

Abstract
The UCSC Genome Browser has been an important tool for genomics and clinical genetics since the sequence of the human genome was first released in 2000. As it has grown in scope to display more types of data it has also grown more complicated. The data, which are dispersed at many locations worldwide, are collected into one view on the Browser, where the graphical interface presents the data in one location. This supports the expertise of the researcher to interpret variants in the genome. Because the analysis of Single Nucleotide Variants (SNVs) and Copy Number Variants (CNVs) require interpretation of data at very different genomic scales, different data resources are required. We present here several Recommended Track Sets designed to facilitate the interpretation of variants in the clinic, offering quick access to datasets relevant to the appropriate scale.
Keywords: Genome Browser, variant interpretation, recommended track set, clinical genetics, Copy Number Variant, Single Nucleotide Variant, CNV, SNV, database
Introduction:
The use of high-throughput technologies for genome sequencing of clinical cases has drastically increased the amount of genomic data produced, making the consistent annotation and organization of data for interpreting genetic variation a matter of critical importance. The large number of genetic variants and associated information continuously discovered in the human genome reside in many data repositories and in scientific publications. Assessing the potential pathogenic effect of a variant on an individual is a complex interpretation process. Automated processes exist for evaluating the many variants in a sample (Hinrichs et al., 2016, McLaren et al., 2016), but ultimately, the candidates identified by such a process must be evaluated manually. The recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) as well as the ClinGen Sequence Variant Interpretation Working Group have become the standard guideline for clinical interpretation of sequence variants in many laboratories worldwide (Richards et al., 2015)(Riggs et al., 2020). They direct that the pathogenicity of a genetic variant must be determined by the body of evidence of genomic data extracted from multiple sources of information. However, the great diversity of data sources, annotations, and limited standardization of terminologies, makes access to and correct use of genomic information challenging.
The UCSC Genome Browser at http://genome.ucsc.edu, hosts a large collection of annotations from multiple data sources and conveniently displays the data as a series of “tracks” aligned with the human genome sequence (Lee et al., 2020). These tracks are updated regularly to keep pace with changes at the underlying data resource and provide sequence, disease-specific, population, and functional information to interpret novel sequence variants, as established by the current guidelines. Based on the evidence criteria and the track data, we have created pre-configured groups of tracks for specific types of analysis. Together, these Recommended Track Sets assist in the interpretation of small variants in coding regions (Clinical SNVs Track Set), large structural variants (Clinical CNVs Track Set), and non-coding variants (Non-coding SNVs Track Set) in Mendelian disease. The new feature provides rapid access to relevant and appropriately configured tracks, allowing visualization and easy comparison of data from multiple sources and supports our clinical users in understanding the relationship between genetic variants and human disease in the course of testing patient specimens.
The UCSC Recommended Track Sets Feature
The UCSC Recommended Track Sets each creates a Browser window with visible groups of tracks that have been selected and pre-configured for a different user scenario. They each allow quick configuration of the Genome Browser graphical display to perform the indicated analysis. The feature, implemented on the hg19/GRCh37 human genome assembly, is accessed from the top blue bar main menu under ‘Genome Browser’ > ‘Recommended Track Sets’. A mouse click on the track set name in the resulting popup box updates the graphical display, replacing existing tracks with tracks useful for interpreting variation data. A button appears to the right of the Genome Assembly information title with the name of the selected track set, from where it is possible to switch between tracks sets as needed or return to the default track collection without changing the locus (Figure 1).
Figure 1: Launching Recommended Track Sets.
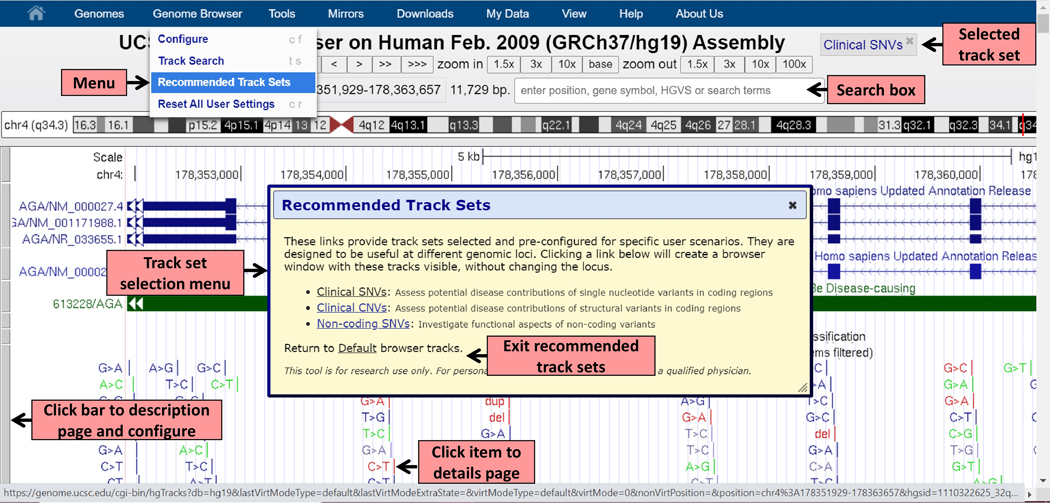
The dropdown menu from the navigation bar at the top allows users to open the Recommended Track Sets selection menu. The search box allows users to navigate to desired genomic locations. Tracks can be configured by clicking on the grey bar to the left of a track. Once a track set is selected, a new button, “Selected track set” appears, identifying the selected track set, from where it is also possible to reopen the selection menu and switch between tracks sets or return to the default tracks without changing the locus. Clicking on a track item opens a page with item-specific information.
From the search box above the Genome Browser track display, the particular genomic position can be specified in a variety of formats (Figure 1), by typing in coordinate ranges, gene names or accession IDs, or using HGVS terms (den Dunnen et al., 2016). HGVS examples include NM_000310.4(PPT1):c.271_287del17insTT, NM_198056.3:c.1654G>T, BRCA1 Ala744Cys. Additional explanation and examples are available here: https://genome.ucsc.edu/goldenPath/help/query.html.
Although the composition of the Recommended Track Sets has been pre-configured by UCSC, users can customize their browser view by adding other tracks selected from the menus below the browser graphic, removing, reordering or reconfiguring tracks to best suit their analysis needs. One option to easily locate additional tracks of interest is the ‘Configure’ option accessed from the main menu in the top blue bar in ‘Genome Browser’ (Figure 1). The ‘Configure’ option opens the full track editing interface that allows searching for tracks, selecting individual tracks or changing track display settings. Once any modification has been made, the “Selected track set” button is automatically removed, indicating that the configuration of the tracks has changed from the original set. The new configuration of tracks can be saved using the ‘Sessions’ tool for future reference or sharing with others. This feature may be accessed via the ‘My Data’ > ‘My Sessions’ link in the top blue navigation bar in any assembly.
The Composition of the Recommended Track Sets
Each Recommended Track Set is made up of key datasets that provide the evidence for variant interpretation; i.e., gene location and gene sequence, gene-disease associations, clinical variation, literature, population variation, and computational and predictive data (Richards et al., 2015). Special emphasis is given to curated datasets. A comprehensive list of all tracks is shown in Table 1.
Table 1.
Genome Browser Clinical SNVs and Clinical CNVs track sets recommended tracks grouped by the type of evidence for the GRCh37/hg19 assembly.
| Type of evidence | Genome Browser Track | Clinical SNVs | Clinical CNVs |
|---|---|---|---|
| Sequence and Genes | NCBI RefSeq genes, curated subset | x | x |
| NCBI RefSeq HGMD subset: transcripts with clinical variants in HGMD | x | x | |
| Locus Reference Genomic (LRG) Fixed Transcript Annotations | x | x | |
| Gene-disease Associations | OMIM Genes | x | x |
| GeneReviews | x | x | |
| ClinGen Gene-Disease Validity Classification | x | x | |
| Panel App | x | x | |
| Orphanet | x | x | |
| Clinical variation | ClinVar Short Variants < 50bp | x | |
| ClinVar SNVs submitted interpretations and evidence | x | ||
| ClinVar Copy Number Variants >= 50 bp | x | ||
| Leiden Open Variation Database (LOVD) short < 50 bp variants and insertions of any length | x | ||
| Leiden Open Variation Database (LOVD) long > 50 bp variants | x | ||
| UniProt/SwissProt amino acid substitutions | x | ||
| DECIPHER CNVs | x | ||
| Literature | Human Gene Mutation Database (HGMD) public variants | x | |
| AVADA Variants extracted from full text publications | x | ||
| Genomenon Mastermind Variants extracted from full text publications | x | ||
| Variation in the general population | Genome Aggregation Database (gnomAD) Exome Variants | x | |
| Genome Aggregation Database (gnomAD) Genome Variants | x | ||
| Genome Aggregation Database (gnomAD) Structural Variants All | x | ||
| NCBI dbVar Curated Common Structural Variants | x | ||
| NCBI dbVar Curated Conflict Structural Variants | x | ||
| Database of Genomic Variants: Structural Variation (CNV, Inversion, In/del) | x | ||
| Computational and predictive data | gnomAD Mean Proportion Expression Across Transcript Scores (pext) | x | |
| gomAD Predicted Loss of Function Constraint Metrics By Gene (pLI) | x | x | |
| gnomAD Predicted Missense Constraint Metrics By Gene (Z-scores) | x | ||
| gnomAD Predicted Regional Missense Constraint Metrics (O/E scores from ExAc Dataset) | x | ||
| ClinGen Dosage Sensitivity Map - Haploinsufficiency | x | ||
| ClinGen Dosage Sensitivity Map - Triplosensitivity | x | ||
| Haploinsufficiency predictions for genes from Decipher | x | ||
| UniProt SwissProt/TrEMBL Protein Annotations | x | ||
| CADD 1.6 Score for all possible single-basepair mutations | x | ||
| CADD 1.6 Score: Deletions | x | ||
| CADD 1.6 Score: Insertions | x | ||
| Vertebrate Multiz alignment and conservation (100 species) | x | ||
| Basewise conservation (phyloP) | x | ||
| Element conservation (phastCons) | x | ||
| REVEL prediction scores | x |
Each annotation track has an associated configuration page that contains a full description of the track (data sources, timestamp of the last data update, methods, credits), and configuration options to fine-tune the information displayed in the track. The description page is accessed from the grey bar to the left of a displayed track (Figure 1) or on the label for the track in the Track Controls section. Clicking a data item (line or box) within the track shows an associated details page. This page generally contains information specific to the item and related links to outside sites, including the data source website, genomic alignments, OMIM, PubMed, etc. (Figure 1).
The Recommended Track sets are primarily designed to provide information for the study of inherited disorders (primarily Mendelian) and should be used with caution. We must emphasize that track selection did not include any special data curation or validation practices. These are only research tools, and in no way should be used to inform medical decisions. UCSC presents these data for use by qualified professionals, and even such professionals should use caution in interpreting the significance of information found here.
Clinical SNV tracks
The Clinical SNV track set includes key data used to assess potential disease contributions of small variants in coding regions. This group is composed of a total of 29 tracks, 12 of which have been created specifically to address this topic (Table 1). Here we discuss the value of each and describe the display features that enhance their utility.
Gene tracks
The primary gene prediction track displays the NCBI RefSeq database (curated subset) (http://www.ncbi.nlm.nih.gov/RefSeq/) (Pruitt et al., 2005). Two more gene tracks, the LRG Transcripts from the Locus Reference Genomic (LRG) database (http://www.lrg-sequence.org) (MacArthur et al., 2014), and the RefSeq HGMD from the Human Gene Mutation Database public version (http://www.hgmd.cf.ac.uk ) (Cooper et al., 1998), show the most used/published reference transcripts for reporting clinically relevant variants to assist users to correctly annotate their variants (Figure 2).
Figure 2: Experimental components of SNV Recommended Track Set.
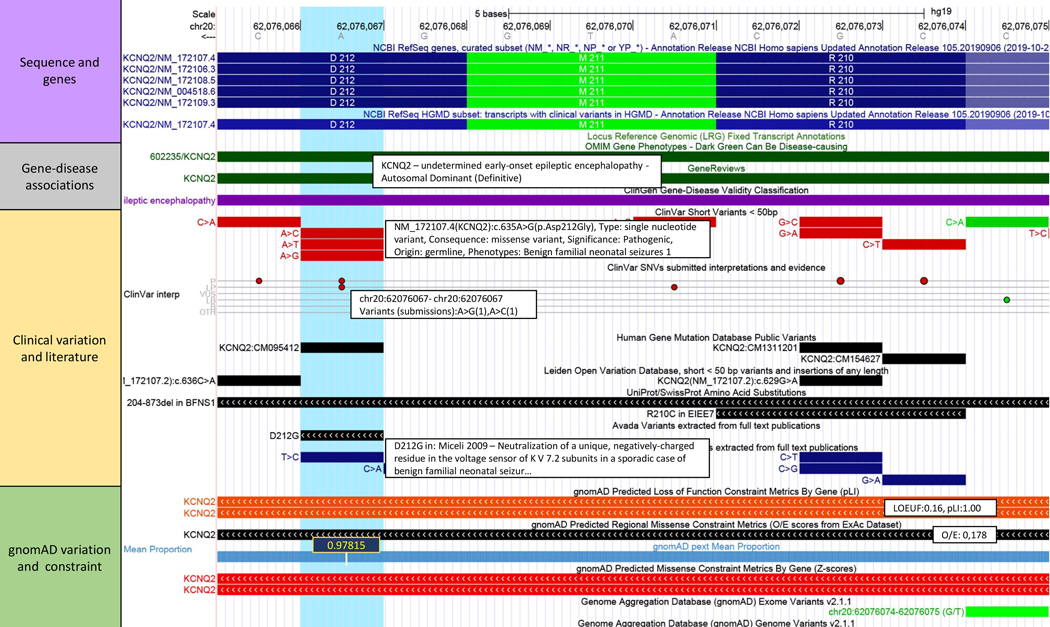
The variant record for NM_172107.4(KCNQ2):c.635A>G(p.Asp212Gly) (blue highlight) is shown, with UCSC Recommended Clinical SNVs track set applied: https://genome.ucsc.edu/s/abenet/HumanMutationFig2. This variant is located in the KCNQ2 gene. This gene has been ‘definitively’ associated with early-onset epileptic encephalopathy, an autosomal dominant inherited disease. The ClinVar Short Variants and SNVs tracks show that all three possible single nucleotide substitutions have been described at this position with pathogenic or likely pathogenic classifications. Each described variant leads to a missense change of the Asp residue. The same amino acid and nearby region is affected by mostly pathogenic variants. These and nearby variants show literature entries in the PubMed and HGMD database. The constraint metrics tracks show that the KCNQ2 gene has a high intolerance to both loss-of-function (LOEUF:0.16, pLI:1.00) and missense variation (Z:4.04), which is consistent with the number of classified pathogenic variants shown in this region of the gene. The pext score shows high isoform expression level across tissues (pext:0.97815). Other prediction data tracks are shown in Figure 3.
Gene-disease association tracks
We created three new tracks showing curated data from expert groups (ClinGen, PanelApp, and Orphanet). Together with the existing OMIM Genes and Gene Reviews data, they provide a wide catalogue of clinically validated gene-disease relationships with well documented penetrance, prevalence, and heterogeneity. Researchers can leverage these tracks in a research setting to carefully evaluate the strength of evidence supporting or refuting the assertion that a variant in a particular gene causes a particular condition.
The ClinGen Gene-Disease Validity track describes gene-disease associations in a semi-quantitative manner (https://clinicalgenome.org/) (Strande et al., 2017). The validated evidence (definitive, strong, moderate, limited, animal model only, no known disease relationship, disputed, or refuted) is shown on the mouseover. This track is part of the existing ClinGen collection: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&clinGenComp=pack&knownGene=pack&singleSearch=knownCanonical&position=PTEN.
The Orphanet track describes gene-disease relationships with extensive phenotype annotations using standardized terms from the Human Phenotype Ontology (HPO) (https://hpo.jax.org/app/) (Köhler et al., 2021). Disease prevalence (if available) are shown in the mouseover and phenotypes (HPO terms) are listed in the details page of each gene (Maiella et al., 2013), https://www.orpha.net/consor/cgi-bin/index.php.
The PanelApp track provides virtual gene panels related to human disorders (https://panelapp.genomicsengland.co.uk/) (Martin et al., 2019). Disease penetrance (if available) is shown in the mouseover and diagnostic-grade genes are colored in green, whereas genes that do not meet enough evidence criteria for disease association are amber or red.
We improved the existing OMIM Genes data by adding the mode of inheritance to the OMIM phenotypes (https://www.omim.org/) (Hamosh et al., 2005). The approved gene and alias symbols from the Human Gene Nomenclature Committee (HGNC), the associated phenotype, and mode of inheritance are shown on mouseover and in the details page for each OMIM gene.
Clinical variation and literature tracks
The new ClinVar Interpretations track, together with the ClinVar SNVs and LOVD short variants, show clinical evaluations and observations of variant changes at a position and nearby, and their effect at the amino acid level. Users can use these tracks to examine if novel variants cause the same amino acid change as an established pathogenic variant, a strong criterion for assertion of pathogenicity according to the ACMG-AMP Variant Interpretation Guidelines (Richards et al., 2015). These data can be overlain with the UniProt SwissProt/TrEMBL protein annotations to enable rapid identification of mutational hotspots and established functional domains in gene regions lacking benign variation, a moderate criterion for pathogenicity that has been often inconsistently applied (Amendola et al., 2020).
ClinVar Interpretations displays the submitted interpretations and evidence to disease from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) (Landrum et al., 2016). A new browser ‘bead’ display sorts variants by clinical significance and number of submissions (Figure 2). The mouseover on each ‘bead’ item summarizes all possible variants starting at that position and the number of submissions with the same classification. The details page lists the supporting information relevant to the clinical classification of the variant for each variant submission: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&clinvarSubLolly=pack&knownGene=pack&singleSearch=knownCanonical&position=FOXE3.
The variant interpretation process also includes revision of scientific and medical literature. Typically, publications are consulted for functional studies of variants and/or to find other individuals with a variant, along with associated phenotypes. The AVADA and MasterMind tracks show variants found in full-text publications by machine learning-based software and give a good overview of how often and in what context a variant has been published (Figure 2). Using these tracks in the browser’s ‘pack’ display mode, users can easily determine if variants and authors overlap in different publications, which is often an indication of duplication of data between studies (Kassirer and Angell, 1995). Also, the evaluation of functional evidence in published studies should be done with caution, as many older publications may not meet the specifications of the ACMG/AMP variant interpretation framework, showing only a moderate level of evidence (Brnich et al., 2019).
The details page of the AVADA Variants data (http://bejerano.stanford.edu/AVADA/) lists all abstracts where a variant and gene was found with external links to the publications (Birgmeier et al., 2020). Note that the variant nomenclature in the mouseover is the original text as the authors of the study described it and may not be valid HGVS.
The MasterMind Variants (https://mastermind.genomenon.com/) are displayed only if a variant has been indexed by the search engine; with links to Genomenon for more details such as references in which the variant was reported (Chun et al., 2020). The intensity of color for an item indicates the frequency of hits of the same amino acid change in papers.
AVADA has demonstrated an accuracy of 61% (Birgmeier et al., 2020) and MasterMind a sensitivity and specificity of more than 98% (Chun et al., 2020) of returned reference matches. Despite these consistent values, variants must be interpreted in the correct research or medical context and publications should always be reviewed manually. These tracks are part of the Variants in Papers collection: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&varsInPubs=pack&knownGene=pack&singleSearch=knownCanonical&position=FGF23.
Population variation tracks
The population datasets can be used to prove the presence or absence of a variant in control populations. We created separated tracks for the gnomAD Exomes and Genomes Variants v2.1 data set (https://gnomad.broadinstitute.org/), showing a more accurate representation of population-specific and population-maximal allele frequencies (PopmaxAF) (Figure 2). Both tracks include single nucleotide variants (SNVs) and small insertion/deletion variants (indels) up to 50 nucleotides in length: https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadSuper.
Clinically relevant variants are expected to be absent or have a very low frequency in population databases. To confirm absence of variants in the gnomAD data, users can enable the gnomAD Coverage tracks to ensure that the read depth in the raw data is sufficient for an accurate call at the variant location. They can also use the sub-population tables in the description pages of the variant to prove that the patient’s ethnicity is represented in the dataset. On the contrary, if the variant under study is present in the gnomAD database, the PopmaxAF can be used, together with the information about disease-specific prevalence, heterogeneity, and penetrance contained in the new Gene-disease association tracks (ClinGen, Orphanet, and PanelApp) to calculate the highest frequency of a possible disease-causing variant in the population (Whiffin et al., 2017). Even so, population data should be taken with caution, as most databases do not provide the phenotype of included individuals and individuals may carry low penetrance pathogenic variants or variants associated with recessive conditions.
Computational and predictive data tracks
One of the main components of the SNV Recommended Track Set is the new gnomAD Constraint data sets, which provide metrics for pathogenicity on a per-gene or -transcript level (Figure 2). These tracks represent the tolerance of a gene to inactivation and are useful for ascertaining if a truncating or missense variant has a deleterious effect on gene function (Karczewski et al., 2020). Constraint metrics can be overlain with the gnomAD pext (proportion of expression of exons across transcripts) track to identify tissue-specific expression patterns and uncover regions in haploinsufficient genes with no evidence of expression, where the presence of loss-of-function variants is unlikely to have a deleterious effect on protein function (Cummings et al., 2020).
The gnomAD LoF Constraint tracks display predicted loss-of-function (pLoF) metrics (probability of loss of function intolerance (pLI) and observed/expected (O/E) scores) and identify genes and transcripts subject to strong selection against heterozygous and homozygous loss-of-function variants (Karczewski et al., 2020). When evaluating how constrained a gene is, high pLI values are an indicator of loss-of-function intolerance, but it is also essential to consider the upper bound fraction of the O/E confidence interval (LOEUF). In contrast to pLI, low LOEUF values are indicative of strong intolerance: https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadPLI.
The gnomAD Missense Constraint and the Regional Missense Constraint (ExAc Dataset) tracks show observed vs. expected missense variant predicted metrics (Z-scores, O/E scores) and identify genes and parts of genes subjected to strong selection against this type of variant (i.e., low rate of benign missense variation).
The gnomAD pext track represents the proportion of expression of exons across transcripts (pext score) calculated from the Genotype Tissue Expression (GTEx) project data (Cummings et al., 2020)(GTEx Consortium et al., 2017). The pext scores quantify isoform expression for variants at the transcript level and can differentiate between weakly and highly conserved exons, an indicator of functional significance: https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadPext).
In-silico tools that predict the effect of a sequence variant at the nucleotide or amino acid level can predict the effect of missense changes, changes in non-coding sequences, or effects on splicing. These software tools tend to have low specificity and it is recommended to consider the combined predictions from multiple tools as a single line of evidence (Richards et al., 2015). Two new tracks, REVEL and CADD 1.6, show computed variant effect predictions that can be viewed in the mouseover of the track items. Together with the Vertebrate Multiz Alignment & Conservation and the UniProt SwissProt/TrEMBL Protein Annotations data, possible deleterious effects of variants that may impair protein functional domains can be deduced (Figure 3).
Figure 3: Computational components of SNV Recommended Track Set.
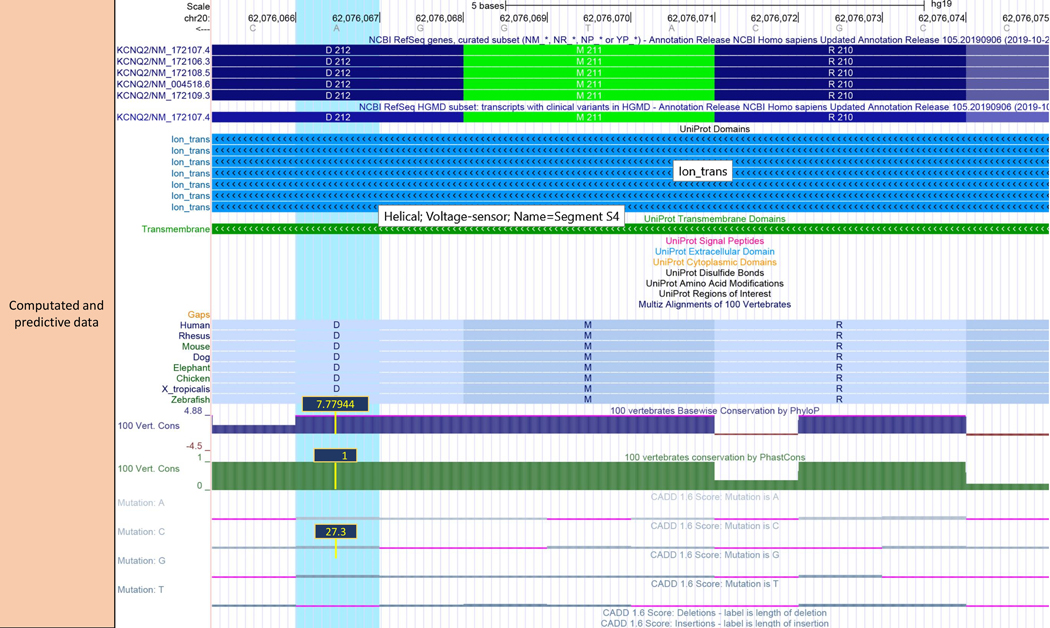
The UCSC Recommended Clinical SNVs computational and predictive tracks are shown for the variant NM_172107.4(KCNQ2):c.635A>G(p.Asp212Gly) (blue highlight): https://genome.ucsc.edu/s/abenet/HumanMutationFig3. The blue and green blocks of the Uniprot Annotations tracks at the top of the image show that the variant is located in a helical transmembrane domain with voltage-sensor function. Clicking on the green transmembrane item will show the details page where users can find more information about the function of this protein, which is a member of the potassium voltage-gated channel subfamily KQT, and outlinks to the Uniprot record. Mouseovers in PhyloP and PhastCons tracks show significant conservation scores at this nucleotide position (~1, 7.7) the aspartic acid residue which is fully conserved across all species of the Multiz 100-species alignment track. A CADD Phred score greater than 20 (C-score:27.3) (range 1 to 100) indicates that this substitution is predicted to be within the 1% most deleterious substitutions in the human genome. Taken together, the evidence extracted from these data suggest that this variant could have an inactivating effect on protein function. This is consistent with the lines of evidence from the clinical, population and constraint data, as described in Figure 2.
The Rare Exome Variant Ensemble Learner (REVEL) scores the pathogenicity of missense variants at the nucleotide level for every possible single nucleotide substitution, based on a combination of pre-computed scores from 13 individual tools including MutPred, FATHMM v2. 3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LRT, GERP++, SiPhy, phyloP, and phastCons (Ioannidis et al., 2016). Scores range from 0 to 1, with variants with higher scores being more likely to be pathogenic: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&revel=pack&knownGene=pack&singleSearch=knownCanonical&position=KCNE4
The Combined Annotation Dependent Depletion (CADD) scores the deleteriousness at nucleotide level for every possible single nucleotide mutation as well as for selected insertions and deletions genome wide. CADD provides a ranking from 0 to 100, with variants with scores over 30 are predicted to be the 0.1% most deleterious possible substitutions in the human genome (Rentzsch et al., 2019): https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&caddSuper=pack&knownGene=pack&singleSearch=knownCanonical&position=KCNE4.
The Clinical CNV tracks
The Clinical CNV track set displays data tracks useful for assessing potential disease contributions of copy-number variations (CNVs) and structural variants (SVs). This group comprises a total of 19 tracks (Table 1), 10 of which show data sets introduced for this project. While the genes and associated disease tracks are the same as those used for the small variants, other tracks focus specifically on CNV and SV data (Figure 4).
Figure 4: Recommended Track Set for CNVs.

The gene record on hg19 for human PTEN exons 6 to 8 (NM_000314.8) is shown with UCSC Recommended Clinical CNVs track set applied: https://genome.ucsc.edu/s/abenet/HumanMutationFig4. The LRG and NCBI RefSeq HGMD tracks show the preferred transcript used for clinical annotation. The PTEN gene shows definitive association with the autosomal dominant PTEN hamartoma tumor syndrome. It is predicted to be an haploinsufficiency gene with sufficient evidence for dosage pathogenicity (ClinGen Dosage score: 3, DECIPHER HI: 0.07%). The clinical variation tracks show pathogenic deletions spanning various exons (dark red) and duplications of benign or unknown significance (light and middle blue). Mouseovers in the ClinVar track show CNV details about clinical significance and phenotypes described for that variant. Thirty-five variants in the ClinVar track and 12 variants in the DECIPHER track spanning the browser window are shown collapsed (Merged items), simplifying the view when zoomed to single-exon events. New colors for new types of variants are shown in the gnomAD Structural Variants track (green: complex, purple: inversion, orange: insertion) and mouseovers in this track show population counts and frequencies.
The implementation of next-generation sequencing-based technologies in the clinical setting has increased the number of copy-number variants reported at the exon level. Clinical databases now contain structural variants ranging from single exons to entire chromosomes, so display of shorter copy-number variants can be compromised by the larger events spanning several megabases of sequence. As detailed in the next section, we have implemented new functionalities which improve the visualization of this type of variant in the Genome Browser. Also, we standardized terminology such as the chromosomal position, genes, and phenotypes (where data are available) in the mouseover of each variant allowing consistent comparison of CNVs and SVs across the variant tracks (Figure 4).
Clinical copy-number variation tracks
The Genome Browser tracks showing ClinVar CNVs, LOVD long variants and DECIPHER CNVs make the largest compendium of structural variant annotations associated with genetic disease reported in clinical studies. All three datasets depict a detailed evidence-based classification of variants and, if available, we show their associated phenotype information. As most CNVs are unique, the interpretation of CNVs depends substantially on the clinical indications and gene content (Riggs et al., 2012). The clinical associations to the genes contained in a CNV can be carefully analysed using the OMIM, Gene Reviews, PanelApp, Orphanet and ClinGen tracks (Figure 4).
We deprecated the ClinGen CNVs tracks by request of the ClinGen team, to avoid redundancy of data in different tracks. The ClinGen CNVs showed microarray data from the ISCA database (Miller et al., 2010) (Kaminsky et al., 2011). This database has now moved to dbVar’s nstd102 and in the Genome Browser can be found in the ClinVar Copy Number Variants (ClinVar CNVs) track, along with all updates: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&clinvar=pack&knownGene=pack&singleSearch=knownCanonical&position=PTEN. Variants from the nstd102 study can still be identified by the dbVar nssv identifier, which is shown to the left of any appropriate data item in the ClinVar CNVs track (in “pack” mode) and on the details page of the variants.
Population variation tracks
We added new structural variant data from population studies, gnomAD v.2.1 and dbVar nstd186 (Figure 4). Together with the Database of Genomic Gold Standard Variants track (http://dgv.tcag.ca/dgv/app/home) (MacDonald et al., 2014), they represent a large reference map of structural variants from genome sequencing and high-resolution microarray data from control individuals with no pathogenic condition. Users should consider that the use of different technologies results in differences in the reported size of identical CNVs and, therefore, the allele frequencies in these databases are not accurate. Furthermore, the majority of CNVs reported from large population studies have not been validated (Haraksingh et al., 2011).
The gnomAD Structural Variants track is the largest publicly-accessible data set providing allele frequencies of SVs across diverse global populations (Collins et al., 2020). Allele frequencies and variant details are shown in the mouseover of each variant (Figure 4). This track is part of the gnomAD track collection: https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadStructuralVariants.
The dbVar Structural Variants tracks show a collection of common variants with curated allele frequencies that have been detected in various project studies (GnomAD, 1000 Genomes, DECIPHER), and pooled in the dbVar nstd186 set (https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd186). Some variants in these studies, from individuals without genetic disease phenotype, overlap with clinical variants of ClinVar casting doubt on their clinical significance. This can result from individuals carrying low penetrance pathogenic variants or variants associated with recessive conditions. These CNVs deserve special attention and have been included in a separated subtrack (dbVar Curated Conflict Variants): https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&dbVarSv=pack&knownGene=pack&singleSearch=knownCanonical&position=EPC2.
Computational and predictive data
The evaluation of copy-number variants also involves the assessment of overlapping genomic content with a focus on genes/regions that may be subject to dosage sensitivity (i.e., increase or decrease in gene copy number is deleterious). It is important to consider the importance of dosage separately for each type of variant, as duplications in some regions may be benign, while deletions in the same position may have a pathogenic effect (Riggs et al., 2018). We created three new tracks presenting a curated map of dosage sensitivity, haploinsufficiency (HI) and triplosensitivity (TS), as follows:
The ClinGen Dosage Sensitivity track, part of the ClinGen track collection, displays curated evidence supporting or refuting dosage sensitivity of genes and genomic regions (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) (Riggs et al., 2012). Evidence for HI and TS is considered separately for each gene and genomic region (Figure 4). Scores corresponding to the strength of the available evidence for HI and TS are provided: https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&hideTracks=1&clinGenComp=pack&knownGene=pack&singleSearch=knownCanonical&position=SLC2A1.
The DECIPHER haploinsufficiency track shows HI predictions for human genes by integrating diverse genomic, evolutionary and functional properties that are characteristic of haploinsufficiency, followed by validation with independent experimental and clinical data (Huang et al., 2010): https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=decipherHaploIns.
These tracks enable the user to visualize single-gene CNVs that overlap with a dosage-sensitive region; or multigene CNVs in which at least one gene is known to be dosage-sensitive and are generally interpreted as “likely pathogenic” or “pathogenic” (Riggs et al., 2018). This tracks can be used together with the gnomAD pLI track to estimate the probability that a gene is LoF-haploinsufficient. However, users should be aware that this metric, based on observations of loss-of-function variants in the general population, may erroneously predict haploinsufficiency in genes in which a loss-of-function pathomechanism causes late-onset phenotypes, such as BRCA1 or LRRK2 (Greene, 1997) (Klein and Westenberger, 2012).
Other lines of evidence may be necessary for a comprehensive interpretation of SVs and CNVs that are not part of the Clinical CNVs Recommended Tracks. For example, the UniProt SwissProt/TrEMBL Protein Annotation tracks provide insight into what extent a deletion spans functional parts of proteins; the ENCODE Regulation tracks provide information on known and predicted non-coding elements of functional significance that may be present in the variant region, or the ClinVar small variants tracks may also show information for LoF variants within the region covered by the observed copy-number event. These tracks are linked to other sets of recommended tracks that can be easily accessed through the selection menu (Figure 1). When switching to another set of tracks, the browser window remains at the same locus, enabling a direct retrieval of information without the need of additional navigation. Users can also customize these tracks as desired and create a new session in ‘My Sessions’. In any case, the flexibility of this feature allows the user to work in a fast, comfortable and complete way.
Non-coding SNV tracks
The third Recommended Track Set, showing data for the evaluation of SNVs in non-protein-coding regions can be used in evaluating candidate SNVs in intergenic regions. The data for evaluating non-coding variants include histone modifications, DNAse hypersensitivity, ATAC-seq, transcription-factor binding, chromatin interactions, RNA binding, evolutionary conservation and others. Because of the paucity of data directly associating non-coding SNVs with disease, these variants do not usually result in definitive assertions of pathogenicity and we will not describe these tracks in detail here.
New display features for variants
In the course of building this utility, we have created new tracks, added new annotations to existing tracks, made secondary data more available on the graphic page, and improved the display of data. The software improvements for track visualization and usability of the Clinical SNVs and Clinical CNVs Recommended Track Sets are described here.
Separate view of short and long variants
We split the variants into two independent tracks according to the length of the variant. In general, the SNV tracks include substitutions, indels shorter than 50 bp, and insertions of any length, and the CNV tracks include copy-number variants (CNVs) of 50 bp or longer (Sudmant et al. 2015). This allows users to manage the two datasets independently in the ClinVar, LOVD, gnomAD and dbVar tracks.
Mouseovers
We have put together relevant biological and medical information for each of the track items in the mouseovers. Holding the mouseover a variant in the ClinVar, LOVD, or gnomAD tracks now shows HGVS annotation, type of variant, molecular consequence, clinical significance, population frequency, and the phenotypes with which the variant has been associated. The available information in a track will vary depending on repository data. The addition of information to mouseovers has simplified the comparison of data between tracks without requiring a click into the details page for each variant. The details pages still provide item-specific information and links to off-site repositories such as PubMed, ClinVar, gnomAD, and OMIM, among others.
A new type of mouseover was applied to tracks with a value associated with position (wiggle or bigWig format) showing calculated scores for each nucleotide position in the genome. This was implemented to show CADD 1.6, gnomAD pext, REVEL, PhyloP and PhastCons scores.
Configuration filters
Filters have been provided on the track configuration page to allow users to create subsets of data based on allele frequency, variation type, clinical significance, allele origin, and molecular consequence. This allows users to configure the variant display according to their specific needs. For example, one might show only variants in coding and splice sites when analyzing exome data, or focus on truncating variants that have been described at very low frequency in the general population, etc. These filters greatly simplify the visualization in tracks that are densely populated with data.
Variant colors
Short variants are colored by clinical significance in the ClinVar tracks (red for pathogenic/ likely pathogenic, dark blue for variant of uncertain significance, green for benign/ likely benign, light blue for conflicting, dark grey for not provided) or by annotation type in the gnomAD tracks (red for pLoF, yellow for missense, green for synonymous, and grey for others), following the conventions on the gnomAD browser.
Structural variants are colored by variant type in the ClinVar, DECIPHER, gnomAD and dbVar tracks (Figure 2). We added more color categories and we used three levels of color intensity to indicate the clinical significance of a variant. The lightest shade indicates “benign/likely-benign”, the darkest shade indicates “pathogenic/likely pathogenic”, and the medium shade indicates “other or no clinical significance has been defined” (https://genome.ucsc.edu/goldenPath/help/hgCnvColoring.html).
Collapse variant items
Structural variants are visually represented by blocks. The new “mergeSpannedItems” feature combines multiple annotations that extend through both sides of the browser window into a single item with the label “Merged [Number] items”, while displaying individually any data items whose start or end coordinates are in the window (Figure 2). Clicking the merged block reverts the display to the default view. A right-click then allows one to remerge the items. This feature reduces the number of block items in the track and dramatically improves the visualization of copy-number variants when viewed at the gene or exon level.
Future Plans
The UCSC Genome Browser team will continue to support the needs of clinical users. We will continue to keep pace with updates to important datasets and incorporate new types of data as they become available. For example, the coming years will see more massively parallel mutation impact assays using CRISPR, such as those conducted on BRCA1 (Findlay et al., 2019) and MSH2 (Jia et al., 2021). We intend to create more Recommended Track Sets addressing other analysis topics such as somatic variation and problematic sequence regions for variant interpretation. We plan to improve the clinical feature set by giving users more options to display only the transcripts most relevant to their work. We want to add links to ClinGen gene curation summaries and support phenotype searches to find relevant variants and genes that are associated with certain diseases. We also want to anticipate new variant-level functionality to allow users to manually curate and annotate their variants of interest. To meet provenance requirements in the clinical setting, we hope to add a “variant report” feature using the Table Browser (Karolchik et al., 2004) that generates a summary of complete details about all variants in all currently displayed tracks. A similar variation of this function, “gene report”, would allow the same type of report for all genes in the current view. We also intend to expand the Recommended Track Sets to include the hg38/GRCh38 assembly as our users continue to migrate to the newer version of the reference genome.
Outreach and contact information
In the past year the Genome Browser’s training team provided >20 seminars and workshops to help users take advantage of the latest features. Outreach is also supported by regular updates to the training documentation (https://genome.ucsc.edu/training/) and blog (http://genome.ucsc.edu/blog/), with videos (https://bit.ly/ucscVideos) and in-depth descriptions of new Browser features. The training documentation also includes information on how to submit a request for a workshop.
General contact information for the UCSC Genome Browser can be found on the website at https://genome.ucsc.edu/contacts.html, including information for accessing our email support list and the archive of previously answered mailing list questions. UCSC also maintains mirrors in Germany and Japan with the gracious assistance of the University of Bielefeld, Germany and the RIKEN Institute of Japan, respectively. Those sites can be found at https://genome-euro.ucsc.edu and https://genome-asia.ucsc.edu.
Data Availability Statement
No newly generated data are presented in this manuscript. All data shown in the Recommended Track Sets are provided by outside data generators. Furthermore, with the exception of OMIM, all data discussed here are downloadable from the UCSC downloads server, https://hgdownload.ucsc.soe.edu.
Acknowledgements
Thank you to all of our users for continuing to use our resource and suggest improvements. Thank you as well to all of the data providers responsible for the annotation that appears in the Browser; it would be an empty shell without your contributions. Finally, thank you to the support staff who keep the computers running and the lights on.
Funding
National Human Genome Research Institute [5U41HG002371 to A.B-P., H.C., M.H., W.J.K., R.M.K., L.R.N, J.N.G., B.J.R., K.R.R., D.S., A.S.Z., 1U41HG010972 to M.H., 5R01HG010329 to M.H., W.J.K.]; National Institutes of Health [U01MH114825 to W.J.K.]; Silicon Valley Community Foundation [2017-171531(5022) to, J.C., M.H., W.J.K., A.S.Z., 2018-194093 to M.D.; California Institute for Regenerative Medicine [GC1R-06673-C to J.C., M.H., W.J.K., B.T.L., M.L.S., A.S.Z., CIRM Match to UCSC Graduate Division to N.M.]; Center for Information Technology Research in the Interest of Society [2020-0000000026 to L.R.N., B.J.R.].
Footnotes
Conflict of interest statement. J.N.G., A.S.Z., D.S., K.R.R., B.J.R., L.R.N., C.M.L., H.C., J.C., R.M.K., M.H. and W.J.K. receive royalties from the sale of UCSC Genome Browser source code, LiftOver, GBiB, and GBiC licenses to commercial entities. W.J.K. owns Kent Informatics.
Web Resources
https://genome.ucsc.edu/s/abenet/HumanMutationFig2
https://genome.ucsc.edu/s/abenet/HumanMutationFig3
https://genome.ucsc.edu/s/abenet/HumanMutationFig4
https://genome.ucsc.edu/goldenPath/help/query.html
http://www.ncbi.nlm.nih.gov/RefSeq/
https://www.orpha.net/consor/cgi-bin/index.php.
https://panelapp.genomicsengland.co.uk/
https://www.ncbi.nlm.nih.gov/clinvar/
http://bejerano.stanford.edu/AVADA/
https://mastermind.genomenon.com/
https://gnomad.broadinstitute.org/
https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadSuper.
https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadPLI
https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadPext
http://dgv.tcag.ca/dgv/app/home
https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=gnomadStructuralVariants.
https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd186
https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/
https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg19&g=decipherHaploIns.
https://genome.ucsc.edu/goldenPath/help/hgCnvColoring.html
https://hgdownload.ucsc.soe.edu
https://genome.ucsc.edu/training/
References
- Amendola LM, Muenzen K, Biesecker LG, Bowling KM, Cooper GM, Dorschner MO, Driscoll C, Foreman A, Golden-Grant K, Greally JM, Hindorff L, Kanavy D, Jobanputra V, Johnston JJ, Kenny EE, McNulty S, Murali P, Ou J, Powell BC, Rehm HL, Jarvik GP (2020). Variant Classification Concordance using the ACMG-AMP Variant Interpretation Guidelines across Nine Genomic Implementation Research Studies. American journal of human genetics, 107(5), 932–941. 10.1016/j.ajhg.2020.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgmeier J, Deisseroth CA, Hayward LE, Galhardo L, Tierno AP, Jagadeesh KA, Stenson PD, Cooper DN, Bernstein JA, Haeussler M, & Bejerano G. (2020). AVADA: toward automated pathogenic variant evidence retrieval directly from the full-text literature. Genetics in medicine: official journal of the American College of Medical Genetics, 22(2), 362–370. 10.1038/s41436-019-0643-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, Tavtigian SV, Wright MW, Harrison SM, Biesecker LG, Berg JS, & Clinical Genome Resource Sequence Variant Interpretation Working Group (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome medicine, 12(1), 3. 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, Watts NA, Solomonson M, O’Donnell-Luria A, Baumann A, Munshi R, Walker M, Whelan CW, Huang Y, Brookings T, Sharpe T, … Talkowski ME (2020). A structural variation reference for medical and population genetics. Nature, 581(7809), 444–451. 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Ball EV, & Krawczak M. (1998). The human gene mutation database. Nucleic acids research, 26(1), 285–287. 10.1093/nar/26.1.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, B. B., Karczewski KJ, Kosmicki JA, Seaby EG, Watts NA, Singer-Berk M, Mudge JM, Karjalainen J, Satterstrom FK, O’Donnell-Luria AH, Poterba T, Seed C, Solomonson M, Alföldi J, Genome Aggregation Database Production Team, Genome Aggregation Database Consortium, Daly MJ, & MacArthur DG (2020). Transcript expression-aware annotation improves rare variant interpretation. Nature, 581(7809), 452–458. 10.1038/s41586-020-2329-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunn LM, Nefcy DC, Scouten RW, Tarpey RP, Chauhan G, Lim MS, Elenitoba-Johnson K, Schwartz SA, & Kiel MJ (2020). Mastermind: A Comprehensive Genomic Association Search Engine for Empirical Evidence Curation and Genetic Variant Interpretation. Frontiers in genetics, 11, 577152. 10.3389/fgene.2020.577152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, et al. (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Human Mutation, 37(6):564–569. 10.1002/humu.22981 [DOI] [PubMed] [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, & Shendure J. (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature, 562(7726), 217–222. 10.1038/s41586-018-0461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene MH (1997). Genetics of breast cancer. Mayo Clinic proceedings, 72(1), 54–65. 10.4065/72.1.54 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, Biospecimen Collection Source Site—RPCI, Biospecimen Core Resource—VARI, Brain Bank Repository—University of Miami Brain Endowment Bank, Leidos Biomedical—Project Management, ELSI Study, Genome Browser Data Integration & Visualization—EBI, Genome Browser Data Integration & Visualization—UCSC Genomics Institute, University of California Santa Cruz, Lead analysts:, Laboratory, Data Analysis &Coordinating Center (LDACC):, NIH program management:, … Montgomery SB (2017). Genetic effects on gene expression across human tissues. Nature, 550(7675), 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, & McKusick VA (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic acids research, 33(Database issue), D514–D517. 10.1093/nar/gki033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraksingh RR, Abyzov A, Gerstein M, Urban AE, & Snyder M. (2011). Genome-wide mapping of copy number variation in humans: comparative analysis of high resolution array platforms. PloS one, 6(11), e27859. 10.1371/journal.pone.0027859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Raney BJ, Speir ML, Rhead B, Casper J, Karolchik D, Kuhn RM, Rosenbloom KR, Zweig AS, Haussler D, Kent WJ. UCSC Data Integrator and Variant Annotation Integrator. Bioinformatics. 2016. May 1;32(9):1430–2. 10.1093/bioinformatics/btv766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, & Hurles ME (2010). Characterising and predicting haploinsufficiency in the human genome. PLoS genetics, 6(10), e1001154. 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, … Sieh W. (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American journal of human genetics, 99(4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Burugula BB, Chen V, Lemons RM, Jayakody S, Maksutova M, & Kitzman JO (2021). Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. American journal of human genetics, 108(1), 163–175. 10.1016/j.ajhg.2020.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, … Martin CL (2011). An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genetics in medicine: official journal of the American College of Medical Genetics, 13(9), 777–784. 10.1097/GIM.0b013e31822c79f9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, & Kent WJ (2004). The UCSC Table Browser data retrieval tool. Nucleic acids research, 32(Database issue), D493–D496. 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassirer J. & Angell M. (1995). Redundant Publication: A Reminder. New England Journal of Medicine, 333:449–450. 10.1056/nejm199508173330709 [DOI] [PubMed] [Google Scholar]
- Klein C, & Westenberger A. (2012). Genetics of Parkinson’s disease. Cold Spring Harbor perspectives in medicine, 2(1), a008888. 10.1101/cshperspect.a008888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, Danis D, Balagura G, Baynam G, Brower AM, Callahan TJ, Chute CG, Est JL, Galer PD, Ganesan S, Griese M, Haimel M, Pazmandi J, Hanauer M, Harris NL, … Robinson PN (2021). The Human Phenotype Ontology in 2021. Nucleic acids research, 49(D1), D1207–D1217. 10.1093/nar/gkaa1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, & Maglott DR (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research, 44(D1), D862–D868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Barber GP, Casper J, Clawson H, Diekhans M, Gonzalez JN, Hinrichs AS, Lee BT, Nassar LR, Powell CC, Raney BJ, Rosenbloom KR, Schmelter D, Speir ML, Zweig AS, Haussler D, Haeussler M, Kuhn RM, & Kent WJ (2020). UCSC Genome Browser enters 20th year. Nucleic acids research, 48(D1), D756–D761. 10.1093/nar/gkz1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur JA, Morales J, Tully RE, Astashyn A, Gil L, Bruford EA, Larsson P, Flicek P, Dalgleish R, Maglott DR, & Cunningham F. (2014). Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence variants. Nucleic acids research, 42(Database issue), D873–D878. 10.1093/nar/gkt1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, & Scherer SW (2014). The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic acids research, 42(Database issue), D986–D992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiella S, Rath A, Angin C, Mousson F, & Kremp O. (2013). Orphanet et son réseau: où trouver une information validée sur les maladies rares [Orphanet and its consortium: where to find expert-validated information on rare diseases]. Revue neurologique, 169 Suppl 1, S3–S8. 10.1016/S0035-3787(13)70052-3 [DOI] [PubMed] [Google Scholar]
- Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, Leong I, Smith KR, Gerasimenko O, Haraldsdottir E, Thomas E, Scott RH, Baple E, Tucci A, Brittain H, de Burca A, Ibañez K, Kasperaviciute D, Smedley D, Caulfield M, McDonagh EM (2019). PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nature genetics, 51(11), 1560–1565. 10.1038/s41588-019-0528-2 [DOI] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. (2016) The Ensembl Variant Effect Predictor. The Ensembl Variant Effect Predictor. Genome Biol 17, 122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, … Ledbetter DH (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American journal of human genetics, 86(5), 749–764. 10.1016/j.ajhg.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, & Maglott DR (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research, 33(Database issue), D501–D504. 10.1093/nar/gki025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic acids research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, & ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine: official journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Andersen EF, Cherry AM et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22, 245–257 (2020). 10.1038/s41436-019-0686-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Church DM, Hanson K, Horner VL, Kaminsky EB, Kuhn RM, Wain KE, Williams ES, Aradhya S, Kearney HM, Ledbetter DH, South ST, Thorland EC, & Martin CL (2012). Towards an evidence-based process for the clinical interpretation of copy number variation. Clinical genetics, 81(5), 403–412. 10.1111/j.1399-0004.2011.01818.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Nelson T, Merz A, Ackley T, Bunke B, Collins CD, Collinson MN, Fan YS, Goodenberger ML, Golden DM, Haglund-Hazy L, Krgovic D, Lamb AN, Lewis Z, Li G, Liu Y, Meck J, Neufeld-Kaiser W, Runke CK, Sanmann JN, … Martin CL (2018). Copy number variant discrepancy resolution using the ClinGen dosage sensitivity map results in updated clinical interpretations in ClinVar. Human mutation, 39(11), 1650–1659. 10.1002/humu.23610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, Wright MW, Milko LV, Cherry JM, Giovanni MA, Murray MF, O’Daniel JM, Ramos EM, Santani AB, Scott AF, Plon SE, … Berg JS (2017). Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. American journal of human genetics, 100(6), 895–906. 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant P, Rausch T, Gardner E. … Corbel J. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin N, Minikel E, Walsh R. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 19, 1151–1158 (2017). 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No newly generated data are presented in this manuscript. All data shown in the Recommended Track Sets are provided by outside data generators. Furthermore, with the exception of OMIM, all data discussed here are downloadable from the UCSC downloads server, https://hgdownload.ucsc.soe.edu.