

Summary
For regenerative cell therapies using pluripotent stem cell (PSC)-derived cells, large quantities of purified cells are required. Magnetic-activated cell sorting (MACS) is a powerful approach to collect target antigen-positive cells; however, it remains a challenge to purify various cell types efficiently at large scale without using antibodies specific to the desired cell type. Here we develop a technology that combines microRNA (miRNA)-responsive mRNA switch (miR-switch) with MACS (miR-switch-MACS) to purify large amounts of PSC-derived cells rapidly and effectively. We designed miR-switches that detect specific miRNAs expressed in target cells and controlled the translation of a CD4-coding transgene as a selection marker for MACS. For the large-scale purification of induced PSC-derived cardiomyocytes (iPSC-CMs), we transferred miR-208a-CD4 switch-MACS and obtained purified iPSC-CMs efficiently. Moreover, miR-375-CD4 switch-MACS highly purified pancreatic insulin-producing cells and their progenitors expressing Chromogranin A. Overall, the miR-switch-MACS method can efficiently purify target PSC-derived cells for cell replacement therapy.
Keywords: MicroRNA switches, magnetic-activated cell sorting, purification, iPSC-derived cells, cell replacement therapy, cardiomyocytes, miR-208a, miR-375
Graphical abstract
Highlights
-
•
MiR-208a-CD4 switch-MACS can purify a large amount of iPSC-CMs in a short time
-
•
MiR-208a switch can purify iPSC-CMs in each subtype-specific protocol
-
•
MiR-375-CD4 switch-MACS can be applied to pancreatic endocrine precursor cells
-
•
MiR-switch-MACS method can be efficient for large-scale target cell purification
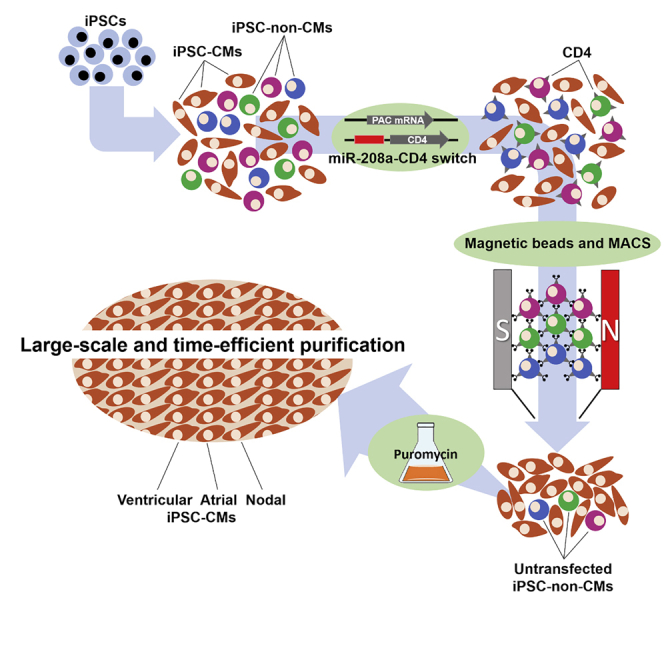
In this article, Tsujisaka, Hatani, et al. demonstrated a novel technology that combines microRNA-responsive synthetic mRNA (miR-switch) with magnetic-activated cell sorting (miR-switch-MACS), which enables large-scale and time-efficient target cell purification. MiR-208a switch-MACS efficiently purified iPSC-derived CMs generated with the ventricular-, atrial-, and nodal-specific protocols. Moreover, miR-375 switch-MACS purified pancreatic endocrine precursor cells, showing the versatility of miR-switch-MACS.
Introduction
Cardiovascular diseases are the leading cause of morbidity and mortality in the world (Virani et al., 2020). To overcome cardiovascular diseases, strenuous efforts to develop new therapies are being made both at the experimental and clinical levels (Sanganalmath and Bolli, 2013). Cardiac replacement therapy using cardiomyocytes (CMs) differentiated from pluripotent stem cells (PSCs), including embryonic stem cells and induced PSCs (iPSCs) have shown many beneficial effects to infarcted hearts in animal models (Chong et al., 2014; Hatani et al., 2018a; Shiba et al., 2016). However, for the clinical application of PSC-derived CMs (PSC-CMs), 1 × 108–1×109 purified PSC-CMs are required (Chong et al., 2014; Hattori and Fukuda, 2012; Shiba et al., 2016). Additionally, the contamination of non-CMs, especially undifferentiated cells, may cause unexpected contingencies and should be avoided. Although cell sorting using a flow cytometer with antibodies against specific cell surface proteins is one promising strategy (Dubois et al., 2011; Osborn et al., 1989), it is time consuming to collect the cells, specific cell surface markers are needed, and contamination of the antibodies on the target cell surface risks an immunogenic reaction and local inflammation after the transplantation, which may lead to graft failure (Dunn et al., 2011).
Magnetic-activated cell sorting (MACS) technology enables time-efficient cell isolation and sorts greater amounts of cells in a shorter time compared with flow cytometers. However, this technology also requires specific antibodies against cell surface proteins and is difficult to apply to cell types that do not have specific cell surface proteins, such as CMs and pancreatic insulin-producing cells.
MicroRNAs (miRNAs) are a class of small non-coding RNAs that regulate gene expressions at the posttranscriptional level by cleaving or translationally repressing their target mRNAs (Ambros, 2001; Bartel, 2004; Nakanishi and Saito, 2019). We previously designed a miRNA-responsive, synthetic mRNA encoding a fluorescent reporter protein and complementary sequences against target miRNA, a technology we call miRNA switch (miR-switch) (Figure 1A) (Miki et al., 2015; Endo et al., 2016; Ohno et al., 2020). This technology changes the translation efficiency of the reporter proteins depending on the expression of the target miRNA in the transfected cells through the hybridization of the synthetic mRNA with the endogenous target miRNA (Figure 1B). Taking advantage of this differential translation, the miR-switch can separate and sort target cells from heterogeneous cell populations using a flow cytometer. We demonstrated that the miR-208a switch specifically separates iPSC-CMs from iPSC-non-CMs with a purity of more than 95% based on troponin T expression. Importantly, the miR-switch degrades in a short time and has no risk of genomic modification, which enables the maintenance of normal gene expressions and miRNA profiles before and after miR-switch transfection (Miki et al., 2015). Thus, miR-switch technology has the potential for clinical application, but its combination with flow cytometric cell sorting is not suitable for collecting a large number of cells in a short time, a condition required in regenerative medicine.
Figure 1.

Schematic illustration of this study
(A) The miR-switch is synthetic mRNA made of two parts. One is the miRNA target site, which is a complementary sequence to the target miRNA, and the other is a protein coding sequence. In this study, CD4 was used as the protein for the selection marker in MACS.
(B) After the transfection of the miR-switch, if the cell expresses the target miRNA, translation from the miR-switch is inhibited, due to the interaction between the miRNA and the miR-switch. In contrast, if the cell does not express the target miRNA, the miR-switch is translated.
(C) Thus, upon transfecting miR-208a-CD4-switch to a heterogeneous cell population, CD4 translation is inhibited in CMs, but not in other cell types. Using CD4 microbeads and MACS, non-CMs bind to the MACS column, and CMs are isolated. By co-transfecting PAC mRNA, untransfected non-CMs are removed after puromycin administration.
Therefore, in this study, we developed a novel purification method of human iPSC-derived cells at large scale by combining the miR-switch and MACS (miR-switch-MACS). We designed miR-switches that encode the CD4 cell surface protein and contain complementary sequences against the target miRNA. The combination of the miR-switch-MACS using microbeads conjugated to monoclonal anti-human CD4 antibody enabled the large-scale purification of iPSC-CMs efficiently, without the attachment of the antibody on the target cell (Figure 1C). We engrafted human iPSC-CMs purified by this method in a mouse model of acute myocardial infarction. Moreover, we also showed that insulin-producing pancreatic progenitor cells (chromogranin A-positive cells) could be purified using miR-375-CD4 switch-MACS. These data indicate that miR-switch-MACS technology can be applied to cell therapies that require large-scale cell preparation.
Results
Transfection of CD4-encoding mRNA to iPSC-derived cells
We aimed to purify iPSC-CMs by removing non-CMs ectopically expressing a cell surface protein (Figure 1C). We first differentiated iPSCs into iPSC-CMs, and, by flow cytometry, investigated the expressions of CD4 and CD271 in differentiated cells, including iPSC-CMs, because MACS antibodies against CD4 and CD271 are available commercially. Some differentiated cells from 201B7 iPSCs expressed CD271 (Figure S1), but none differentiated from three different iPSC lines (201B7, 409B2, and 692D2) expressed CD4 (Figure 2A). Because differentiated cells should not express the surface protein endogenously, we selected CD4 for the following experiments. After the co-transfection of two mRNAs coding for CD4 and BFP, we found these proteins were expressed efficiently at a similar level (Figure 2B). We then compared the expression level of CD4 in CMs (GFP positive) and non-CMs (GFP negative) differentiated from the cardiac iPSC reporter line 201B7-MYH6-EGFP (Funakoshi et al., 2016). We found similar CD4 expression levels between GFP positive and GFP-negative cells, indicating that mRNA can be transfected and translated in both CM and non-CM populations (Figure 2C). We also assessed the time course of the protein expression of BFP, finding transfected BFP mRNA expression was high in the first 3 days, but decreased gradually thereafter, and fell to less than one-half within 5 days (Figure 2D and Table S1).
Figure 2.

CD4 mRNA and miR-208a-CD4-switch transfection in differentiated cells
(A) Differentiated 201B7, 409B2, and 692D2 iPSCs showed no CD4 expression on the cell surface. Representative data from two biologically independent experiments are shown.
(B) Co-transfection of CD4 mRNA and BFP mRNA to differentiated 409B2 and 692D2 iPSCs. Representative data from two biologically independent experiments are shown. In the left panel, BFP expression was 80.0%, and CD4 expression was 79.5%. In the right panel, the percentages were 73.2 and 73.2%, respectively.
(C) Co-transfection of CD4 mRNA and BFP mRNA to differentiated 201B7-MYH6-EGFP iPSCs. Representative data from two biologically independent experiments are shown. EGFP-positive cells, which are CMs, were 69.5% of the cell population (red). EGFP-negative cells, which are non-CMs, were 30.5% of the cell population (blue). In the upper right panel, BFP expression was 83.0% and CD4 expression was 82.7%. In the lower panel, BFP expression was 82.2% and CD4 expression was 81.5% in EGFP negative cells and 83.3 and 83.2%, respectively, in EGFP positive cells.
(D) Time course of the mRNA expression as assessed by the flow cytometric analysis of BFP-positive cells from 12 h to 9 days after the transfection of 201B7 differentiated cells. The values were acquired from three biologically independent measurements. The actual values are shown in Table S1.
(E) Transfection of 300 ng of miR-208a-CD4 switch to differentiated 201B7-MYH6-EGFP iPSCs. Representative data from three biologically independent experiments are shown. Red shows EGFP-positive cells (CMs) and blue shows EGFP-negative cells (non-CMs). The analysis before MACS is shown in the left panel, and the analysis after MACS is shown in the right panel. After MACS sorting, CD4-positive cells were eliminated.
Transfection of miR-208a-CD4 switch
Next, we investigated whether we could modulate ectopic CD4 expression levels by the endogenous and specific miRNA expression in iPSC-CMs. As a marker for CM-specific miRNA, we chose miR-208a, because the fluorescent protein expression difference between CMs and non-CMs was largest using the miR-208a switch (Miki et al., 2015). Thus, we designed miR-208a-responsive CD4-coding mRNA (miR-208a-CD4 switch) to selectively repress CD4 expression in iPSC-CMs. We transfected different amounts of miR-208a-CD4 switch (100, 200, 300, 600, and 900 ng) and 300 ng of control BFP mRNA to 201B7-MYH6-EGFP-differentiated cells and compared expression profiles (Figure S2 and Table S2). Flow cytometric analysis showed the CD4 signal intensity of iPSC-CMs and non-iPSC-CMs increased with higher quantities of transfected miR-208a-CD4 switch. After MACS, CD4-positive cells were clearly eliminated. We used 300 ng miR-208a-CD4 switch to collect large quantities of iPSC-CMs, because we could efficiently purify iPSC-CMs among transfected cells (>95% purity), and retain more than 50% of iPSC-CMs based on the calculation of the number of CMs before and after MACS (Figures 2E, S2, and Table S2).
Purification of iPSC-CMs with miR-208a switch-MACS
Since untransfected cells do not express CD4, the cells sorted by MACS still contained untransfected cells. To eliminate these cells, we co-transfected miR-208a-CD4 switch and puromycin N-acetyltransferase (PAC) mRNA instead of BFP mRNA (Figure 3A). Flow cytometric analysis using an antibody against troponin T revealed that iPSC-CMs were highly purified after MACS and the subsequent 48 h administration of puromycin (Figures 3B, 3C, and Table S3). Indeed, we found that iPSC-CMs were purified at more than 97% in three different iPSC lines. Immunostaining also showed that iPSC-CMs were highly purified (Figure 3D). In addition, purified iPSC-CMs spontaneously beat and were characterized by ventricular action potentials having a long plateau phase and a typical action potential duration (APD) ratio (APD90/APD50 < 1.4) (Burridge et al., 2014) (Figure S3A and Table S4). We also confirmed that the final purified iPSC-CM population did not express CD4, indicating that transfected miR-208a-CD4 switch in iPSC-CMs did not induce the translation of CD4 after MACS (Figure 3E).
Figure 3.

Purification of iPSC-CMs using miR-208a-CD4 switch and MACS
(A) Schematic procedure for the purification of iPSC-CMs using miR-208a-CD4 switch and MACS. To eliminate untransfected cells, PAC mRNA and puromycin were used.
(B) Representative data of troponin T-positive differentiated 201B7, 409B2, and 692D2 iPSCs before MACS, after MACS, and after purification with puromycin (final). Representative data are shown from three biologically independent experiments. The boxed areas indicate troponin T-positive cells.
(C) The percentage of troponin T-positive differentiated 201B7, 409B2, and 692D2 iPSCs before MACS, after MACS, and after purification. The values are from three biologically independent measurements and denoted as mean ± standard deviation. ∗p < 0.015 versus the corresponding sample before MACS, ∗∗p < 0.01 versus the corresponding sample before MACS, ∗∗∗p < 0.005 versus the corresponding sample after MACS, ∗∗∗∗p < 0.005 versus the corresponding sample before MACS, ∗∗∗∗∗p < 0.001 versus the corresponding sample after MACS, ∗∗∗∗∗∗p < 0.001 versus the corresponding sample before MACS. The data were analyzed by the Bonferroni-Dunn test. Actual values are shown in Table S3.
(D) Fluorescence immunostaining of differentiated 201B7 iPSCs before MACS, after MACS, and after the final purification (n = 3). Representative data from three biologically independent experiments are shown. Red, troponin T; blue, Hoechst. Scale bars, 100 μm.
(E) CD4 expression of the final purified CMs using the miR-208a switch-MACS method. Flow cytometric analysis found no CD4 expression in the final purified CMs after application of the miR-208a-CD4 MACS method and subsequent 48 h administration of puromycin.
RNA expression analysis of purified iPSC-CMs
Next, we compared the gene expression profiles of the iPSC-CMs purified by the miR-switch-MACS method and those sorted by a conventional antibody-based method (CD172a/b positive, CD31 negative, CD49a negative, CD90 negative, and CD140b negative). Hierarchical clustering analysis of the samples revealed that iPSC-CMs induced in the first experiment (lot #1) and iPSC-CMs induced in the second experiment (lot #2) were classified in the same clustering, indicating differences in the expression profiles of the iPSC-CMs sorted by the two methods were smaller than those between the two lots (Figure 4). These data suggest that the cellular characteristics of iPSC-CMs purified by the miR-switch-MACS method are similar to those purified by the conventional method.
Figure 4.
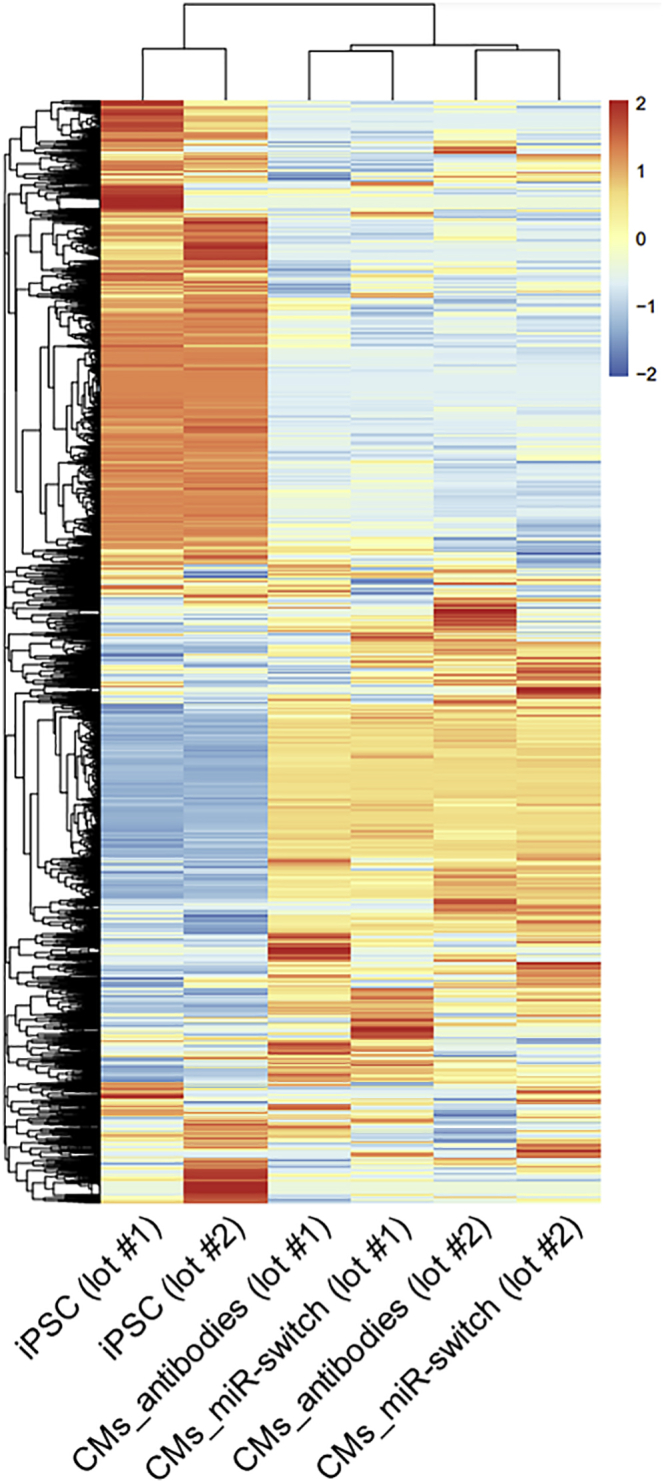
Hierarchical clustering of gene expression profiles
Hierarchical clustering of the gene expression profiles of 201B7 iPSCs (control), 201B7 iPSC-CMs purified by antibodies (CD172a/b positive, CD31 negative, CD49a negative, CD90 negative, and CD140b negative), and 201B7 iPSC-CMs purified by the miR-208a-CD4-switch and MACS. The data are shown from two biologically independent experiments. Color indicates scaled values of log2 (normalized counts).
Transplantation of purified iPSC-CMs
We next investigated the viability of the sorted iPSC-CMs in vivo. We differentiated iPSC-CMs from 201B7-luc iPSCs (Funakoshi et al., 2016) and purified them using the miR-switch-MACS method. Thereafter, we transplanted the cells by direct injection into immunodeficient NOG mouse hearts with myocardial infarction. Hearts were extracted from the mice three months after the transplantation and fixed with 4% paraformaldehyde in PBS. Immunostaining against troponin T and luciferase confirmed that 201B7-luc iPSC-CMs purified using the miR-switch-MACS method were engrafted efficiently and substantially in the mouse heart (Figure 5). High-magnification images revealed that the transplanted iPSC-CMs formed sarcomere structures (Figure S4A).
Figure 5.

Engraftment of iPSC-CMs purified using the miR-208a switch-MACS method
Anti-luciferase immunostaining (red) demonstrated that a substantial amount of iPSC-CMs was engrafted in the host mouse left ventricular wall with myocardial infarction three months after the transplantation. Representative data are shown from nine biologically independent experiments. Immunostaining of troponin T (green) marks CMs.
(A) The relatively thin anterior wall of the LV indicates an infarcted area. Scale bar, 1000 μm.
(B) A high magnification of the region of interest indicated by the white square in (A). The troponin T-negative area indicates the scar of the myocardial infarction. Scale bars, 100 μm.
Echocardiography showed that left ventricular diastolic dimensions (LVDd) and left ventricular systolic dimensions (LVDs) were increased and the fractional shortening (FS) was decreased just after the myocardial infarction in both groups. However, at 1 week after the injection, the FS was significantly higher in the transplanted group compared with the control. Similarly, the LVDd at 2 weeks after the injection was significantly lower in the transplanted group. These differences were maintained for the 3-month follow-up period. Thus, LV remodeling and reduced LV function after the myocardial infarction were relatively mild in mice with transplanted iPSC-CMs compared with control mice, and the cardiac function was significantly better with iPSC-CM transplantation (Figure S4B).
Collectively, iPSC-CMs purified by the miR-208a switch-MACS method could engraft in vivo and improved cardiac function, suggesting the versatility for regenerative cell transplantation therapy.
Time and efficiency for the purification of iPSC-CMs
To compare the time efficiency of the miR-switch-MACS method and conventional antibody-based flow cytometric cell sorting, we measured the time to obtain a large scale of purified iPSC-CMs. We finally obtained 1.85 ± 0.14×108 purified cells after 28 min of MACS and the following 48 h of puromycin administration. In contrast, we obtained only 1.15 ± 0.042×106 purified cells after 27 min of fluorescence-activated cell sorting (FACS) (Table S5). Based on these results, we assume that FACS would take approximately 68 h to obtain the equal amount of the final purified cells using miR-switch-MACS method. In contrast, although the miR-switch-MACS method requires the additional 48 h of puromycin selection, it does not require continuous FACS runs. Thus, the miR-switch-MACS method is a highly efficient and simple approach compared with FACS-based methods.
To bypass the flow cytometric cell sorting process, in our previous work, we used Bim as another reporter of miR-switch and demonstrated that miR-208a-Bim induced apoptosis selectively in non-iPSC-CMs to purify the iPSC-CM population, although the purification efficiency of iPSC-CMs was approximately 90% (Miki et al., 2015). Here we compared the efficiency of miR-208a-Bim and miR-208a-CD4 by transfecting both of them. The miR-208a-CD4 switch resulted in a significantly higher purity than the miR-208a-Bim switch (p < 0.001, by two-way ANOVA with repeated measures) (Figure S5 and Table S6).
Purification of subtype-specific iPSC-CMs with miR-208a switch
To investigate further application of the miR-208a switch-MACS method, we transfected miR-208a switch into cells differentiated from iPSCs using CM subtype-specific protocols.
Our standard iPSC differentiation protocol favors the development of ventricular iPSC-CMs (ventricular protocol). To confirm this, we differentiated 201B7 iPSCs, purified them using miR-208a switch, and immunostained the final cell product with antibodies against troponin T and MLC2V, which is a ventricular-specific myosin isoform. Immunostaining showed that almost all cells were troponin T positive and MLC2V positive (Figure 6A). Next, we differentiated 201B7 iPSCs using the atrial protocol and purified them as above. Before purification, the percentage of troponin T positive cells and the percentage of cells that reacted to miR-208a switch were similar (Figure 6B). Moreover, flow cytometric analysis after the purification showed that the percentage of troponin T-positive cells was approximately 95% (95.4 ± 3.3%) (Figure 6B). These findings suggest that a high purification of iPSC-CMs was achieved in the atrial protocol as well. Thereafter, we differentiated iPSC-CMs using both protocols. A qPCR analysis of the differentiated cells before purification showed that ventricular-related genes (MYL2 and IRX4) were expressed at significantly higher levels when using the ventricular protocol compared with the atrial protocol. The converse was true for atrial-related genes (COUPTF-Ⅱ and MYL7). These gene expression differences were maintained in the final purified cell population by the miR-208a switch method as well as the conventional antibody-based method (CD172a/b positive, CD31 negative, CD49a negative, CD90 negative, and CD140b negative) (Figure 6C). Immunostaining of the final purified cell population using both protocols confirmed these differences (ventricular CMs, COUPTF-Ⅱ negative; atrial CMs, COUPTF-Ⅱ positive) (Figure 6D). The final purified iPSC-CMs with the atrial protocol spontaneously beat and were characterized by atrial action potentials having absence of a prominent plateau phase and a typical APD ratio (APD90/APD50 > 1.7) (Burridge et al., 2014) (Figure S3B and Table S4).These findings revealed that the miR-208a switch method is capable of purifying atrial iPSC-CMs.
Figure 6.

Flow cytometric, immunostaining and qPCR analysis of cells in subtype-specific iPSC-CMs
(A) Immunostaining of cells differentiated using the standard protocol and purified using miR-208a-switch.
(B and E) Flow cytometric analysis of cells differentiated using the atrial (B) and nodal (E) protocols. (Left) The percentage of troponin T-positive cells and cells that reacted to miR-208a switch before purification. (right) Troponin T-positive cells were significantly increased (B, 95.4 ± 3.3%; and E, 95.2 ± 2.4%) after the final purification. The values were acquired from three biologically independent measurements.
(C and F) qPCR analysis of ventricular-related (MYL2 and IRX4), atrial-related genes (COUPTF-Ⅱ and MYL7) (C) and nodal-related genes (SHOX2 and ISL1) (F) in cells before purification, after purification with the conventional antibody-based method, and after purification with the miR-208a-switch. The values were acquired from three biologically independent measurements.
(D and G) Immunostaining of purified iPSC-CMs using miR-208a switch with the atrial (D) and nodal protocols (G). All scale bars, 100 μm. The values are denoted as means. All error bars represent the standard deviation. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, by a paired t-test.
Next, we compared the nodal and ventricular protocols by flow cytometric analysis, qPCR, and immunostaining. Before purification, the percentage of cells reacting to the miR-208a switch was similar with that of troponin T-positive cells. After the purification, 95.2 ± 2.4% of the final purified cell population was troponin T positive (Figure 6E), confirming a high purification of iPSC-CMs when using the nodal protocol. Compared with the ventricular protocol, the differentiated cells from the nodal protocol had a significantly lower expression of ventricular-related genes (MYL2 and IRX4) and significantly higher expression of nodal-related genes (SHOX2 and ISL1). These gene expression profiles were similar after purification using the miR-208a-switch method and the conventional antibody-based method (Figure 6F). In the nodal protocol, immunostaining showed that most of the final purified cell populations were troponin T positive and NKX2.5 negative, which is characteristic of nodal CMs. In contrast, using the ventricular protocol, most of the cells were troponin T positive and NKX2.5 positive, which is characteristic of ventricular CMs (Figure 6G). The final purified iPSC-CMs spontaneously beat relatively faster than those using the ventricular and atrial protocols. Nodal action potentials with slow upstroke, prominent phase four depolarization, and a typical APD ratio (1.4 < APD90/APD50 < 1.7) (Burridge et al., 2014) were recorded (Figure S3C and Table S4). Hence, nodal iPSC-CMs can also be purified using the miR-208a-switch method.
Purification of insulin-producing cells with miR-375 switch
Finally, to show the versatility of the miR-switch-MACS method, we aimed to purify and collect other types of cells. We chose miR-375, an insulin-producing cell-expressing miRNA, to collect insulin-producing cells. We transfected miR-375-CD4 switch and BFP mRNA simultaneously into differentiated cells derived from iPSCs (585A1) and confirmed that CD4-positive cells were eliminated using MACS (Figure 7A). Next, we co-transfected miR-375-CD4-switch and PAC mRNA instead of BFP mRNA for purification. Flow cytometric analysis using antibodies against chromogranin A, a marker for pancreatic endocrine precursor cells, and C-peptide showed insulin-producing cells were highly purified after MACS and puromycin selection (Figures 7B, 7C, and Table S7). Immunostaining showed that the final purified cell population included C-peptide positive-cells, glucagon positive cells, and some somatostatin positive cells (Figure 7D). Furthermore, some of the C-peptide positive cells were also positive for PDX1, a marker for functional β cells, suggesting the final purified cell population were fetal-type endocrine cells, including immature β cells.
Figure 7.

Purification of pancreatic cells using miR-375-CD4 switch and MACS
(A) The transfection of 300 ng miR-375-CD4-switch to differentiated 585A1 iPSCs. Representative data from three biologically independent experiments are shown. After MACS sorting, CD4-positive cells were eliminated.
(B) Representative data of chromogranin A- and C-peptide positive differentiated 585A1 iPSCs before MACS, after MACS, and after purification with puromycin (final) (n = 3). Representative data from three biologically independent experiments are shown. The boxed areas indicate chromogranin A-positive cells (upper panels) and C-peptide-positive cells (lower panels).
(C) The percentage of chromogranin A- and C-peptide positive differentiated 585A1 iPSCs before MACS, after MACS, and after purification. Data were collected from three biologically independent experiments. The values are denoted as mean ± standard deviation. ∗p < 0.001 versus the corresponding sample before MACS, ∗∗p < 0.001 versus the corresponding sample after MACS, ∗∗∗p < 0.001 versus the corresponding sample before MACS, ∗∗∗∗p < 0.015 versus the corresponding sample after MACS, ∗∗∗∗∗p < 0.005 versus the corresponding sample before MACS. These were analyzed by the Bonferroni-Dunn test. The actual values are shown in Table S7.
(D) Immunostaining of the purified insulin-producing cells using the miR-375 switch-MACS method. Scale bars, 100 μm.
Discussion
In the present study, we developed a novel and efficient purification method for iPSC-CMs and insulin-producing cells that is applicable to large-scale cell preparation by combining miR-switch-MACS technologies.
Although antibody-based purification methods for CMs using a cell sorter is one promising strategy to prepare the cells for regenerative medicine, this approach is time consuming when collecting the large number of cells needed for cell transplantation. A comparison of the time to purify iPSC-CMs showed that the miR-switch-MACS method could obtain at least 100 times more iPSC-CMs than the conventional cell sorting method using antibodies, despite similar procedure times (Table S5). Even including the subsequent 48 h for puromycin selection after MACS (Figure 3A), the miR-switch-MACS method notably decreases the time and effort needed for standard cell sorting methods. Furthermore, conventional antibody-based purification methods risk contaminating antibodies, which may cause an immunogenic reaction and local inflammation, resulting in graft failure (Dubois et al., 2011; Osborn et al., 1989). In the miR-switch-MACS method, there is the possibility that the low of level ectopic CD4 expression, which cannot be trapped by MACS, causes an immunogenic reaction in the host heart after transplantation. We did not assess the immunogenic reaction or local inflammation of the transplanted mice. Further study is needed to compare the miR-switch-MACS method and conventional antibody-based methods from an immunological perspective. However, this concern is mitigated by the rapid degradation of the transfected mRNA and the absent CD4 expression in the final product of purified iPSC-CMs (Figures 2D, 3E, and Table S1). Additionally, the miR-switch-MACS method purifies cells by negative selection, which needs no genetic modification of the cell (Figure 1C). Thus, this method is assumed to be safe for clinical applications.
We previously reported the miR-Bim switch as another option to bypass the flow cytometry sorting process, as this switch uses pro-apoptotic Bim to automatically eliminate non-target cells after the miR-switch induction (Miki et al., 2015). However, we found that this method was inferior to the miR-switch-MACS method in terms of iPSC-CM purification efficiency (Figure S5 and Table S6). The reason might be that more steps are needed for the Bim-induced apoptosis, whereas the MACS-based selection simply depends on CD4 expression.
We also demonstrated that the miR-switch-MACS method can purify another cell type: insulin-producing cells. Notably, few methods have reported the efficient purification of this cell type with specific antibodies. The combination of miR-375-CD4 switch and -MACS showed more than 99% purification of chromogranin A-positive cells and approximately 60% of C-peptide-positive cells after eliminating untransfected cells by puromycin selection (Figures 7B, 7C, and Table S7). MiR-375 is one of the most abundant miRNAs in insulin-secreting β cells; however, it is also expressed in immature pancreatic endocrine precursor cells (Eliasson, 2017). Chromogranin A is also expressed widely in immature pancreatic endocrine precursor cells, which have polyhormonal aspects (Karlsson, 2001; Rieck et al., 2012), as well as in mature endocrine cells. In addition, immunostaining showed that some purified cells expressed PDX1 (Figure 7D). PDX1 is expressed in functional β cells and essential for maintaining mature β cell function (Ahlgren et al., 1998; Wang et al., 2001). Thus, these findings suggest that the miR-375-switch-MACS method could purify endocrine precursor cells at a high efficiency, and importantly the purified cells include insulin-producing β cells. Since some studies have reported pancreatic cells are mature in the kidney subcapsule of immunodeficient mice after transplantation, pancreatic endocrine precursor cells purified by the miR-375-switch-MACS method may be a potential candidate for regenerative cell therapy for patients with diabetes (Toyoda et al., 2015). To isolate more mature C-peptide-positive insulin-producing cells, an miR-switch that senses miRNAs specifically expressed in mature insulin-producing cells is desired.
In the present study, we demonstrate that the miR-switch-MACS method is applicable to two types of cells (CMs and insulin-producing cells). However, miR-switch technology may be limited to cell types that uniquely have a miRNA specifically present or absent in such cell types. One solution to this problem is to use multiple miR-switches (Endo et al., 2016, 2019; Fujita et al., 2022) or miR-switch-based logic circuits, such as AND, OR, NAND, NOR, and XOR gates (Matsuura et al., 2018). Such multiple miR-switches and circuits may assist in purifying complicated cell populations. As for the purification of iPSC-CMs, cultivation under a glucose-depleted and lactate-abundant condition is also an option (Burridge et al., 2014; Tohyama et al., 2013). This type of metabolic selection may also be applicable to other types of cells. It is important to use a proper purification method for each type of cell.
For large-scale purification by the miR-switch-MACS method, the efficient delivery of synthetic RNAs into large amounts of cells must be addressed. We thus optimized the lipofection method and eventually achieved more than 70% transfection efficiency (Figures 2E, S2, and Table S2). The development of a cost-efficient transfection method with higher introduction efficiency (near 100%) would benefit the clinical application of this RNA-based technology. Additionally, puromycin selection after transfecting puromycin-resistant gene-coding mRNA can eliminate untransfected cell population before transplantation. Although RNA transfection could affect some cells, our transcriptome analysis showed only negligible differences in RNA expression profiles between ours and conventional purification methods (Figure 4). Moreover, iPSC-CMs purified by the miR-switch-MACS method were robustly engrafted in vivo and improved cardiac function according to acute myocardial infarction mouse models (Figures 5 and S4). These findings suggest the versatility of our method for regenerative cell transplantation therapies. Before future clinical application, however, larger animal models, such as pigs and monkeys, are needed.
In addition, the miR-switch-MACS method could be applied to atrial and nodal iPSC-CMs as well as ventricular ones. It has been reported that mimicking the developmental signal pathway by decreasing the concentration of BMP4 and activin A and adding retinoic acid or inhibiting fibroblast growth factor signaling can generate atrial or nodal CMs from iPSCs (Lee et al., 2017; Protze et al., 2017). Our subtype-specific protocols successfully differentiated atrial and nodal iPSC-CMs, and miR-208a switch could purify them. Disease modeling and future clinical application of atrial and nodal iPSC-CMs, such as atrial fibrillation and biological pacemakers, ideally require a pure CM population. However, miR-208a switch could not purify each subtype from one another. Future work should aim to produce subtype-specific miR-switches.
In conclusion, we developed a novel method for large-scale cell purification that combines miR-switch-MACS technologies. We designed a miR-switch that encodes the CD4 cell surface protein tagged with complementary sequences against the targeted miRNA. Using CD4 microbeads and MACS, we could easily and efficiently purify iPSC-CMs of several subtypes and insulin-producing cells at large scale. We also demonstrated that human iPSC-CMs purified by this method could be engrafted in a mouse model of acute myocardial infarction.
Experimental procedures
Human iPSC lines, cell culture and cardiac differentiation
Experiments with human iPSCs were approved by the ethics committee of the Department of Medicine and Graduate School of Medicine, Kyoto University. We used three human iPSC lines (201B7, 409B2, and 692D2), a fourth line into which a human MYH6 promoter driven-EGFP reporter cassette was integrated (201B7-MYH6-EGFP) (Funakoshi et al., 2016; Ohnuki et al., 2009), and a fifth line into which a CAG promoter-driven luciferase-expressing cassette was integrated (201B7-luc) (Funakoshi et al., 2016).
Human iPSCs were maintained as previously reported (Hatani et al., 2018b). Briefly, iPSCs were maintained on SNL feeders in primate ES cell medium (ReproCELL, cat. no. RCHEMD001) supplemented with 4 ng/mL recombinant human basic FGF (rhbFGF) (Wako, cat. no. 060–04543).
iPSCs were induced to differentiate into CMs using a modified embryoid body method as previously described (Funakoshi et al., 2016; Hatani et al., 2018b; Yang et al., 2008). Briefly, on day 0, iPSCs were dissociated, and EBs were generated in 10-cm low-attachment plates (Corning, cat. no. 3262) with StemPro-34 media (Life Technologies, cat. no. 10639-011) in the presence of 2 mM L-glutamine (Life Technologies), 4 × 10-4 M monothioglycerol (MTG; Sigma-Aldrich), 50 μg/mL ascorbic acid (AA; Sigma-Aldrich), 150 μg/mL transferrin (Roche), 10 μM Y-27632 (Wako, cat. no. 253–00513), and 2 ng/mL rhBMP4 (R&D Systems, cat. no. 314-BP). After 24 h, the EBs were cultured in StemPro-34 media containing 2 mM L-glutamine, 4 × 10-4 M MTG, 50 μg/mL AA, 150 μg/mL transferrin, 10 ng/mL rhBMP4, 5 ng/mL rhbFGF (R&D Systems, cat. no. 233-FB), and 6 ng/mL rh activin A (R&D Systems, cat. no. 338-AC) for ventricular differentiation. In the protocol for atrial differentiation, rhBNP4 and rh activin A were 3 ng/mL and 4 ng/mL, respectively. In the protocol for nodal differentiation, rhBNP4 and rh activin A were 3 ng/mL and 1 ng/mL, respectively. On day 4, the EBs were suspended in StemPro-34 media supplemented with 2 mM L-glutamine, 4 × 10-4 M MTG, 50 μg/mL AA, 150 μg/mL transferrin, 10 ng/mL rhVEGF (R&D Systems, cat. no. 293-VE), and 1 μM Wnt inhibitor IWP-3 (Stemgent, cat. no. 04–0035) for 3 days. For the atrial protocol, 2 μM all-trans-retinoic acid (RA; Wako, cat. no. 182–01116) were added. For the nodal protocol, 2 μM RA, 0.25 ng/mL rhBNP4, 5.4 μM SB431542 (Sigma-Aldrich, cat. no. S4317-5MG), and 200 nM FGFR inhibitor PD 173074 (Chemscene, cat. no. CS-0182) were added. On day 7, the media were changed to StemPro-34 media supplemented with 2 mM L-glutamine, 4 × 10-4 M MTG, 50 μg/mL AA, 150 μg/mL transferrin, 5 ng/mL rhbFGF, and 10 ng/mL rhVEGF. Cultures were maintained in these media with media change every 2–3 days.
Template DNA for in vitro transcription of mRNAs and miR-switches
Template DNAs were prepared as previously described (Miki et al., 2015). Briefly, each sequence encoding the protein was duplicated from each vector (Table S8). 5′untranslated region (UTR) and -3′UTR were also amplified by PCR. For the template DNAs of the mRNAs, 5′UTR, which does not encode miRNA target sequences, the protein encoding region, and 3′UTR were integrated and amplified by PCR with the T7FwdG3C primer and the Rev120A primer. For the template DNAs of the miR-switches, 5′UTRtemp_Txxx, the protein encoding region, and 3′UTR were integrated and amplified by PCR with the T7FwdB primer and the Rev120A primer. KOD-Plus-Neo (Toyobo, cat. no. KOD-401) was used as DNA polymerase.
The PCR products amplified from the plasmids were treated with DpnI restriction enzyme (Toyobo, cat. no. DPN-101) for 30 min at 37 °C. The PCR products were purified using a MinElute PCR Purification Kit (QIAGEN, cat. no. 28006) according to the manufacturer’s instructions. Complete sequences of the template DNAs for the mRNAs and miR-switches used are listed in Table S8.
Synthesis and purification of mRNAs and miR-switches
mRNAs and miR-switches were synthesized as previously described (Miki et al., 2015; Warren et al., 2010). Briefly, they were generated with a MEGAscript T7 Transcription Kit (Ambion, cat. no. AMB13345) and a modified protocol. Template DNAs, T7 enzyme, ATP, guanosine triphosphate, pseudo-UTP (Tri-Link Bio Technologies, cat. no. N-1019-10), 5-methyl-CTP (Tri-Link Bio Technologies, cat. no. N-1014-10) and Anti Reverse Cap Analog (Tri-Link Bio Technologies, cat. no. N-7003-10) were reacted at 37 °C for 4 h. Adding TURBO DNase, the reacted products were further incubated at 37 °C for 30 min. The resulting mRNAs and miR-switches were incubated with Antarctic Phosphatase (New England Biolabs, cat. no. M0289S) at 37 °C for 30 min. The RNeasy MinElute Cleanup Kit (QIAGEN, cat. no. 74204) was used to purify the mRNAs and miR-switches.
Transfection of mRNAs and miR-switches, MACS, and puromycin selection
mRNAs and miR-switches were transfected into day 15 iPSC-differentiated cells using Lipofectamine MessengerMAX Transfection Reagent (Invitrogen, cat. no. LMRNA008) according to the manufacturer’s protocol with modification. The amount of mRNA or miR-switch was 300 ng, the transfected cell number was 1×106 according to a TC20-automated cell counter (Bio-Rad), and the volume of Lipofectamine MessengerMAX Transfection Reagent was 7.5 μL. After 24 h from the transfection of the mRNAs and miR-switches, MACS was done according to the manufacturer’s protocol. MACSelect 4 MicroBeads (Miltenyi Biotec, cat. no. 130-070-101; 1:7.5), which are magnetic microbeads conjugated to monoclonal anti-human CD4 antibodies, were used. MidiMACS Separator (Miltenyi Biotec, cat. no. 130-042-302) was used to collect non-magnetic cells by trapping magnetically labeled cells on LD Columns (Miltenyi Biotec, cat. no. 130-042-901). In the experiments with a large number of cells, the amount of mRNAs, miR-switches and Lipofectamine MessengerMAX Transfection Reagent were increased in proportion to the number of applied cells, and the columns and separator described above were used for MACS. (According to the manufacturer’s protocol, the maximum number of applied cells before MACS is 5×108 and the maximum number of purified cells after MACS is 1×108, per column.) After MACS purification, 2 μg/mL puromycin was administered for 48 h to exclude untransfected cells.
RNA sequencing and analysis
For the RNA expression analysis, we used 201B7 iPSCs and 201B7 iPSC-CMs purified by antibodies (CD172a/b positive, CD31 negative, CD49a negative, CD90 negative, and CD140b negative) (Dubois et al., 2011) or purified by a miR-208a-CD4 switch and MACS. The cells were lysed with QIAzol Lysis Reagent (QIAGEN, cat. no. 79306), and total RNA was extracted using a RNeasy Mini Kit (QIAGEN, cat. no. 74104). The library construction and sequencing were performed using the TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold Set A (48 samples) and Set B (48 samples) (Illumina, cat. no. RS-122-2301 and RS-122-2302) and NextSeq 500/550 High Output Kit v2 (75 cycles) (Illumina, cat. no. TG-160-2005). The quality of raw single-end reads were evaluated using RSeQC (ver. 2.6.4) (Wang et al., 2012). Before mapping, raw reads were trimmed with cutadapt (ver. 1.14) (Martin, 2011), and the removed reads were mapped in human rRNA and tRNA by bowtie2 (ver. 2.2.5) (Langmead and Salzberg, 2012). Trimmed reads were mapped to the human genome (GRCh38) using STAR (ver. 2.5.3a) (Dobin et al., 2013). Aligned reads were assigned using HTSeq (ver. 0.9.1) with GENCODE annotation (ver. 27) (Anders et al., 2015). In R (ver. 3.4.3), the counts were normalized by DESeq2 (ver. 1.18.1) (Love et al., 2014), and a heatmap with clustering was produced by pheatmap (ver. 1.0.12).
Data and code availability
The RNA sequence data are available at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE153714.
Author contributions
Y.T. and T.H. equally contributed to this work as co-first authors. Y.T., T.H., K.C., T.T., K.O., T.K., H.S., and Y.Y. contributed to the design of the study. Y.T., T.H., and C.O. carried out the experiments. R.I. and A.K. performed the pancreatic differentiation. Y.T., T.H., M.N., and C.O. extracted and processed the RNA and analyzed the RNA data. Y.T., T.H., H.S., and Y.Y. wrote the manuscript.
CONFLICTS OF INTEREST
Y.Y. receives grants from Takeda Pharmaceutical Company. H.S. and Y.Y are the investigators of a record listed on the patent application (PCT/JP2016/062710 filed by Kyoto University on 22/04/2016, and PCT/JP2015/058466 filed by Kyoto University on 20/03/2015) related to this work. Y.Y. and S.F. are scientific advisors for Orizuru Therapeutics. H.S. owns shares and has an outside director of aceRNA Technologies Ltd.
Acknowledgments
We greatly appreciate colleagues at our laboratories and the Department of Cardiovascular Medicine, Kyoto University, for their critical comments. We thank Peter Karagiannis for critical reading of the manuscript, and Misato Nishikawa and Azusa Inagaki for technical support. We would like to express our heartfelt gratitude to Yoko Uematsu and Kaoru Shimizu for their administrative support. Funding was provided by The Leducq Foundation (18CVD05) (Y.Y.), the Japan Society for the Promotion of Science (JSPS) KAKENHI No. 15H05722 (H.S.), No. 20H05626 (H.S.), No. 17H04176 (Y.Y.), and No. 21H02912 (Y.Y.), Research Center Network for Realization of Regenerative Medicine, Japan Agency of Medical Research and Development (AMED) (JP19bm0104001, JP19bm0204003, JP19bm0804008, and JP20bm0804022) (Y.Y., H.S.), the Research on Regulatory Science of Pharmaceuticals and Medical Devices, AMED (JP19mk0104117) (Y.Y.), the Research Project for Practical Applications of Regenerative Medicine, AMED (JP19bk0104095) (Y.Y.), the iPS Cell Research Fund (Y.Y., H.S., and T.H.) and the SECOM Science and Technology Foundation (Y.Y.).
Published: June 9, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2022.05.003.
Contributor Information
Hirohide Saito, Email: hirohide.saito@cira.kyoto-u.ac.jp.
Yoshinori Yoshida, Email: yoshinor@cira.kyoto-u.ac.jp.
Supplemental information
References
- Ahlgren U., Jonsson J., Jonsson L., Simu K., Edlund H. β-Cell-specific inactivation of the mouseIpf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–1768. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Burridge P.W., Matsa E., Shukla P., Lin Z.C., Churko J.M., Ebert A.D., Lan F., Diecke S., Huber B., Mordwinkin N.M., et al. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.J.H., Yang X., Don C.W., Minami E., Liu Y.W., Weyers J.J., Mahoney W.M., Van Biber B., Cook S.M., Palpant N.J., et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois N.C., Craft A.M., Sharma P., Elliott D.A., Stanley E.G., Elefanty A.G., Gramolini A., Keller G. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat. Biotechnol. 2011;29:1011–1018. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn T.B., Noreen H., Gillingham K., Maurer D., Ozturk O.G., Pruett T.L., Bray R.A., Gebel H.M., Matas A.J. Revisiting traditional risk factors for rejection and graft loss after kidney transplantation. Am. J. Transplant. 2011;11:2132–2143. doi: 10.1111/j.1600-6143.2011.03640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson L. The small RNA miR-375 - a pancreatic islet abundant miRNA with multiple roles in endocrine beta cell function. Mol. Cell. Endocrinol. 2017;456:95–101. doi: 10.1016/j.mce.2017.02.043. [DOI] [PubMed] [Google Scholar]
- Endo K., Hayashi K., Saito H. High-resolution identification and separation of living cell types by multiple microRNA-responsive synthetic mRNAs. Sci. Rep. 2016;6:21991. doi: 10.1038/srep21991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo K., Hayashi K., Saito H. Numerical operations in living cells by programmable RNA devices. Sci. Adv. 2019;5:eaax0835. doi: 10.1126/sciadv.aax0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y., Hirosawa M., Hayashi K., Hatani T., Yoshida Y., Yamamoto T., Saito H. A versatile and robust cell purification system with an RNA-only circuit composed of microRNA-responsive ON and OFF switches. Sci. Adv. 2022;8:eabj1793. doi: 10.1126/sciadv.abj1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi S., Miki K., Takaki T., Okubo C., Hatani T., Chonabayashi K., Nishikawa M., Takei I., Oishi A., Narita M., et al. Enhanced engraftment, proliferation, and therapeutic potential in heart using optimized human iPSC-derived cardiomyocytes. Sci. Rep. 2016;6:19111. doi: 10.1038/srep19111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatani T., Funakoshi S., Deerinck T.J., Bushong E.A., Kimura T., Takeshima H., Ellisman M.H., Hoshijima M., Yoshida Y. Nano-structural analysis of engrafted human induced pluripotent stem cell-derived cardiomyocytes in mouse hearts using a genetic-probe APEX2. Biochem. Biophys. Res. Commun. 2018;505:1251–1256. doi: 10.1016/j.bbrc.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatani T., Miki K., Yoshida Y. Induction of human induced pluripotent stem cells to cardiomyocytes using embryoid bodies. Methods Mol. Biol. 2018;1816:79–92. doi: 10.1007/978-1-4939-8597-5_6. [DOI] [PubMed] [Google Scholar]
- Hattori F., Fukuda K. Strategies for replacing myocytes with induced pluripotent stem in clinical protocols. Transplant. Rev. 2012;26:223–232. doi: 10.1016/j.trre.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Karlsson E. The role of pancreatic chromogranins in islet physiology. Curr. Mol. Med. 2001;1:727–732. doi: 10.2174/1566524013363294. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Protze S.I., Laksman Z., Backx P.H., Keller G.M. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell. 2017;21:179–194.e4. doi: 10.1016/j.stem.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Matsuura S., Ono H., Kawasaki S., Kuang Y., Fujita Y., Saito H. Synthetic RNA-based logic computation in mammalian cells. Nat. Commun. 2018;9:4847. doi: 10.1038/s41467-018-07181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki K., Endo K., Takahashi S., Funakoshi S., Takei I., Katayama S., Toyoda T., Kotaka M., Takaki T., Umeda M., et al. Efficient detection and purification of cell populations using synthetic MicroRNA switches. Cell Stem Cell. 2015;16:699–711. doi: 10.1016/j.stem.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Nakanishi H., Saito H. Mammalian gene circuits with biomolecule-responsive RNA devices. Curr. Opin. Chem. Biol. 2019;52:16–22. doi: 10.1016/j.cbpa.2019.04.013. [DOI] [PubMed] [Google Scholar]
- Ohno H., Akamine S., Saito H. Synthetic mRNA-based Systems in mammalian cells. Adv Biosyst. 2020;4:e1900247. doi: 10.1002/adbi.201900247. [DOI] [PubMed] [Google Scholar]
- Ohnuki M., Takahashi K., Yamanaka S. Generation and characterization of human induced pluripotent stem cells. Current Protoc. Stem Cell Biol. 2009;Chapter 4 doi: 10.1002/9780470151808.sc04a02s9. Unit 4A 2. [DOI] [PubMed] [Google Scholar]
- Osborn L., Hession C., Tizard R., Vassallo C., Luhowskyj S., Chi-Rosso G., Lobb R. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- Protze S.I., Liu J., Nussinovitch U., Ohana L., Backx P.H., Gepstein L., Keller G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017;35:56–68. doi: 10.1038/nbt.3745. [DOI] [PubMed] [Google Scholar]
- Rieck S., Bankaitis E.D., Wright C.V. Lineage determinants in early endocrine development. Semin. Cell Dev. Biol. 2012;23:673–684. doi: 10.1016/j.semcdb.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanganalmath S.K., Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 2013;113:810–834. doi: 10.1161/circresaha.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba Y., Gomibuchi T., Seto T., Wada Y., Ichimura H., Tanaka Y., Ogasawara T., Okada K., Shiba N., Sakamoto K., et al. Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature. 2016;538:388–391. doi: 10.1038/nature19815. [DOI] [PubMed] [Google Scholar]
- Tohyama S., Hattori F., Sano M., Hishiki T., Nagahata Y., Matsuura T., Hashimoto H., Suzuki T., Yamashita H., Satoh Y., et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Toyoda T., Mae S.I., Tanaka H., Kondo Y., Funato M., Hosokawa Y., Sudo T., Kawaguchi Y., Osafune K. Cell aggregation optimizes the differentiation of human ESCs and iPSCs into pancreatic bud-like progenitor cells. Stem Cell Res. 2015;14:185–197. doi: 10.1016/j.scr.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Virani S.S., Alonso A., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Delling F.N., et al. Heart disease and stroke statistics-2020 update: a report from the American heart association. Circulation. 2020;141:e139–e596. doi: 10.1161/cir.0000000000000757. [DOI] [PubMed] [Google Scholar]
- Wang H., Maechler P., Ritz-Laser B., Hagenfeldt K.A., Ishihara H., Philippe J., Wollheim C.B. Pdx1 level defines pancreatic gene expression pattern and cell lineage differentiation. J. Biol. Chem. 2001;276:25279–25286. doi: 10.1074/jbc.M101233200. [DOI] [PubMed] [Google Scholar]
- Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Warren L., Manos P.D., Ahfeldt T., Loh Y.H., Li H., Lau F., Ebina W., Mandal P.K., Smith Z.D., Meissner A., et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Soonpaa M.H., Adler E.D., Roepke T.K., Kattman S.J., Kennedy M., Henckaerts E., Bonham K., Abbott G.W., Linden R.M., et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequence data are available at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE153714.