | SMARCB1– | 1999 | Nagashima (Yokohama City University School of Medicine) | Null | WT | Het gene loss | Low (0.21×) | 24 |
| +cytokeratin, +EMA/MUC1, +vimentin, +ALK, +NSE, +TH | |||||||||||
|
VA‐ES‐BJ (CF‐00750) |
41 | M | Bone marrow mets | SMARCB1–/– | 1995 | Helson (St. Agnes Hospital) | Null | WT | Diploid | Low (0.53×) | 33 |
| Primary, vertebral | +AE1/AE3, +vimentin, +EMA/MUC1 | ||||||||||
| PCB‐00490‐5 | 22 | F | Primary, shoulder | SMARCB1– | 2018 | Keller (Children's Cancer Therapy Development Institute) | Null | N/A | Het gene loss, Hom intragenic loss (100 bp) | Mid (1.07×) | 8 |
| +ABL1, +NOTCH1, +MDM4, +PAK4, +MAP4K5 | |||||||||||
| CF‐00442‐2 | 26 | M |
Pulmonary mets Primary, right hand |
SMARCB1– | 2019 | Keller | Trace | Gene fusion | Het gene loss | High (2.57×) | N/A |
|
Epi‐544 (CF‐00979) |
‐ | ‐ | Primary, foot | SMARCB1– | 2011 | Lev (MD Anderson Cancer Center) | Trace | WT | Diploid | High (3.16×) | 34 |
| 0 | |||||||||||
|
ESX (CF‐02018) |
73 | F | Primary, left thigh | Hetero SMARCB1– | 2013 | Sato (Sapporo Medical University School of Medicine) | Present | Inframe deletion (c.10_12 delATG) | Diploid | High (8×) | 23 |
|
+AE1/AE3, +vimentin S100–, CD34–, CA125– | |||||||||||
| PCB495 | ‐ | M | ‐ | SMARCB1– | 2018 | Keller | ‐ | Splice acceptor variant (c.629‐1G>A) | Hom intragenic loss (425 bp) | ‐ | 8 |
| SJSTS046147_X1 | 18 | M | Primary, spine | Not reported | Unknown | St. Jude's Children's Research Hospital | ‐ | ‐ | ‐ | ‐ | N/A |
| CF‐01311 | 37 | F | Primary, vulva | Not reported | 2019 | Keller | Trace | ‐ | ‐ | ‐ | N/A |
Note: RNA status factor is ratio between tumour expression and median normal skeletal muscle expression from GTExII cohort (median value 25.5 TPM). Low represents statistically low expression, High represents statistically high expression, and Mid representing no significant difference between the expression in the sample and the expression in the GTExII data set.
Hom = homozygous, Het = heterozygous, ‐ = data not available.
TABLE 2.
Summary of epithelioid sarcoma deidentified patient data
| SMARCB1 status this report | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sample ID | Age (years) | Sex | Location | Clinical SMARCB1 status | EPS tumour type | Mutation status | CNV status | RNA status |
| CF‐00442 | 26 | M | Right arm, metastatic | SMARCB1– | Distal | Potential LOF fusion | Het gene loss | High (2.57×) |
| CF‐00463 | 29 | M | Vertebral primary, multiple locations metastatic | SMARCB1– | Proximal | WT | Het gene loss | Low (0.26×) |
| CF‐00477 | 30 | M | Left arm primary | SMARCB1– | Distal | Cancer Predisposition Mut (c.897G>A) | Diploid | High (1.49×) |
| CF‐00488 | 15 | M | Right thumb primary | SMARCB1– | Distal | Cancer Predisposition Mut (c.897G>A) | Diploid | ‐ |
| CF‐00490 | 42 | M | ‐ | ‐ | ‐ | ‐ | ‐ | Low (0.49×) |
| CF‐00982 | 15 | F | Right thumb primary | SMARCB1– | Distal | N/A | Het gene loss; Hom intragenic loss (328 bp) | Mid (1.07×) |
| CF‐00983 | 8 | M | Left hand finger primary | SMARCB1– | Distal | N/A | Het gene loss; Hom intragenic loss (200 bp) | High (1.6×) |
| CF‐01427 | 50 | M | Buttock | ‐ | Proximal | LOF Mut (c.510_515delTTCCGC) | Diploid | Low (0.62×) |
| CF‐01428 | 33 | M | Thigh | ‐ | Distal | Disruptive INDEL (c.829_830insTCC) | Diploid | High (2.46×) |
| CF‐01429 | 35 | M | Perineum | ‐ | Proximal | Disruptive Frameshift (c.222_223insCGTG) | Het gene loss; Hom intragenic loss (266 bp) | Low (0.43) |
| CF‐01430 | 18 | M | Groin | ‐ | Proximal | Missense Mut (c.673G>A) | Het gene loss; Hom intragenic loss (66 bp) | Low (0.64×) |
| CF‐01431 | 64 | M | Shoulder | Proximal | LOF Mut (c.508_509insGAGA) | Het gene loss | High (2.11×) | |
| CF‐01432 | 29 | F | Forearm | ‐ | Distal | LOF Mut (c.219A>C) | Diploid | High (1.82×) |
| CF‐01433 | 49 | F | Shoulder | ‐ | Proximal | WT | Diploid | High (2.02×) |
| CF‐01434 | 28 | M | Arm | p.Leu297_Gly302del p.Leu306_Gly311del p.Leu251_Gly256del p.Leu288_Gly293del | Distal | Disruptive Frameshift (c.445_457delACAACCATCAACA) | Het gene loss | Low (0.34×) |
| CF‐01435 | 73 | F | Left labia majorus | ‐ | Proximal | N/A | Hom gene loss; Hom intragenic loss (128 bp) | Low (0.65×) |
| CF‐01436 | 37 | M | Thigh | ‐ | Distal | WT | Diploid | High (1.93×) |
| CF‐01437 | 77 | F | Perineum | ‐ | Proximal | N/A | Hom gene loss; Hom intragenic loss (97 bp) | Low (0.33×) |
| CF‐01438 | 40 | M | Thigh | ‐ | Distal | Disruptive Frameshift (c.306_307delCA) | Diploid | Mid (1.01×) |
| CF‐01439 | 32 | F | Thigh | Distal | LOF Mut (c.219A>C) | Diploid | High (2.66×) | |
| CF‐01425 | 39 | F | Vulva | Proximal | ‐ | ‐ | ‐ | |
Note: RNA status factor is ratio between tumour expression and median normal skeletal muscle expression from GTExII cohort (median value 25.5 TPM). Low represents statistically low expression, High represents statistically high expression, and Mid representing no significant difference between the expression in the sample and the expression in the GTExII data set.
Mut = mutation, Hom = homozygous, Het = heterozygous, LOF = loss of function, ‐ = data not available.
3. RESULTS
3.1. Molecular characterisation of SMARCB1 status in EPS samples
For the distal and proximal subtypes of EPS (Figure 1A–D), we sought to collect and characterise all available biopsies/surgical resection samples and cell lines. Sources of biopsy tissues included a US‐based cohort (Keller Lab) and a Germany‐based cohort (Grünewald lab). To confirm SMARCB1 status in publicly available EPS cell lines and primary culture resources, we performed protein studies of SMARCB1 via western blot (Figure 2A, Tables 1 and 2) and compared expression against SMARCB1 levels in SMARCB1 wild type (WT) a normal cell line (HEK‐293) and SMARCB1 null rhabdoid tumour cell lines (G401, BT‐12). Protein studies confirmed SMARCB1 expression was absent or present only at trace levels in ten of eleven (10/11) EPS cell models tested. Only the ESX cell line demonstrated SMARCB1 levels comparable to the SMARCB1 WT normal cell lines. In EPS cell lines with SMARCB1 expression we investigated expression and localisation of SMARCB1 in EPS cell models via immunocytochemistry (ICC) (Figure 2C), demonstrating no nuclear SMARCB1 expression in VA‐ES‐BJ or PCB‐490‐5, but retained nuclear SMARCB1 expression in CF‐01311, ESX, Epi‐544 and CF‐00442‐2, consistent with western blotting. To further define the SMARCB1 status of EPS cell models and patient samples, we performed DNA whole exome and RNA whole transcriptome sequencing of the EPS sample cohort to identify pathogenic SMARCB1 aberrations (Tables 1 and 2, Figure 3A, Tables S1 and S2). Identified aberrations include heterozygous gene loss events (three cell models, six patient samples), homozygous gene loss (one cell model, four patient samples), homozygous focal region loss (two cell models, six patient samples), reduced RNA expression (four cell models, eight patient samples) and disruptive genomic regions (three cell models, thirteen patient samples) (Tables 2 and 3). Across the EPS cohort, 7 cell models and 11 patient samples demonstrated strong evidence of SMARCB1 loss (null protein expression or evidence of homozygous inactivation of SMARCB1), 3 cell models and 9 patient samples demonstrated weak evidence of SMARCB1 loss (trace protein expression or evidence of heterozygous inactivation of SMARCB1) and 1 cell culture and 1 patient sample lacked evidence of SMARCB1 alteration (Tables 1 and 2). Among published cell lines with nuclear SMARCB1 expression, ESX was found to have a heterozygous in frame deletion (p.Met4del), while no SMARCB1 alterations were identified in Epi‐544.
FIGURE 2.
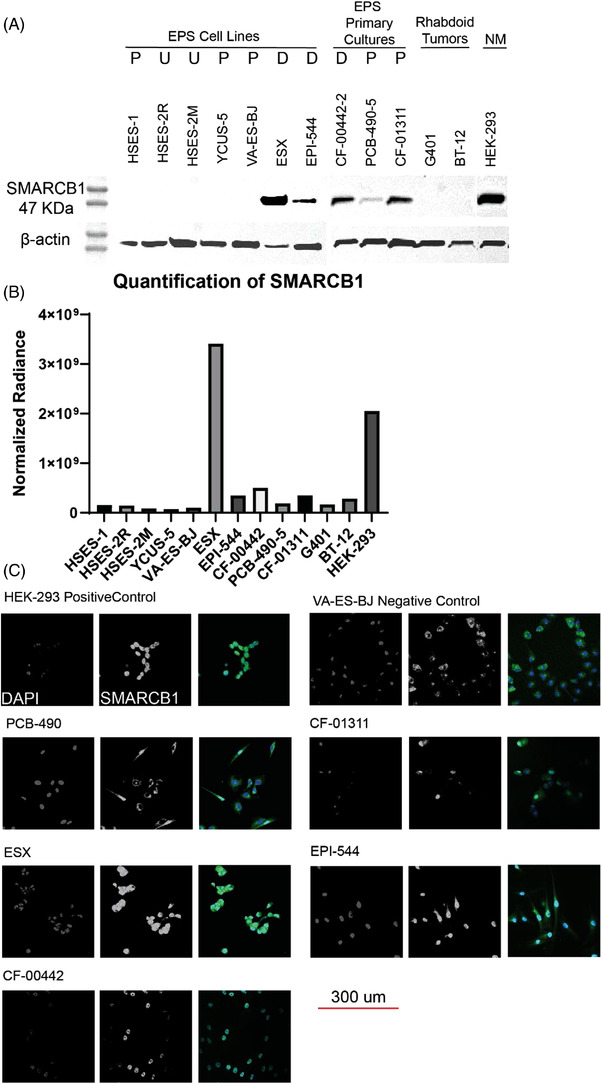
Protein analysis and immunocytochemistry of SMARCB1 in EPS cultures. (A) Immunoblot of SMARCB1. (B) The quantification of the previous panel with all samples normalised to HSES‐2M β‐actin. (C) SMARCB1 is labelled with Alexa fluor 488 (green), and the nucleus is stained with DAPI (blue). The scale bar is 300 μm and scale is the same for all of the cell lines. Nuclear staining of SMARCB1 is demonstrated in HEK‐293, ESX, CF‐00442 and EPI‐544, while absent or mostly absent in PCB‐490, VA‐ES‐BJ and CF‐01311. Alexa fluor 488‐positive staining of cellular cytoplasm is likely a result of non‐specific antibody binding given that SMARCB1 is considered to be a nuclear protein. P = proximal, D = distal, U = unknown, NM = normal. Proximal versus distal location is given as anatomical, not histological
FIGURE 3.
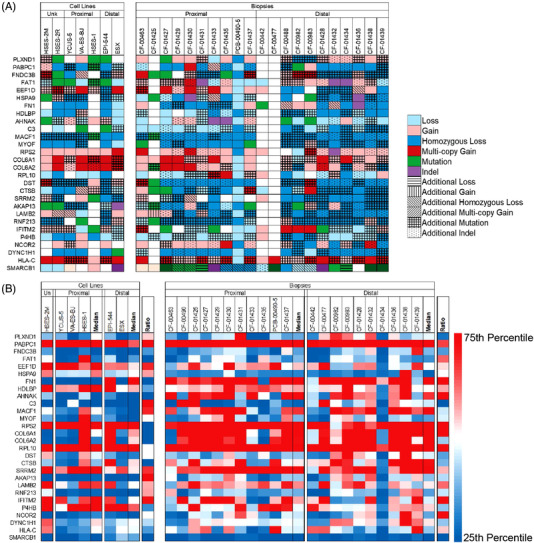
DNA and RNA expression. (A) Recurrent gene mutation, insertions/deletions or copy number changes prioritised by internal molecular analysis pipeline. (B) RNA expression of prioritised genes in panel A
TABLE 3.
Focused compound selection (version 5)
| Target | Drug name | Clinical status |
|---|---|---|
| RNA transcription initiation | Triptolide | Phase I |
| HDAC‐pan | Panobinostat* | Phase III |
| 20S Proteasome | Bortezomib | Phase III |
| mTORC1,2 | INK128/sapanisertib/tak228 | Phase I/II |
| HDAC1/2/3/10, PI3K | Cudc907 | Phase I/II |
| CDK1/2/4/6/9 | At7519 | Phase I/II |
| Proteasome | Ixazomib | Phase III |
| STAT 4 | Napabucasin | Phase III |
| HSP90 | Cudc305 | Phase I |
| AKT, ERK | Onc201 | Phase I/II |
| ALK, BMP | Ldn‐212854 | preclinical |
| ATF1/CREB | 666‐15 | preclinical |
| ATR | Azd6738 | Phase I/II |
| Beta‐blocker | Propranolol HCL | Phase I/II |
| B‐Raf | Vemurafenib | Phase I/II |
| BRD2,3,4 and BRD‐T | Gsk525762/i‐bet‐762 | Phase I/II |
| BRD4, PI3k | Ly294002 | preclinical |
| BRD4, PLK1, TAF1, TAF1L, CBP, P300 | Bi2536 | Phase I/II |
| CDC25a; iron chelation | Ciclopirox | FDA approved |
| CDK 4/6 | Ribociclib | Phase III |
| CDK8/19, Wnt | Cct251545 | preclinical |
| CENP‐E | Gsk923295 | Phase I |
| CHK1/2 | Prexasertib/LY2606368 | Phase I/II |
| c‐MET, ALK | Crizotinib | Phase III |
| CPB, CREBBP/EP300 | Sgc‐cbp30 | preclinical |
| CRM‐1/XPO‐1 | Selinexor | Phase III |
| EGFR (allosteric inhibitor) | Eai045 | preclinical |
| EGFR‐PI3K | Mtx‐211 | preclinical |
| EZH2 | Tazemetostat | Phase I/II |
| FGFR1‐4 | Ly2874455 | Phase I |
| HDAC1/2, EGFR, HER2 | Cudc101 | Phase I |
| HDAC1/3 | Entinostat | Phase III |
| IAP | Cudc427 | preclinical |
| IGF1R | Bms‐754807 | Phase I/II |
| IKBKE | Amlexanox | Phase II |
| JAK1/2 | Ruxolitinib | Phase III |
| JMJD | Gsk‐j4 | preclinical |
| JAK1/2, TBK, IKKε | Cyt387 | Phase III |
| LSD1 | Sp‐2509 | preclinical |
| MCL‐1 | S63845 | preclinical |
| Mdm2 | Amg232 | Phase I/II |
| MEK1/2 | Cobimetinib | Phase III |
| MITF | Ml329 | preclinical |
| Mitochondrial energy metabolism | Cpi‐613 | Phase III |
| mTOR | Sirolimus/rapamycin | Phase 4 |
| mTOR, PI3K | BKM120/buparlisib | Phase I/II |
| Multiple RTK, PDGFR, SRC, EphB4 | Dasatinib | Phase I/II |
| Multiple RTK, VEGFR, PDGFR, RAF | Sorafenib | Phase III |
| p300 | C646 | preclinical |
| PI3Ka/d, NFkB | Pictilisib | Phase I/II |
| PIM 1‐2‐3‐ Kinase | Azd1208 | Phase I |
| Proton pump | Esomeprazole | FDA approved |
| ROCK | Fasudil | Phase III |
| S100A9; microenvironment | Tasquinimod | Phase III |
| S100B | Pentamidine | Phase I/II |
| TAR RNA‐binding protein 2 | Enoxacin/penetrex | FDA approved |
| TNFa induction | Dmxaa | Phase III |
| TP53 (mutant) | Apr‐246 | Phase III |
| pan‐TrkA/B/C, ROS1, ALK | Entrectinib | Phase II |
| TrkA, TrkB, TrkC | Larotrectinib | FDA approved |
| Wee1 | MK‐1775/adavosertib | Phase I/II |
is used a a positive control.
3.2. Molecular status of EPS samples
Having confirmed SMARCB1 expression in EPS cell models, we expanded molecular characterisation to include DNA whole exome and RNA whole transcriptome sequencing of EPS cell lines, cell cultures and patient‐origin tumour tissues (Figure 3), identifying recurrent variations in a subset of 26 genes including SMARCB1 (Figure 3A) and corresponding gene expression of the frequently altered genes (Figure 3B). Notable genomic DNA features included PABC1 and RPS2 stochastic gains (but uniformly high RNA expression), yet no consistent secondary genomic event was observed.
3.3. Eigengene analysis of proximal versus distal EPS
We applied small‐cohort eigengene analysis to the EPS whole transcriptome sequencing data to identify functional gene modules differentially expressed between proximal and distal EPS subtypes (Figure 4). We identified potentially differentially regulated modules via unsupervised clustering of the eigengenes. DAVID analysis of the largest modules (from a total of 8) resulting from eigengene analysis were significantly enriched (FDR < 5%) for generic signalling and signal peptide ontology terms. Module 1 showed significant enrichment for Zinc finger and KRAB domain proteins, suggesting a possible role in or loss of function of epigenetic regulation. 14 Module 2 showed notable but non‐significant (FDR ∼5%) enrichments for LDL receptor associated genes (Figure S1). Module 3 showed notable but non‐significant (FDR ∼11%) enrichments for DNA repair‐related terms and hypermutation of immunoglobin genes (GO:0016446). Module 4 showed significant enrichment for MYB‐related genes and suggestive enrichment for cell cycle regulation. Module 5 also showed significant or notable enrichment for a number of specific signalling pathways, including GNRH signalling, oxytocin signalling and antigen processing and presentation. Modules 6 and 7 were both enriched for extracellular matrix and cell adhesion terms, suggesting genes in these modules could be related to tumour microenvironment and adhesion differences between subtypes.
FIGURE 4.
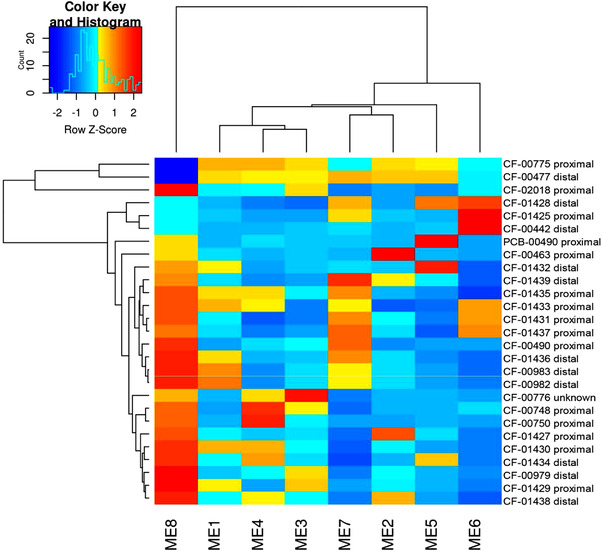
Eigengene proximal versus distal gene expression analysis. Eigengene modules showed enrichment for zinc finger and KRAB domain proteins in module 1; LDL receptors in module 2; DNA repair‐related processes, and hypermutation of immunoglobin genes in module 3; MYB‐related genes in module 4; cell cycle regulation, extracellular matrix and cell adhesion processes in module 6 and 7 and specific signalling pathways including GNRH signalling, oxytocin signalling, antigen processing, and presentation in module 5
3.4. Supervised machine learning analysis of proximal versus distal EPS
We applied a supervised machine learning approach to the whole exome and whole transcriptome sequencing data sets to identify gene features and associated biological processes differentially present in proximal versus distal EPS (Figure 5A). The genes prioritised in the heatmap define a connected network centred around SMARCB1, NFIB, CXCL12, VCAN, TGFBR2, CD34 and CD44 (Figure 5B). Ontology analysis of the prioritised genes identified ontology processes differentially present in distal versus proximal, which focus on angiogenesis‐associated processes, epithelial and epithelium migration and cell‐substrate and cell‐matrix adhesion (Figure 5C). Overall, results suggest a significant increase in angiogenesis‐associated activity and vasculature development in distal EPS (consistent with anecdotal clinical reports of response to pazopanib 15 , 16 ), as well as processes associated with cell migration and extracellular matrix activity.
FIGURE 5.
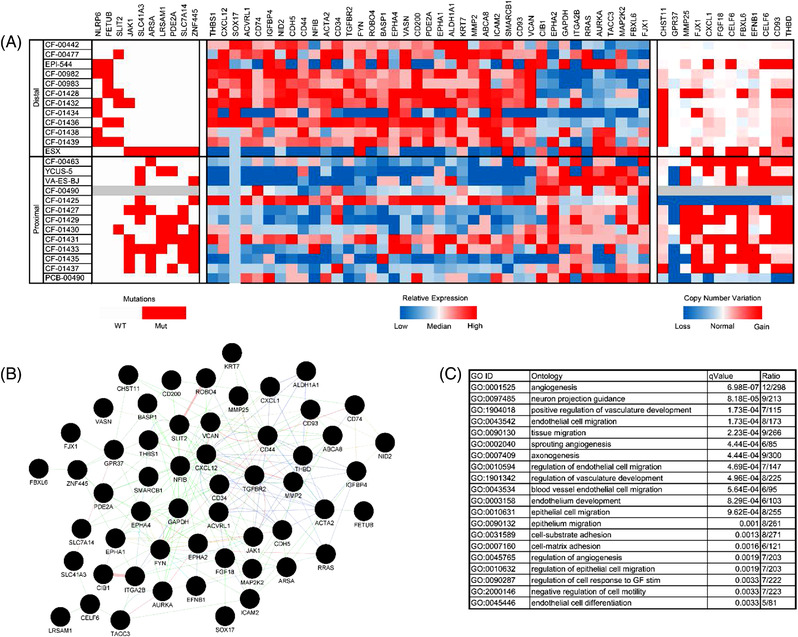
Proximal versus distal supervised learning analysis. Whole exome and whole transcriptome sequencing data from the EPS sample cohort were integrated in a supervised learning framework to identify molecular features differentiating proximal versus distal EPS subtypes with cohesive biological relevance. (A) Heatmap of mutation, copy number variation and gene expression features differentially occurring in proximal vs distal EPS samples. (B) Interaction and connection network of genes identified in differential EPS subtype analysis. (C) Gene ontology analysis of genes identified during differential analysis. The top 20 ontology classes are reported here. The full list is provided in Table S3. Note: CXCR4, the receptor for CXCL12, is the mechanistic target of plerixafor and BL‐8040
Multiple biologically relevant genes were identified through the supervised learning analysis, including several genes significantly upregulated in distal versus proximal EPS (Figure S2) and significantly upregulated in proximal versus distal EPS (Figure S3), as well as genes with elevated expression in distal EPS (Figure S4) and proximal EPS (Figure S5). Top distal overexpressed genes include NID2 (basement membrane glycoprotein with a role in extracellular matrix interactions, 7× overexpression), KRT7 (keratin family member gene with intracellular roles, 17.8× overexpression), FYN (Src family proto‐oncogene known to be inhibited by dasatinib at clinically relevant concentrations, 3.3× overexpression), SMARCB1 (3.1× overexpression), CXCL12 (CXCR4 cytokine ligand implicated in tumour growth and metastasis, 6.5× overexpression), MMP2 (extracellular matrix protein, 7.4× overexpression), BASP1 (membrane‐bound protein abundantly expressed in the brain, 2.5× overexpression) and VASN (binds and regulates TGFβ, 2.6× overexpression). Top proximal overexpressed genes include SHC1 (Src homology‐containing adapter protein that couples activated growth factor receptors to signalling pathways, 2.8× overexpression), C19orf33 (also called H2RSP, involved in single‐ and double‐stranded DNA binding, 6.5× overexpression), RIPK4 (serine‐threonine kinase involved in PKCδ and NF‐κB signalling pathways, 12.2× overexpression), EPHA2 (involved in numerous processes including cancer development and progression, 2.4× overexpression), KRT8 and KRT18 (KRT8 and KRT18 dimerise and are involved in signal transduction and cellular differentiation, 4.6× overexpression and 6.6× overexpression, respectively).
3.5. Gene expression in EPS samples
Analysis of the RNA expression data from the EPS cohort identified the highest overall median expression occurred in RNA processing genes and mitochondrial genes (Figure 6A). None of the highest‐expressing genes demonstrated significantly different expression between proximal and distal biopsy cohorts from two independent laboratories (Keller Lab and Grünewald Lab), before or after correcting for multiple comparisons.
FIGURE 6.
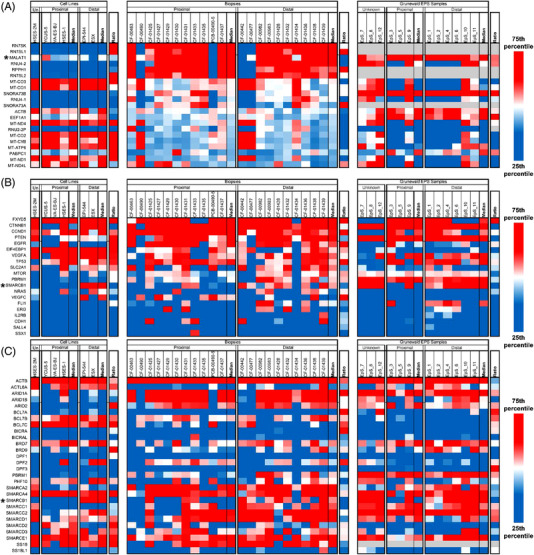
Disease‐wide expression of gene cohorts of interest. Median = median of associated samples, ratio = ratio between proximal and distal median of the associated samples. (A) Expression of genes with the highest median expression across EPS samples. The long non‐coding RNA MALAT1 has consistently high expression in biopsy‐origin samples and lower expression in cell lines. (B) Expression of previously summarised potentially actionable biomarkers. Only SMARCB1 expression significantly differs between distal and proximal samples. (C) Expression of the BAF, PBAC and ncBAF complex components. Only SMARCB1 expression significantly differs between distal and proximal samples
We also compared gene expression of twenty (20) genes previously summarised as potential EPS biomarkers (Figure 6B). 6 Three of the twenty biomarker genes were upregulated in distal EPS samples (SMARCB1, 3.1× overexpression, SALL4, 2× overexpression and FLI1, 1.7× overexpression) and one gene upregulated in proximal EPS samples (CCND1, 1.1× overexpression). However, the significance identified did not hold true under multiple comparison testing.
SMARCB1 expression was also significantly upregulated in distal cell lines versus proximal cell lines (p < .01) but significance did not hold true under multiple comparison testing. SMARCB1 was also statistically upregulated in Keller Lab origin tissue samples (p < .05) and all Keller Lab origin distal samples versus proximal samples (p < .01). Notably, SMARCB1 and in all distal samples (biopsy and cell line together) versus proximal samples (p < .001), which was significant even after multiple comparison testing. Consistent differential expression of SMARCB1 coupled with the importance of SMARCB1 to EPS aetiology suggests there may be fundamental differences between the two subtypes connected to, or beyond, site of origin.
We also analysed expression of the individual members of the BAF, PBAC and ncBAF due to critical roles in tumourigenesis of EPS (Figure 6C). Beyond SMARCB1, only ACTL6A showed significantly different expression (1.4× overexpression in proximal biopsy samples, p < .05), but the result was not significant after multiple comparison testing.
Given previous studies suggesting the importance of the extracellular matrix (ECM) in sarcoma, 17 we investigated the expression patterns of extracellular matrix genes in EPS (Figure S6). Four ECM genes were significantly overexpressed in distal EPS (COL4A1, 3.3× overexpression, COL5A, 4.2× overexpression, COL12A1, 2.5× overexpression, COLEC12, 1.5× overexpression), but significance did not hold true under multiple comparison testing.
Finally, a previous study had identified the MYC pathway as overexpressed in proximal EPS versus distal EPS, and the Sonic Hedgehog (SHH) and Notch pathways as overexpressed in distal EPS versus proximal EPS. 18 In the SHH pathway, only DHH 3.2× overexpression) was differentially expressed in distal versus proximal EPS after multiple comparison correction (Figure 7A). In the Notch and MYC pathways, no genes were differentially expressed in distal versus proximal EPS after multiple comparison testing (Figures 7B and 8). Overall, the three previously identified signalling pathways were not significantly differentially expressed within the current cohort of EPS samples.
FIGURE 7.
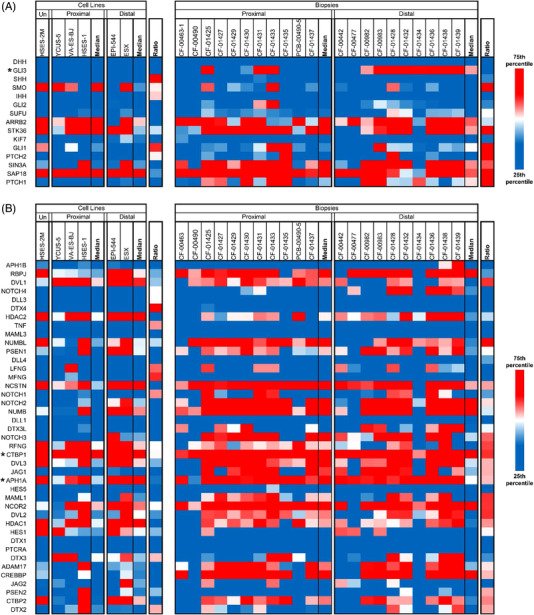
Expression of Sonic Hedgehog and notch pathway genes. (A) Expression of Sonic Hedgehog (SHH)‐associated genes. GLI3 is over‐expressed in distal versus proximal biopsies, but not in cell lines. (B) Expression of Notch‐associated genes. CTBP1 is consistently overexpressed and is the target of small molecule NSC95397. The gamma secretase subunit APHA1A is also consistently expressed in both subtypes. Nirogacestat, a clinically investigated gamma secretase inhibitor, may be a viable therapeutic.
FIGURE 8.

Expression of MYC pathways genes. THBS1 (thrombospondin 1) is more highly expressed in distal than proximal biopsy samples; however, significance was not present adjusted for multiple comparison testing. All other MYC pathway genes were not significantly differentially expressed
All statistical analyses are presented in Table S3. The complete set of DNA whole exome and RNA whole transcriptome sequencing data are provided as supplementary data.
3.6. Targeting PABPC1 in EPS
As stated earlier, gene expression analysis also identified PABPC1 as a highly expressed target in EPS, as are four additional PABP‐family genes: PABPN1, PABPC4, PABPC1L and PABPC1P3 (Figure S7A). PABPC1 promotes ribosome recruitment/translation and is critical in the first step of mRNA decay. 19 Correspondingly, we investigated pharmacological effect of PABPC1 siRNA knockdown in combination with autophagy inhibitor hydroxychloroquine (HCQ) or nuclear export inhibitor selinexor (Figure S7B and C). We confirmed PABPC1 knockdown following siRNA exposure via protein quantification (Figure S7D). While both agents induced changes in cell growth in a concentration‐dependent manner for EPS cell line VA‐ES‐BJ, drug effect was independent of PABPC1 siRNA knockdown suggesting that PABPC1 targeting in conjunction with autophagy or nuclear export inhibition may not be an effective therapeutic strategy for EPS.
3.7. Therapeutic compound screen and validation studies on EPS cell models
Having confirmed SMARCB1 status of EPS cell models, we proceeded to screen eight EPS cell models, two SMARCB1‐null rhabdoid cell models and two normal cell lines against a custom compound library (Figure S8) consisting of 61 pre‐clinical and clinical agents selected based on relevance to biological pathways implicated in sarcoma and targets currently under investigation as therapeutic mechanisms in sarcoma (Table 3). Overall results from the compound screen measuring 72‐h cell viability demonstrated a lack of response specificity to either EPS cell models versus non‐EPS cell models, and a lack of specificity to SMARCB1‐null cell models versus SMARCB1–expressing cell models (Figure S8). Nonetheless, we identified a subset of non‐differentially active agents and pursued further validation experiments (Figure S9A–D) using a set of PI3K pathway inhibitors (BEZ235, XL765, INK128, BKM120 and BYL719) and multi‐kinase inhibitors (dasatinib, sunitinib and sorafenib). The follow‐on validation studies did not demonstrate consistent sensitivity for a single agent across all tested cell models, although dasatinib (multi‐kinase inhibitor) and BEZ235 (PI3K/mTOR inhibitor) performed comparatively better than other pathway inhibitors. Correspondingly, we performed a checkerboard concentration study to determine synergy between BEZ235 and dasatinib, which demonstrated synergy at clinically relevant concentrations (dasatinib C max ≈ 160 nM, BEZ235 C max ≈ 457 nM) 20 , 21 (Figure S9E)
3.8. Tazemetostat‐focused combination studies in EPS cell models
Following the lack of single agents active at clinically feasible concentrations for the high‐throughput in vitro chemical screen, and given the recent clinical trials demonstrating tazemetostat efficacy, we investigated combinations built around tazemetostat pre‐treatment as potential EPS treatments. Given previously published synergy between EZH2 inhibitors (tazemetostat) and bromodomain inhibitors (BRDi), 22 we investigated the effect of tazemetostat pre‐treatment on response of bromodomain inhibitors (+)‐JQ1 and UNC0642 (Figure S10). While pre‐treatment generally decreases in vitro BRDi IC50 concentrations, tazemetostat pre‐treatment does not significantly alter the response of either BRDi agent.
The lack efficacy of BRDi small molecule inhibitor agents led us to confirm the biological effect of the BRD7/9 PROTACs VZ185 and cis‐VZ185 (Figure S11). Functional response of PROTAC monotherapy demonstrates response only at high concentrations, suggesting the monotherapy PROTAC is unlikely to be an effective therapy for EPS. Similarly, the combination of tazemetostat with phosphodiesterase agents modulating oxidative phosphorylation had no demonstrable effect on EPS tumour cell growth (Figure S12).
4. DISCUSSION
Herein we report the most comprehensive study to date of the functional genomic landscape of EPS. Across the largest cohort of EPS cell line models and patient‐origin biopsy tissues to date, no consistent secondary mutation was found. DNA variations and RNA expression showed PABPC1 as a potential target, but in our interference experiment, we did not observe an effect on autonomous cell viability. Additional functional validations were equally non‐informative, underlining the complexity of EPS and the need for deeper and more wide‐ranging functional studies in a larger chemical space.
A key finding in this study was the identification of biological differences between distal EPS (common in children and young adults, often having a favourable diagnosis) and proximal EPS (more common in adults). Specifically, molecular sequencing and immunocytochemical staining of SMARCB1 generally demonstrates deletion in proximal EPS samples, while distal EPS samples show a pattern of retained and/or presumably dysfunctional SMARCB1. While distal and proximal EPS generally demonstrate similar gene expression patterns across the broader transcriptome, the fundamental biological difference observed in the pathogenic initiator of EPS corresponds with differential expression patterns in genes associated with and connected to SMARCB1. Differentially expressed genes include directly or indirectly actionable molecular targets, such as FYN, GLI3 and CXCL12. Retained expression in distal EPS enables therapeutic targeting of SMARCB1 through protein degradation platforms. While traditionally viewed as a tumour suppressor gene in the context of rhabdoid tumours and EPS, targeting of retained SMARCB1 merits functional investigation.
5. CONCLUSION
Overall results of our functional genomic investigation of EPS highlight the complexity of the disease and the current limited knowledge of the optimal chemical space for therapeutic advancement. Nonetheless, the subtle differences in the initiating pathogenic alteration between the two EPS subtypes highlights the biological disparities between younger and older EPS patients and emphasises the need to approach the two subtypes as molecularly and clinically distinct diseases. Long non‐coding RNA's and miRNA's may be an area of further exploration.
CONFLICT OF INTEREST
The authors declare no direct conflicts of interest with respect to these studies. CK has sponsored research agreements with Eli Lily, Roche‐Genentech and Cardiff Oncology as well as research collaborations with Novartis, and is co‐founder of Tio Companies. Artisan Biopharma is a wholly owned subsidiary of cc‐TDI.
Supporting information
Supplementary information
ACKNOWLEDGEMENTS
This manuscript was written in honour of Connor Webb, Cory Norton, Jaya Gupta, Ella Engeström and Elijah Hughes. We are grateful to Drs. William Tap, Robert Maki, Lia Gore, Carrye Cost, Margaret Macy and Robin Jones for assistance with therapeutic agent selection. We thank Dr. Torsten Nielsen for early discussions of this project. We thank Mattie Clark for help with graphics. This work was supported by the direct contributions from EPS families and crowdfunding contributions made from families as well as by the Sam Day Foundation and Andrea Bolzicco. The laboratory of T.G.P.G. was supported by the SMARCB1 association and the Barbara and Wilfried Mohr Foundation.
Rasmussen SV, Jin J, Bickford LR, et al. Functional genomic analysis of epithelioid sarcoma reveals distinct proximal and distal subtype biology. Clin Transl Med. 2022;12:e961. 10.1002/ctm2.961
Contributor Information
Robin L. Jones, Email: robin.jones@rmh.nhs.uk.
Paul H. Huang, Email: paul.huang@icr.ac.uk.
Thomas G. P. Grünewald, Email: t.gruenewald@dkfz.de.
Charles Keller, Email: noah@cc-tdi.org, Email: charles@cc-tdi.org.
REFERENCES
- 1. Jawad MU, Extein J, Min ES, Scully SP. Prognostic factors for survival in patients with epithelioid sarcoma: 441 cases from the SEER database. Clin Orthop Relat Res. Nov 2009;467(11):2939‐2948. 10.1007/s11999-009-0749-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jones RL, Constantinidou A, Olmos D, et al. Role of palliative chemotherapy in advanced epithelioid sarcoma. Am J Clin Oncol. Aug 2012;35(4):351‐357. 10.1097/COC.0b013e3182118cf7 [DOI] [PubMed] [Google Scholar]
- 3. Enzinger FM. Epitheloid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer. Nov 1970;26(5):1029‐1041. 10.1002/1097-0142(197011)26:5<1029::aid‐cncr2820260510>3.0.co;2‐r [DOI] [PubMed] [Google Scholar]
- 4. Chbani L, Guillou L, Terrier P, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol. Feb 2009;131(2):222‐227. 10.1309/ajcpu98abipvjaiv [DOI] [PubMed] [Google Scholar]
- 5. Chase DR, Enzinger FM. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. Apr 1985;9(4):241‐263. [PubMed] [Google Scholar]
- 6. Noujaim J, Thway K, Bajwa Z, et al. Epithelioid sarcoma: opportunities for biology‐driven targeted therapy. Front Oncol. 2015;5:186. 10.3389/fonc.2015.00186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jamshidi F, Bashashati A, Shumansky K, et al. The genomic landscape of epithelioid sarcoma cell lines and tumours. J Pathol. Jan 2016;238(1):63‐73. 10.1002/path.4636 [DOI] [PubMed] [Google Scholar]
- 8. Berlow NE, Rikhi R, Geltzeiler M, et al. Probabilistic modeling of personalized drug combinations from integrated chemical screen and molecular data in sarcoma. BMC Cancer. Jun 17 2019;19(1):593. 10.1186/s12885-019-5681-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zaffaroni N, Frezza AM, Zuco V, et al. Doxorubicin (D), gemcitabine (G), ifosfamide (I) and the EZH2 inhibitor EPZ‐011989 in epithelioid sarcoma (ES): a comparison of different regimens in a patient‐derived xenograft (PDX) model. J Clin Oncol. 2018;36(15_suppl):11578. 10.1200/JCO.2018.36.15_suppl.11578 [DOI] [Google Scholar]
- 10. Gounder M, Schöffski P, Jones RL, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open‐label, phase 2 basket study. Lancet Oncol. Nov 2020;21(11):1423. 10.1016/s1470-2045(20)30451-4 [DOI] [PubMed] [Google Scholar]
- 11. Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol. Mar 2013;26(3):385‐392. 10.1038/modpathol.2012.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakayama RT, Pulice JL, Valencia AM, et al. SMARCB1 is required for widespread BAF complex‐mediated activation of enhancers and bivalent promoters. Nat Genet. Nov 2017;49(11):1613‐1623. 10.1038/ng.3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isakoff MS, Sansam CG, Tamayo P, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A. Dec 6 2005;102(49):17745‐17750. 10.1073/pnas.0509014102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lupo A, Cesaro E, Montano G, Zurlo D, Izzo P, Costanzo P. KRAB‐zinc finger proteins: a repressor family displaying multiple biological functions. Curr Genomics. 2013;14(4):268‐278. 10.2174/13892029113149990002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Urakawa H, Kawai A, Goto T, et al. Phase II trial of pazopanib in patients with metastatic or unresectable chemoresistant sarcomas: a Japanese Musculoskeletal Oncology Group study. Cancer Sci. 2020;111(9):3303‐3312. 10.1111/cas.14542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irimura S, Nishimoto K, Kikuta K, et al. Successful treatment with pazopanib for multiple lung metastases of inguinal epithelioid sarcoma: a case report. Case Rep Oncol. 2015;8(3):378‐384. 10.1159/000439427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lian X, Bond JS, Bharathy N, et al. Defining the Extracellular matrix of rhabdomyosarcoma. Front Oncol. 2021;11:601957. 10.3389/fonc.2021.601957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Czarnecka AM, Sobczuk P, Kostrzanowski M, et al. Epithelioid sarcoma‐from genetics to clinical practice. Cancers (Basel). 2020;12(8) 10.3390/cancers12082112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mattijssen S, Kozlov G, Fonseca BD, Gehring K, Maraia RJ. LARP1 and LARP4: up close with PABP for mRNA 3' poly(A) protection and stabilization. RNA Biol. 2021;18(2):259‐274. 10.1080/15476286.2020.1868753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Demetri GD, Lo Russo P, MacPherson IR, et al. Phase I dose‐escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res. Oct 1 2009;15(19):6232‐6240. 10.1158/1078-0432.Ccr-09-0224 [DOI] [PubMed] [Google Scholar]
- 21. Wise‐Draper TM, Moorthy G, Salkeni MA, et al. A Phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target Oncol. 2017;12(3):323‐332. 10.1007/s11523-017-0482-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti‐cancer agents. Br J Cancer. 2021;124(9):1478‐1490. 10.1038/s41416-021-01321-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emori M, Tsukahara T, Murase M, et al. High expression of CD109 antigen regulates the phenotype of cancer stem‐like cells/cancer‐initiating cells in the novel epithelioid sarcoma cell line ESX and is related to poor prognosis of soft tissue sarcoma. PLoS One. 2013;8(12):e84187. 10.1371/journal.pone.0084187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goto H, Takahashi H, Funabiki T, Ikuta K, Sasaki H, Nagashima Y. Brief report: neural differentiation of a novel cell line, YCUS‐5, established from proximal‐type epithelioid sarcoma of a child. Med Pediatr Oncol. 1999;33(2):137‐138. 10.1002/(sici)1096-911x(199908)33:2<137::aid‐mpo18>3.0.co;2‐n [DOI] [PubMed] [Google Scholar]
- 25. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- 26. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinfo. 2008;9:559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1‐13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc: Ser B (Methodol). 1995;57(1):289‐300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 29. Berlow NE. Probabilistic Boolean modeling of pre‐clinical tumor models for biomarker identification in cancer drug development. Curr Protoc. 2021;1(10):e269. 10.1002/cpz1.269 [DOI] [PubMed] [Google Scholar]
- 30. Bacher JW, Flanagan LA, Smalley RL, et al. Development of a fluorescent multiplex assay for detection of MSI‐High tumors. Dis Markers. 2004;20(4‐5):237‐250. 10.1155/2004/136734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics. 2014;30(7):1015‐1016. 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sonobe H, Ohtsuki Y, Sugimoto T, Shimizu K. Involvement of 8q, 22q, and monosomy 21 in an epithelioid sarcoma. Cancer Genet Cytogenet. 1997;96(2):178‐180. 10.1016/s0165‐4608(96)00412‐8 [DOI] [PubMed] [Google Scholar]
- 33. Helson C, Melamed M, Braverman S, Traganos F, Preti R, Helson L. Va‐es‐bj – an epithelioid sarcoma cell‐line. Int J Oncol. 1995;7(1):51‐56. 10.3892/ijo.7.1.51 [DOI] [PubMed] [Google Scholar]
- 34. Sakharpe A, Lahat G, Gulamhusein T, et al. Epithelioid sarcoma and unclassified sarcoma with epithelioid features: clinicopathological variables, molecular markers, and a new experimental model. Oncologist. 2011;16(4):512‐522. 10.1634/theoncologist.2010-0174 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information