

Abstract
Reduced muscle tone, muscle weakness, and physical fatigue can impact considerably on quality of life for children with neurofibromatosis type 1 (NF1). Human muscle biopsies and mouse models of NF1 deficiency in muscle show intramyocellular lipid accumulation, and preclinical data have indicated that L‐carnitine supplementation can ameliorate this phenotype. The aim of this study is to examine whether daily L‐carnitine supplementation is safe and feasible, and will improve muscle strength and reduce fatigue in children with NF1. A 12‐week Phase 2a trial was conducted using 1000 mg daily oral levocarnitine tartrate supplementation. Recruited children were between 8 and 12 years old with a clinical diagnosis of NF1, history of muscle weakness and fatigue, and naïve to L‐carnitine. Primary outcomes were safety (self‐reporting, biochemical testing) and compliance. Secondary outcomes included plasma acylcarnitine profiles, functional measures (muscle strength, long jump, handwriting speed, 6‐minute‐walk test [6MWT]), and parent‐reported questionnaires (PedsQL™, CBCL/6–18). Six children completed the trial with no self‐reported adverse events. Biochemical tests for kidney and liver function were normal, and the average compliance was 95%. Plasma acylcarnitine levels were low, but within a range not clinically linked to carnitine deficiency. For strength measures, there was a mean 53% increase in dorsiflexion strength (95% confidence interval [CI] 8.89–60.75; p = 0.02) and mean 66% increase in plantarflexion strength (95% CI 12.99–134.1; p = 0.03). In terms of muscle performance, there was a mean 10% increase in long jump distance (95% CI 2.97–16.03; p = 0.01) and 6MWT distance (95% CI 5.88–75.45; p = 0.03). Comparison with the 1000 Norms Project data showed a significant improvement in Z‐score for all of these measures. Parent reports showed no negative impact on quality of life, and the perceived benefits led to the majority of individuals remaining on L‐carnitine after the study. Twelve weeks of L‐carnitine supplementation is safe and feasible in children with NF1, and a Phase 3 trial should confirm the efficacy of treatment.
Keywords: children, fatigue, L‐carnitine, muscle weakness, neurofibromatosis type 1, NF1
1. INTRODUCTION
Neurofibromatosis type 1 (NF1) is the most common autosomal dominant genetic disorder, with a birth incidence of 1:3000 globally (Friedman, 2002). NF1 is caused by inactivating mutations in the NF1 gene located on chromosome 17q11.2 (Shen et al., 1996). The NF1 gene encodes neurofibromin, a RAS‐specific GTPase activating protein that modulates the biological activity of RAS proteins (Shen et al., 1996), and thus NF1 is classified as a tumor suppressor gene. While tumors are often the focus of clinical management at all stages of life, children with NF1 can be challenged by reductions in lean tissue mass, global muscle weakness, and problems in fine and gross motor functioning (Cornett et al., 2015; Dulai et al., 2007; Summers et al., 2015). They also express higher levels of physical and cognitive fatigue (Vassallo et al., 2020). In a study of self‐concept, approximately 30% of children and adolescents with NF1 reported a low self‐concept for physical and sporting abilities (Barton & North, 2007). There are currently no effective interventions for managing the physical limitations associated with NF1.
Summers et al. (2018) showed that double inactivation of Nf1 in murine muscle leads to intramyocellular lipid accumulation, which was also observed in NF1 patient muscle biopsies Summers et al., 2018). This phenotype was reminiscent of metabolic myopathies, a series of conditions that are often managed by L‐carnitine supplementation and/or dietary enrichment with medium‐chain fatty acids. Treatment of the Nf1 Prx1 −/− mouse with this intervention led to a decrease in the accumulation of long‐chain fats in the muscle, leading to a 45% increase in grip strength following a 12‐week treatment.
L‐carnitine is a vital molecular component of several energy producing pathways (Bremer, 1983). Greater than 95% of the body's total carnitine is localized in skeletal muscle, where it is necessary for the transport of long‐chain fatty acids through the mitochondrial membrane for beta‐oxidation (Stephens et al., 2007). Normally, the body's requirements for carnitine are met by the consumption of meat, but endogenous synthesis and increased renal absorption efficiency can contribute to whole‐body carnitine homeostasis. Impairments in L‐carnitine synthesis, transport or metabolism can result in primary or secondary deficiencies, which can in turn lead to elevated levels of intramyocellular lipid in muscle biopsies (Vasiljevski et al., 2018). Carnitine deficiency often results in muscle weakness and increased physical fatigue.
L‐carnitine supplementation is frequently recommended to patients with carnitine deficiency syndromes. Primary carnitine deficiency responds dramatically to oral carnitine therapy, with a complete reversal of clinical symptoms within a month (Al‐sharefi & Bilous, 2015; Tomlinson et al., 2018). Patients with other disorders that feature a secondary carnitine deficiency, such as kidney disease and dialysis patients, and very long chain acyl‐CoA dehydrogenase deficiency can also receive benefits from carnitine replacement therapy (Ahmad, 2001; Touma et al., 2001).
L‐carnitine supplementation has never been examined as a clinical treatment for muscle weakness or physical fatigue in the context of NF1. Hence, this study represents the first proof‐of‐concept trial to examine compliance, safety, and efficacy of levocarnitine tartrate treatment in children with NF1‐associated muscle weakness and fatigue. While prior studies have tested L‐carnitine for pediatric and adult conditions, deficiencies in fatty acid metabolism in NF1 muscle identified by preclinical studies justify safety testing in this specific patient group. To explore evidence for a secondary carnitine deficiency or other metabolic deficit, patients were assessed in terms of their plasma acylcarnitine profile. Our hypotheses were that: (1) daily 1000 mg levocarnitine tartrate supplementation (two divided doses) in children with NF1‐associated muscle weakness and fatigue would be safe, feasible, and acceptable to families; (2) changes in strength and endurance measures may be detectable; and (3) plasma acylcarnitine profiling may show evidence of a secondary carnitine deficiency.
2. SUBJECTS AND METHODS
2.1. Study design and participants
This open‐label, single‐arm, single center, Phase 2a clinical trial was designed to assess the safety and compliance of L‐carnitine supplementation in children with NF1. The trial was registered on the Australian New Zealand Clinical Trials Registry with ACTRN number 12618002021257 (Study protocol: https://anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12618002021257). The study was approved and monitored by the Sydney Children's Hospital Network Human Research Ethics Committee (reference no. HREC/18/SCHN/288).
Participants were children between 8 and 12 years of age that fulfilled: (1) the National Institutes of Health Consensus Conference diagnostic criteria for NF1, (2) reported a history of muscle weakness and/or physical fatigue, and (3) were naïve to L‐carnitine supplementation. The clinic provides NF1 specialist services to children in the Greater Sydney Metropolitan Region. Prior to the initial assessment, a medical history of all participants was obtained from the parent(s), and the participant's medical file. Participants were excluded from the study if they met any of the following criteria: (1) severe cognitive impairment, (2) insufficient English, (3) seizures, (4) skeletal abnormalities, e.g. tibial bowing and pseudarthrosis, acute foot or lower limb injuries, e.g. fracture and ankle sprain, or (5) incapacity to comply with a research protocol, e.g. prolonged absence. Written informed consent was obtained from all parents and assent from children as developmentally appropriate.
2.2. Procedures
All participants were allocated a daily dose of 1000 mg Levocarnitine tartrate (Musashi, Vitaco Health Australia Pty Ltd). Hard capsules (500 mg) were consumed twice daily for 12 weeks. The families were instructed to provide the children with the capsules at breakfast and dinner time, roughly 10 h apart.
L‐carnitine is an over‐the‐counter nutraceutical supplement taken to improve fatty acid oxidation and energy production. L‐carnitine supplementation is used to treat primary carnitine deficiency. However, as L‐carnitine supplementation had never been previously clinically trialed in NF1, we started the first three participants 1 month apart. As there were no adverse events in these children, the remaining three participants were started on L‐carnitine supplementation at fortnightly intervals.
L‐carnitine was dispensed from the Pharmacy Department of The Children's Hospital at Westmead. Functional assessments and questionnaires were carried out at the Kids Research Clinical Research Centre, The Children's Hospital at Westmead. A trained clinical evaluator took all functional assessments in the same order at baseline (0 weeks) and end of study (12 weeks). Questionnaires were completed by the parents at baseline and 12‐weeks posttreatment. Blood was collected and analyzed by the Pathology Department of The Children's Hospital at Westmead. Blood samples were collected at baseline and 12‐weeks posttreatment, and urine was collected at 12 weeks.
Participants were given the option to continue L‐carnitine supplementation at their own cost after the study endpoint (12 weeks), and were followed up after 3 months. The participants were asked whether they chose to continue L‐carnitine treatment, and what their regimen was. They were also asked to complete the same set of questionnaires after this period.
2.3. Outcome measures
The primary outcome measures for this study were safety and compliance. Safety was primarily analyzed by adverse event self‐reporting. A weekly phone call was made to each participant's family to ensure any safety concerns or adverse events were expressed. Additionally, there were three in‐person consultations for each participant, which were scheduled prior to each functional assessment. Biochemical safety assessments included plasma liver function (Supporting Table 1), urine chemistry (Supporting Table 2), circulating triglycerides and cholesterols, and acylcarnitine profiling (Supporting Table 3). At the final consultation, participants returned any remaining capsules. The numbers of remaining capsules were counted to assess their compliance. The intervention was declared safe and feasible if: (1) no more than one of the six participants withdrew due to experiencing an adverse event attributable to treatment, and (2) at least four of the six participants were able to complete at least 75% of the prescribed dose of treatment and comply with study requirements.
Secondary outcomes were functional assessments, including body fat, measures of muscle strength (grip, dorsiflexion and plantarflexion), gait, power (long jump), fine motor function (hand writing speed test), gross motor function (6 minute‐walk‐test [6MWT]) and quality of life (Pediatric Quality of Life and Child Behavior Checklist for ages 6–18). Body fat was measured using the MC‐780MA Tanita Tokyo body composition analyzer. The MC‐780MA body composition analyzer divides the human body into five sections; left leg, right leg, trunk, left arm, and right arm, measuring impedance by a tetrapolar 8‐point tactile electrode at 50 kHz. Maximal isometric strength of three muscle groups involved in prime movements, including grip strength, ankle dorsiflexion, and plantarflexion were tested using hand‐held dynamometry by a trained clinical evaluator (Citec; CIT Tehcnics, Harren, the Netherlands). To meet the independence requirements for statistical analysis, the measurements from only the dominant limb were included for each participant, and this was kept consistent for every study visit (Menz, 2004). Three repetitions were performed per muscle group, and an average measurement of strength was determined from this. Gait was assessed by heel and toe walking, which was scored on a 3‐point scale of difficulty: “no,” “some,” and “yes” to further examine dorsiflexion and plantarflexion strength. Power was measured by a standing long jump on a padded mat. Fine motor endurance was evaluated by the Handwriting Speed Test (Wallen et al., 1996) that gives a raw score in letters per minute, and gross motor endurance by the 6MWT, which was completed barefoot on a point‐to‐point, 25‐m long, flat, straight, hard surfaced track. Quality of life was assessed by parent reported questionnaires, including the Pediatric Quality of Life (PedsQL™) Generic 4.0 and Neuromuscular (3.0) modules, and The Child Behavior Checklist for ages 6–18 or CBCL/6‐18 (©ASEBA 2020). The PedsQL™ Generic 4.0 module consists of physical, psychosocial, and total summary scores, and the PedsQL™ Neuromuscular 3.0 module consists of neuromuscular disease, communication, family resources, and neuromuscular total summary scores. Both are scored on a scale ranging from 0 to 100, with lower scores indicating worse health. The CBCL/6‐18 (©ASEBA 2020) consists of 113 questions, scored on a 3‐point Likert scale (0 = absent, 1 = occurs sometimes, 2 = occurs often) that tests for empirically based syndrome scales. To fall within the clinical range of a syndrome scale means that the parent‐reported results from that questionnaire indicate their child clinically manifest the syndrome.
2.4. Statistical analysis
We analyzed the treatment effect by calculating average % change from baseline compared to 12‐weeks posttreatment for all outcome measures for each participant (n = 6), and confidence intervals for improvement were also calculated. Spearman's rank‐order correlation efficiency was applied to determine a weight specific dosage effect. Participant functional assessments from baseline and 12‐weeks posttreatment were compared to reference data to generate Z‐score changes (negative Z‐score is indicative of motor impairment based on age and sex‐matched normative data). Hand‐dynamometry strength measures, long jump, and 6MWT were compared to reference data obtained from the 1000 Norms Project, an observational study investigating outcome measures of self‐reported health and physical function in 1000 healthy individuals aged 3 to 101 years (McKay et al., 2016). Handwriting speed was compared to normative data adapted from Cermak (1989) and Wechsler (1974). Height, weight, and body mass index (BMI) Z‐scores were generated using USCDC2000 reference data.
2.5. Role of the funding source
This study was funded by the Children's Tumor Foundation (US). The funding source had no involvement in the following: study design, collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
3. RESULTS
Between June and August 2019, a total of six participants were recruited at the Neurogenetics/Neuromuscular clinic located in The Children's Hospital at Westmead. All participants fulfilled clinical diagnostic criteria for NF1, a medical history of muscle weakness and physical fatigue and were naïve to L‐carnitine supplementation. Demographics and clinical characteristics for the six participants are summarized in Table 1. Four (67.7%) were male and two (33.3%) were female. The mean age was 10.7 years (SD 1.2), and BMI Z‐score was −0.32. All participants were assigned the predetermined daily dose of 1000 mg levocarnitine tartrate (n = 6). The mean calculated daily weight‐specific dose was 31.6 mg/kg/day (SD 10.5), based on weight measurements collected at baseline. All participants completed the predetermined treatment duration of 12 weeks. All bottles of L‐carnitine supplements were returned at the completion of the study.
TABLE 1.
Baseline characteristics of participants
| Participant number (#) | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | Mean (SD) | |
| Gender | Male | Female | Male | Male | Female | Male | NA |
| Age (year) | 11 | 10 | 9 | 12 | 12 | 10 | 10.7 (1.2) |
| Height (cm) | 135.0 | 150.9 | 127.6 | 143.8 | 157.2 | 123.5 | 139.7 (12.1) |
| Height Z‐score | −1.38 | 1.23 | −1.24 | −0.75 | 0.35 | −2.43 | −0.7 (1.2) |
| Weight (kg) | 28.8 | 42.8 | 21.5 | 35.2 | 56.5 | 25.8 | 35.1 (11.8) |
| Weight Z‐score | −1.45 | 0.81 | −2.34 | −0.80 | 1.14 | −1.44 | −0.7 (1.3) |
| BMI Z‐score | −0.80 | 0.54 | −2.54 | −0.39 | 1.19 | 0.11 | −0.3 (1.2) |
| Calculated dose (mg/kg/day) | 34.7 | 23.4 | 46.5 | 28.4 | 17.7 | 38.8 | 31.6 (10.5) |
| Treatment duration (weeks) | 12 | 12 | 12 | 12 | 12 | 12 | 12 (0) |
There were no side effects or adverse events reported throughout the duration of this study. Kidney and liver function were normal, and there were no clinically significant changes in circulating fats following 12 weeks of L‐carnitine supplementation. There were no withdrawals, and all participants completed ≥84% of the treatment course (calculated as a percentage; number of capsules taken/168*100), with a mean compliance of 95 ± 6.2%. Therefore, 1000 mg/daily levocarnitine tartrate treatment was deemed safe and feasible.
Acylcarnitine profiling revealed no abnormalities in plasma acylcarnitine levels (Table 2). Mean baseline levels of acylcarnitines included total carnitine 39.3 (Ref: 5–106, SD 9.2), free carnitine 31.8 (Ref: 3–60, SD 9.6), acetyl‐carnitine 6 (Ref: 2–39, SD 1.5), propionylcarnitine 0.4 (Ref: 0.12–0.97, SD 0.2), and isovalerylcarnitine 0.1 (Ref: 0.00–0.22, SD 0.1). Following 12 weeks of L‐carnitine supplementation, levels of acylcarnitines were total carnitine 54.5 (Ref: 5–106, SD 6.8), free carnitine 44.5 (Ref: 3–60, SD 5.6), acetyl‐carnitine 8.3 (Ref: 2–39, SD 1.8), propionylcarnitine 0.7 (Ref: 0.12–0.97, SD 0.3), and isovalerylcarnitine 0.2 (Ref: 0.00–0.22, SD 0.1).
TABLE 2.
Acylcarnitine profile test results were all within reference range. Total carnitine, free carnitine, acetyl‐carnitine, propionylcarnitine, and isovalerylcarnitine results are representatively displayed for each of the participants (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w). The mean and SD were calculated at baseline and 12 weeks of supplementation
| Procedure | Reference range | 1 | 2 | 3 | 4 | 5 | 6 | Mean (SD) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0w | 12w | 0w | 12w | 0w | 12w | 0w | 12w | 0w | 12w | 0w | 12w | 0w | 12 | ||
| Total carnitine (C1) | 5–106 | 30 | 42 | 48 | 64 | 31 | 54 | 54 | 54 | 32 | 53 | 41 | 60 | 39.3 (9.16) | 54.5 (6.83) |
| Free carnitine (C2) | 3–60 | 19 | 34 | 42 | 53 | 24 | 46 | 46 | 44 | 27 | 44 | 33 | 46 | 31.83 (9.62) | 44.5 (5.59) |
| Acetyl‐carnitine (C3) | 2–39 | 9 | 7 | 5 | 9 | 6 | 7 | 6 | 8 | 4 | 7 | 6 | 12 | 6.00 (1.53) | 8.33 (1.8) |
| Propionylcarnitine (C4) | 0.12–0.97 | 0.18 | 0.42 | 0.62 | 1.21 | 0.22 | 0.32 | 0.68 | 1.00 | 0.40 | 0.48 | 0.35 | 0.60 | 0.41 (0.19) | 0.67 (0.32) |
| Isovalerylcarnitine (C8) | 0.00–0.22 | 0.08 | 0.10 | 0.10 | 0.21 | 0.07 | 0.09 | 0.23 | 0.22 | 0.19 | 0.18 | 0.01 | 0.19 | 0.11 (0.08) | 0.17 (0.05) |
Bioelectrical impedance analysis was performed to assess the effect of L‐carnitine supplementation on body fat mass (kg) or amount (% body weight), due to its primary role in fat metabolism. There was a mean reduction in fat mass by 0.8% (SD 4.5, p = 0.76), and mean reduction in fat amount by 2.8% (SD 4.4, p = 0.21), neither statistically significant (Figure 1a). The average BMI Z‐score decreased from −0.32 at baseline to −0.41 at 12‐weeks posttreatment.
FIGURE 1.

Percentage change following 12 weeks of L‐carnitine supplementation. Percentage change from baseline group mean in (a) body fat, (b) strength measures, and (c) other functional outcomes, including long jump, 6 minute walk test (MWT), and handwriting speed test. n = 6 NF1 children, at 12 weeks one child did not complete the 6MWT due to abdominal cramping. Percentage change calculated by (12 weeks value – baseline value)/baseline value × 100. Data presented as group mean + SD. p‐Values were assessed by paired T test of baseline values and 12‐weeks posttreatment values. *p < 0.03. Each symbol denotes participants 1–6
Following 12 weeks of L‐carnitine supplementation, there was no significant improvement in grip strength (95% CI −2.49 to 10.15; p = 0.18); however, there was a mean 53% increase in dorsiflexion strength (95% CI 8.89–60.75; p = 0.02) and a mean 66% increase in plantarflexion strength (95% CI 12.99–134.1; p = 0.03) (Figure 1b). Standing long jump distance was significantly greater on average by 10% after the 12‐week treatment course (95% CI 2.97–16.03; p = 0.01) (Figure 1c). A mean 10% improvement was also observed for the 6MWT after 12‐weeks of L‐carnitine supplementation (95% CI 5.88–75.45; p = 0.03). To note, the 6MWT of participant two was excluded due to cramping during testing time. Furthermore, handwriting speed was increased by 15% on average; however, due to the considerable variability in participant performance, this failed to reach statistical significance (95% CI −7.38 to 20.24; p = 0.28) (Figure 1c).
Functional outcome measures collected throughout the study were compared to age and sex matched normative data. NF1 participants performed below average on every outcome measure at baseline, with the exception of participant six who generated a positive Z‐score for dorsiflexion strength and handwriting speed, and participants three and four who also had a positive Z‐score for handwriting speed (Figure 2). After 12 weeks of L‐carnitine supplementation, there was an average Z‐score improvement of dorsiflexion strength from −1.57 to −0.32 (95% CI 0.17–2.33; p = 0.03) and plantarflexion strength from −1.98 to −0.15 (95% CI 0.08–3.59; p = 0.04). Long jump Z‐score improved from −2.05 to −1.5 (95% CI 0.15–0.95; p = 0.02), and 6MWT from −3.2 to −2.38 (95% CI 0.34–1.3; p = 0.01) (Figure 2; Supporting Table 4).
FIGURE 2.

Z‐score analysis of patient outcome measures compared at baseline and 12‐weeks posttreatment to age and gender matched normative data. Z‐score comparison of (a) grip strength, (b) dorsiflexion strength, (c) plantarflexion strength, (d) long jump, (e) 6 minute walk (MWT), and (f) handwriting speed. (a–d, f) N = 6 NF1 children, (e) N = 5 NF1 children. Z‐score calculated by sample value – normative (age and gender matched) mean/SD. Data present as group mean + SD. p‐Values were assessed by paired T test. *p < 0.05 and **p < 0.01. Normative data were collected through the 1000 Norms project. n = 8 10‐year male, n = 8 10‐year female, n = 8 11‐year male, n = 8 12‐year male, n = 8 12‐year female, n = 10 9‐year male. Normative data were adapted from Cermak (1989) and Wechsler (1974) for (f). Dotted line at 0 represents where NF1 children would have a comparable Z‐score to age and sex matched normative data. Each symbol denotes participants 1–6
Comparison of PedsQL™ scores between baseline and 12‐weeks posttreatment showed a trend toward improvement on the physical health summary domain of the generic module (median 46.9, interquartile range [IQR] 42.2–60.9 to median 71.9, IQR 51.6–87.5) (Figure 3; Supporting Table 5). Due to varied parent perception when scoring their child, there was large variability in the starting scores resulting in statistical significance being unattainable for this outcome measure. Other domains, including psychosocial health summary, neuromuscular disease summary, communication summary, and family resources summary scores showed no significant differences suggesting that L‐carnitine supplementation does not interfere with participant's quality of life.
FIGURE 3.
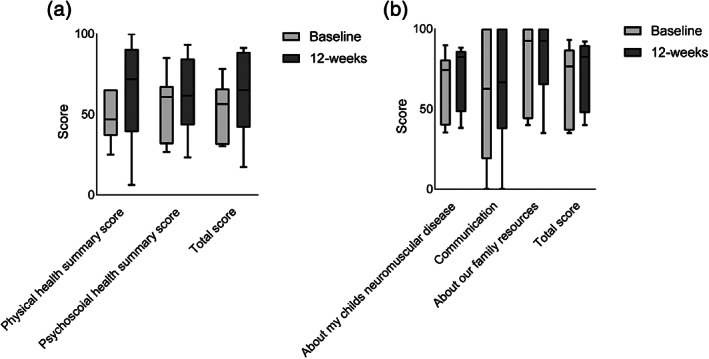
Box plots of PedsQL™ domain scores. (a) Generic 4.0 core module domains, including physical health summary, psychosocial health summary, and total scores and (b) neuromuscular 3.0 module domains, including about my child's neuromuscular disease, communication, about our family resources, and total scores. Data are presented as median and interquartile range at baseline and 12‐weeks posttreatment, n = 6
Participants were scored to the CBCL/6‐18 empirically based syndrome scales, including anxious/depressed, withdrawn/depressed, somatic complaints, social problems, thought problems, attention problems, rule‐breaking behavior, and aggressive behavior at baseline and 12‐weeks posttreatment. Majority of the children (4/6) fell within the clinical range of >2 syndrome scales at baseline. This was reduced to 2/6 children following 12‐weeks posttreatment (Supporting Table 6). However, no statistically significant differences could be detected due to sample size and interparticipant variability (Figure 4).
FIGURE 4.

CBCL/6‐18 syndrome scale scores. Raw scores of (a) anxious/depressed, (b) withdrawn/depressed, (c) somatic complaints, (d) social problems, (e) thought problems, (f) attention problems, (g) rule‐breaking, and (h) aggressive behavior. Data are presented for each participant at baseline and 12‐weeks posttreatment. Each symbol denotes participants 1–6 (n = 6)
All participants were followed up 3 months after completion of the trial. Three families continued supplementation with L‐carnitine. One family ceased treatment, however recommenced after fatigue symptoms returned. Two families did not continue treatment, with both families recognizing that NF1 cognitive and social behaviors were confounding their view of L‐carnitine supplementation for their child's muscle weakness and fatigue, and one family awaiting the published trial results to reconsider L‐carnitine.
4. DISCUSSION
The primary goal of this Phase 2a, proof‐of‐concept clinical trial was to establish the safety and feasibility of L‐carnitine as a therapeutic intervention for NF1 muscle weakness and physical fatigue. Critically, there were no side effects of L‐carnitine supplementation or adverse events reported and no abnormalities seen in liver and kidney function tests. These data are consistent with L‐carnitine being well tolerated in both children and adolescents, as it is part of a normal diet (albeit in lower amounts). The safety of L‐carnitine supplementation has previously been demonstrated in a number of different pediatric cohorts, including primary carnitine deficiency, autism spectrum disorder (ASD), and Rett syndrome (Ellaway et al., 1999; Geier et al., 2011; Kilic et al., 2012). However, this represents its first trial in a pediatric NF1 population. The high compliance rate of 95%, with no withdrawals suggesting that daily L‐carnitine supplementation is a well‐received therapeutic approach to treat muscle weakness and fatigue in NF1 children.
A daily dose of 1000 mg was predetermined for all participants. Future studies could better control for weight‐specific dose, which varied from 17.7 to 46.5 mg/kg/day in our cohort. Correlation analysis revealed no association between dose and functional outcomes, although this analysis had limited power. In prior pediatric studies, a 50 mg/kg/day dose of L‐carnitine supplementation is commonly practiced with high safety and proven efficacy and this likely represents a suitable starting dose for those naïve to the therapy. Anecdotal reports and clinical studies suggest that dose escalation could merely increase the incidence and severity of side effects, such as nausea and vomiting (Geier et al., 2011; Goin‐Kochel et al., 2019). There were clinical concerns that kidney or liver function had the potential to be particularly affected in NF1 children as these organs are sensitive to metabolic changes; however, no evidence for this was seen.
The use of L‐carnitine in NF1 children with muscle weakness who suffer from seizures remains a question for future study. Such individuals were excluded from recruitment due to contraindications with L‐carnitine, although these risks are poorly substantiated. A review of encephalopathy patients on valproic acid found no data suggesting that seizures were worsened by L‐carnitine supplementation (Zeiler et al., 2016). Therefore, future studies may include removing this as an exclusion criterion.
It has been suggested that NF1 could benefit from being described as a lipid‐storage metabolic myopathy (11). This concept is supported by the potential efficacy of L‐carnitine seen in this trial. The efficacy of L‐carnitine supplementation has already been demonstrated in some secondary mitochondrial disorders (Ellaway et al., 1999; Malaguarnera & Cauli, 2019; Zhang et al., 2010). This category of conditions includes spinal muscular atrophy (Harpey et al., 1990), Parkinson's disease (Park et al., 2018), Rett syndrome (Shulyakova et al., 2017), and ASD (Siddiqui et al., 2016). Further mechanistic studies and mitochondrial function analysis may reveal that NF1 has features of a secondary mitochondrial disorder, although it will be important to rule out a primary mitochondrial disease) in cases of NF1 by gene panel testing of oxidative phosphorylation‐related genes.
It was speculated that individuals enrolled in the study may show clinical deficiency in carnitine that could explain their muscle weakness and/or fatigue. While plasma acylcarnitine profiling revealed no clinical deficiency, many individuals were toward the lower end of the normal range. Comparison to a control cohort would be necessary to assess a secondary carnitine deficiency in NF1 children. More importantly, plasma carnitine concentrations do not always reflect the carnitine concentration observed in skeletal muscle. For example, hemodialysis patients frequently exhibit normal plasma carnitine, but have a low muscle carnitine concentration (Moorthy et al., 1983; Savica et al., 1983). However, muscle biopsies are highly invasive and would represent a major barrier to trial recruitment.
Our study has several limitations. Children with NF1 and muscle weakness and fatigue are a poorly defined subcohort yet represent the precise group that would be most engaged with finding a muscle‐targeted therapy. Thus, while this trial design did not use a randomized or placebo‐controlled design, it captures a stratification of the NF1 community most likely to adopt routine L‐carnitine supplementation. Moreover, this limitation was balanced by comparing individual cases before and after therapy, and to a normative group from the 1000 Norms Project data set. As previously noted, the lack of an L‐carnitine dose normalized to weight is another limitation, but one that was necessary within the practicalities of commercially available carnitine preparations. Indeed, this has proved advantageous to families looking to sustain their supplement use after the study endpoint. Finally, from the initial conception this study aimed to examine safety and compliance within a small cohort and was not designed to completely accommodate the genetic and phenotypic heterogeneity of children with NF1 and was not powered toward functional outcomes. Therefore, it is difficult to distinguish whether function improvement is an effect of the treatment, a placebo effect or the effect of a natural history. Further, sex‐specific effects/caveats of L‐carnitine supplementation may be a consideration. Hence, it was always anticipated that this would represent a precursor to a larger multicenter trial.
In summary, our data demonstrate that 1000 mg daily levocarnitine tartrate is safe and feasible in children with NF1‐associated muscle weakness and fatigue. Efficacy data suggest possible improvements in muscle strength and energy levels. However, we propose that a multisite, randomized, double‐blind placebo‐controlled trial with a consistent dosage regimen of 50 mg/kg/day would be the optimal approach to firmly establish the efficacy of L‐carnitine supplementation. A greater childhood age range, which could be compared to age‐, sex‐, height‐, and weight‐matched controls, would also improve future study data.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
EV, JB, PB, and AS contributed to the design of the study. EV, GD, AM, KJ, AB, and CM participated in recruitment and collection and assessment of data, and JB, JNB, and MM contributed the 1000 Norms data. EV did the statistical analysis, with support from Sydney Children's Hospital Network (SCHN) Statistician, Ms. Liz Barnes. All authors participated in the writing and editing of the manuscript.
Supporting information
Supplementary Table 1 Liver function test procedures with reference range and units
Supplementary Table 2: Urine chemistry tested with units
Supplementary Table 3: Acylcarnitines included in the acylcarnitine profile test with reference range and units
Supplementary Table 4: Absolute change of functional outcome measures for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w).
Supplementary Table 5: PedsQL domain scores. Scores are displayed for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w). 0 = low score and 100 = high score. Median (interquartile range) have been displayed to better represent the data.
Supplementary Table 6: CBCL syndrome scores. Scores are displayed for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w). Score reduction indicates normalization. C = clinical range, B = borderline clinical range.
ACKNOWLEDGMENTS
We would like to acknowledge the Sydney Children's Hospital Network (SCHN) Statistician, Ms. Liz Barnes for her support in statistical design and analysis. We would also like to acknowledge the Clinical Research Centre of Kids Research, and the Pharmacy, Pathology and Biochemical Genetics Departments of The Children's Hospital at Westmead who assisted with start‐up, day‐to‐day running, biochemical testing, and analysis of results. We acknowledge Sally Maspero for her assistance during recruitment. Further, we acknowledge the US Children's Tumor Foundation (CTF) for their ongoing financial support. Finally, we acknowledge the six families involved in the clinical trial. Without their engagement and active cooperation, this research would not have been possible. Funding for this clinical trial was from the US Children's Tumor Foundation (CTF) (https://www.ctf.org/) Clinical Research Award scheme (2018‐10‐001), with EV receiving stipend support from a Research Training Program (Australian Government) PhD scholarship.
Vasiljevski, E. R. , Burns, J. , Bray, P. , Donlevy, G. , Mudge, A. J. , Jones, K. J. , Summers, M. A. , Biggin, A. , Munns, C. F. , McKay, M. J. , Baldwin, J. N. , Little, D. G. , & Schindeler, A. (2021). L‐carnitine supplementation for muscle weakness and fatigue in children with neurofibromatosis type 1: A Phase 2a clinical trial. American Journal of Medical Genetics Part A, 185A:2976–2985. 10.1002/ajmg.a.62392
Funding information Research Training Program (Australian Government); US Children's Tumor Foundation (CTF), Grant/Award Number: 2018‐10‐001
DATA AVAILABILITY STATEMENT
Data collected for the study will not be made available to others.
REFERENCES
- Ahmad, S. (2001). L‐carnitine in dialysis patients. Seminars in Dialysis, 14(3), 209–217. [DOI] [PubMed] [Google Scholar]
- Al‐sharefi, A. , & Bilous, R. (2015). Reversible weakness and encephalopathy while on long‐term valproate treatment due to carnitine deficiency. BMJ Case Reports, 2015, bcr2015210727. 10.1136/bcr-2015-210727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, B. , & North, K. (2007). The self‐concept of children and adolescents with neurofibromatosis type 1. Child: Care, Health and Development, 33(4), 401–408. [DOI] [PubMed] [Google Scholar]
- Bremer, J. (1983). Carnitine—Metabolism and functions. Physiological Reviews, 63(4), 1420–1480. [DOI] [PubMed] [Google Scholar]
- Cermak, S. (1989). Norms and Scores. In: Developing Norm‐Referenced Standardized Tests (Chapter 5, pp. 91–123). England: Hawthorne Press. 10.4324/9781315859811-5. [DOI] [Google Scholar]
- Cornett, K. M. , North, K. N. , Rose, K. J. , & Burns, J. (2015). Muscle weakness in children with neurofibromatosis type 1. Developmental Medicine and Child Neurology, 57(8), 733–736. [DOI] [PubMed] [Google Scholar]
- Dulai, S. , Briody, J. , Schindeler, A. , North, K. N. , Cowell, C. T. , & Little, D. G. (2007). Decreased bone mineral density in neurofibromatosis type 1: Results from a pediatric cohort. Journal of Pediatric Orthopaedics, 27(4), 472–475. [DOI] [PubMed] [Google Scholar]
- Ellaway, C. , Williams, K. , Leonard, H. , Higgins, G. , & Christodoulou, J. (1999). Rett syndrome: Randomized controlled trial of L‐carnitine. Journal of Child Neurology, 14(3), 162–167. [DOI] [PubMed] [Google Scholar]
- Friedman, J. M. (2002). Neurofibromatosis 1: Clinical manifestations and diagnostic criteria. Journal of Child Neurology, 17(8), 548–554 discussion 71–2, 646–51. [DOI] [PubMed] [Google Scholar]
- Geier, D. A. , Kern, J. K. , Davis, G. , King, P. G. , Adams, J. B. , Young, J. L. , & Geier, M. R. (2011). A prospective double‐blind, randomized clinical trial of levocarnitine to treat autism spectrum disorders. Medical Science Monitor, 17(6), PI15–PI23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goin‐Kochel, R. P. , Scaglia, F. , Schaaf, C. P. , Berry, L. N. , Dang, D. , Nowel, K. P. , Laakman, A. L. , Dowell, L. R. , Minard, C. G. , Loh, A. , & Beaudet, A. L. (2019). Side effects and behavioral outcomes following high‐dose carnitine supplementation among young males with autism spectrum disorder: A pilot study. Global Pediatric Health, 6, 2333794X19830696‐2333794X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpey, J.‐P. , Charpentier, C. , Paturneau‐Jouas, M. , Renault, F. , Romero, N. , & Fardeau, M. (1990). Secondary metabolic defects in spinal muscular atrophy type II. The Lancet, 336(8715), 629–630. [DOI] [PubMed] [Google Scholar]
- Kilic, M. , Ozgül, R. K. , Coşkun, T. , Yücel, D. , Karaca, M. , Sivri, H. S. , Tokatli, A. , Sahin, M. , Karagöz, T. , & Dursun, A. (2012). Identification of mutations and evaluation of cardiomyopathy in Turkish patients with primary carnitine deficiency. JIMD Reports, 3, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaguarnera, M. , & Cauli, O. (2019). Effects of l‐carnitine in patients with autism spectrum disorders: Review of clinical studies. Molecules (Basel, Switzerland), 24(23), 4262. 10.3390/molecules24234262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, M. J. , Baldwin, J. N. , Ferreira, P. , Simic, M. , Vanicek, N. , Hiller, C. E. , Nightingale, E. J. , Moloney, N. A. , Quinlan, K. G. , Pourkazemi, F. , Sman, A. D. , Nicholson, L. L. , Mousavi, S. J. , Rose, K. , Raymond, J. , Mackey, M. G. , Chard, A. , Hübscher, M. , Wegener, C. , … 1000 Norms Project Consortium . (2016). 1000 norms project: Protocol of a cross‐sectional study cataloging human variation. Physiotherapy, 102(1), 50–56. [DOI] [PubMed] [Google Scholar]
- Menz, H. (2004). Two feet, or one person? Problems associated with statistical analysis of paired data in foot and ankle medicine. The Foot, 14, 2–5. [Google Scholar]
- Moorthy, A. V. , Rosenblum, M. , Rajaram, R. , & Shug, A. L. (1983). A comparison of plasma and muscle carnitine levels in patients on peritoneal or hemodialysis for chronic renal failure. American Journal of Nephrology, 3(4), 205–208. [DOI] [PubMed] [Google Scholar]
- Park, J.‐S. , Davis, R. L. , & Sue, C. M. (2018). Mitochondrial dysfunction in Parkinson's disease: New mechanistic insights and therapeutic perspectives. Current Neurology and Neuroscience Reports, 18(5), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savica, V. , Bellinghieri, G. , di Stefano, C. , Corvaja, E. , Consolo, F. , Corsi, M. , Maccari, F. , Spagnoli, L. G. , Villaschi, S. , & Palmieri, G. (1983). Plasma and muscle carnitine levels in haemodialysis patients with morphological‐ultrastructural examination of muscle samples. Nephron, 35(4), 232–236. [DOI] [PubMed] [Google Scholar]
- Shen, M. H. , Harper, P. S. , & Upadhyaya, M. (1996). Molecular genetics of neurofibromatosis type 1 (NF1). Journal of Medical Genetics, 33(1), 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulyakova, N. , Andreazza, A. C. , Mills, L. R. , & Eubanks, J. H. (2017). Mitochondrial dysfunction in the pathogenesis of Rett syndrome: Implications for mitochondria‐targeted therapies. Frontiers in Cellular Neuroscience, 11, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui, M. F. , Elwell, C. , & Johnson, M. H. (2016). Mitochondrial dysfunction in autism Spectrum disorders. Autism Open Access, 6(5), 1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, F. B. , Constantin‐Teodosiu, D. , & Greenhaff, P. L. (2007). New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. The Journal of Physiology, 581(Pt 2), 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers, M. A. , Quinlan, K. G. , Payne, J. M. , Little, D. G. , North, K. N. , & Schindeler, A. (2015). Skeletal muscle and motor deficits in Neurofibromatosis type 1. Journal of Musculoskeletal & Neuronal Interactions, 15(2), 161–170. [PMC free article] [PubMed] [Google Scholar]
- Summers, M. A. , Rupasinghe, T. , Vasiljevski, E. R. , Evesson, F. J. , Mikulec, K. , Peacock, L. , Quinlan, K. G. , Cooper, S. T. , Roessner, U. , Stevenson, D. A. , Little, D. G. , & Schindeler, A. (2018). Dietary intervention rescues myopathy associated with neurofibromatosis type 1. Human Molecular Genetics, 27(4), 577–588. [DOI] [PubMed] [Google Scholar]
- Tomlinson, S. , Atherton, J. , & Prasad, S. (2018). Primary carnitine deficiency: A rare, reversible metabolic cardiomyopathy. Case Reports in Cardiology, 2018, 3232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma, E. H. , Rashed, M. S. , Vianey‐Saban, C. , Sakr, A. , Divry, P. , Gregersen, N. , & Andresen, B. S. (2001). A severe genotype with favourable outcome in very long chain acyl‐CoA dehydrogenase deficiency. Archives of Disease in Childhood, 84(1), 58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljevski, E. R. , Summers, M. A. , Little, D. G. , & Schindeler, A. (2018). Lipid storage myopathies: Current treatments and future directions. Progress in Lipid Research, 72, 1–17. [DOI] [PubMed] [Google Scholar]
- Vassallo, G. , Mughal, Z. , Robinson, L. , Weisberg, D. , Roberts, S. A. , Hupton, E. , Eelloo, J. , Wright, E. M. B. , Garg, S. , Lewis, L. , Gareth Evans, D. , & Stivaros, S. M. (2020). Perceived fatigue in children and young adults with neurofibromatosis type 1. Journal of Paediatrics and Child Health, 56(6), 878–883. 10.1111/jpc.14764. [DOI] [PubMed] [Google Scholar]
- Wallen, M. A. , Bonney, M.‐A. , & Lennox, L. (Eds). (1996). The Handwriting Speed Test. Adelaide SA: Helios Art and Book Co. [Google Scholar]
- Wechsler, D. (1974). Wechsler intelligence scale for children‐Revised edition. San Antonio, TX: The Psychological Corporation. [Google Scholar]
- Zeiler, F. A. , Sader, N. , Gillman, L. M. , & West, M. (2016). Levocarnitine induced seizures in patients on valproic acid: A negative systematic review. Seizure, 36, 36–39. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Jia, H. , Liu, J. , Ao, N. , Yan, B. , Shen, W. , Wang, X. , Li, X. , Luo, C. , & Liu, J. (2010). Combined R‐alpha‐lipoic acid and acetyl‐L‐carnitine exerts efficient preventative effects in a cellular model of Parkinson's disease. Journal of Cellular and Molecular Medicine, 14(1–2), 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Liver function test procedures with reference range and units
Supplementary Table 2: Urine chemistry tested with units
Supplementary Table 3: Acylcarnitines included in the acylcarnitine profile test with reference range and units
Supplementary Table 4: Absolute change of functional outcome measures for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w).
Supplementary Table 5: PedsQL domain scores. Scores are displayed for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w). 0 = low score and 100 = high score. Median (interquartile range) have been displayed to better represent the data.
Supplementary Table 6: CBCL syndrome scores. Scores are displayed for each participant (1–6) at baseline (0w) and following 12 weeks of L‐carnitine supplementation (12w). Score reduction indicates normalization. C = clinical range, B = borderline clinical range.
Data Availability Statement
Data collected for the study will not be made available to others.