

Abstract
Background and Aims
Wilson’s disease (WD) is an autosomal‐recessive disorder caused by ATP7B gene mutations leading to pathological accumulation of copper in the liver and brain. Adoption of initial treatments for WD was based on empirical observations. These therapies are effective, but there are still unmet needs for which treatment modalities are being developed. An increase of therapeutical trials is anticipated.
Approach and Results
The first Wilson Disease Aarhus Symposium (May 2019) included a workshop on randomized clinical trial design. The authors of the article were organizers or presented during this workshop, and this article presents their consensus on the design of clinical trials for WD, addressing trial population, treatment comparators, inclusion and exclusion criteria, and treatment endpoints. To achieve adequate recruitment of patients with this rare disorder, the study groups should include all clinical phenotypes and treatment‐experienced as well as treatment‐naïve patients.
Conclusions
The primary study endpoint should be clinical or a composite endpoint until appropriate surrogate endpoints are validated. Standardization of clinical trials will permit pooling of data and allow for better treatment comparisons, as well as reduce the future numbers of patients needed per trial.
Abbreviations
- ALF
acute liver failure
- ALT
alanine aminotransferase
- AST
asparate aminotransferase
- CuEXC
exchangeable copper
- FIB‐4
Fibrosis‐4 index
- KF
Kayser‐Fleischer
- MELD
Model of End‐Stage Liver Disease
- NCC
non‐ceruloplasmin‐bound copper
- RCT
randomized clinical trial
- SOC
standard of care
- UWDRS
Unified Wilson’s Disease Rating Scale
- WD
Wilson’s disease
Wilson’s disease (WD) is an autosomal‐recessive disorder of reduced biliary copper excretion attributable to mutations in the ATPase copper‐transporting beta gene (ATP7B) leading to pathological copper accumulation in liver, brain, and other tissues.( 1 , 2 , 3 ) Symptom onset is generally in adolescence to early adulthood, but may occur at any age.
WD requires lifelong therapy to prevent, reduce, or stabilize symptoms.( 1 , 2 ) Current treatments were introduced without controlled studies. The chelators (D‐penicillamine and trientine) that increase urinary copper excretion and zinc salts that decrease enteric copper absorption have raised WD patient survival to near‐normal for age‐matched populations with good adherence and initiation before severe organ damage.( 4 , 5 , 6 , 7 ) However, excellent outcomes are not universal. Up to 45% of patients have poor long‐term medication adherence, with risk of disease progression.( 1 , 6 , 8 ) Incomplete resolution of symptoms is common.( 4 , 9 , 10 ) Medication side effects lead to cessation in many.( 11 ) Drug‐induced paradoxical neurological deterioration may occur during initial treatment.( 4 , 12 , 13 , 14 ) Thus, developing new treatments for WD is necessary (Table 1).
TABLE 1.
Medical Needs in WD
|
The European Medicines Agency (EMA) and the U.S. Food and Drug Administration support development of drugs for rare diseases and published guidelines for trial design and analytical method development.( 16 , 17 ) Currently, only one controlled, partly blinded, short trial in neurological WD has been reported.( 14 ) The interpretation of other studies is limited by nonuniform definitions of outcomes.( 11 ) With more randomized studies expected in the future, using uniform definitions of outcomes will facilitate study comparisons.
The first Wilson Aarhus Symposium (May 2019) included a workshop on randomized clinical trial (RCT) design. A diverse group of international experts contributed. In premeetings, Drs. Ott, Ferenci, Weiss, and Schilsky defined the most important issues and invited experts to address them at the meeting. This article summarizes conclusions from the proceedings aimed at providing guidance for design and conduct of future WD phase 2 and phase 3 clinical trials. Study populations, outcome measures, and needs for research are identified.
Study Population
Clinical Presentation
The clinical phenotype of WD patients at presentation is variable and includes acute and chronic symptoms (Fig. 1). Given that nonacute phenotypes are not clearly separated,( 3 , 15 ) studies should include all phenotypes except those with acute liver failure (ALF) or end‐stage disease refractory to medical therapy (see Exclusion Criteria below).
FIG. 1.
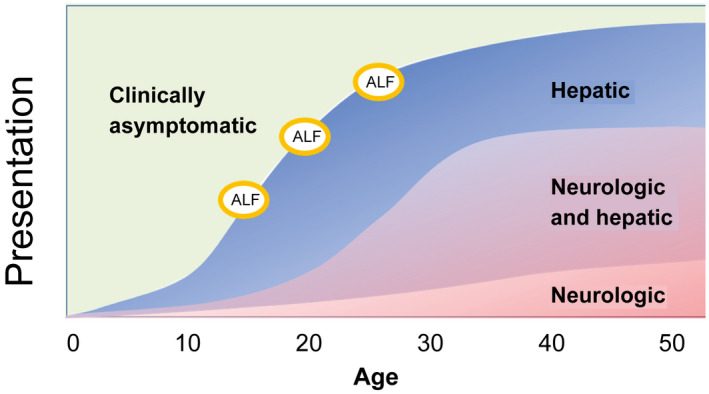
Clinical course of WD. After a subclinical period, WD presents with hepatic (mean age, 17.6 years) and/or neurological (mean age, 23.4 years) symptoms. Approximately 60% have both. Three percent to 5% present with acute hepatic failure (ALF), which is fatal without liver transplantation. In the remaining patients, the medical treatment aims at preventing or stopping disease progression and, if possible, inducing a regression of symptoms.
Clinically asymptomatic siblings of WD patients are effectively identified by genetic testing. Though appearing healthy, affected persons have elevated hepatic copper and progress to overt disease without treatment. They can be included in clinical trials after clinical and metabolic characterization.
As preconception, prenatal, and newborn genetic testing becomes more widespread, fetuses and newborn infants will be diagnosed before development of pathological copper overload. Treatment will aim to prevent development of injury and disease.( 16 ) Drug selection and age for treatment initiation is unclear and require further study.
Given that disease course and underlying pathophysiology is similar, children ≥12 years old and adults can be included in the same RCTs after appropriate bioethical considerations and dosing modification. Separate studies are needed for younger children.
Patient Genotype
Only approximately half of reported ATP7B mutations are considered pathogenic or likely pathogenic.( 17 ) Several studies failed to demonstrate clear genotype‐phenotype correlations.( 1 , 15 , 18 ) Siblings and even monozygotic twins may have diverse phenotypes.( 19 ) Thus, stratification based on genotype is not reasonable, except in future trials for gene repair.
Treatment Status
Ideally, an RCT includes only treatment‐naïve patients. However, the recruitment phase may be unacceptably long, even if patients treated for <28 days are included. Most current studies therefore included both treatment‐naïve and ‐experienced patients. The ratio should be balanced given that clinical and biochemical improvement is more likely in the treatment naïve. It is reasonable to stratify analysis of treatment‐experienced patients to <3 or ≥3 years because biochemically and clinical stability is more likely after 3 years.( 20 ) A run‐in period on current treatment is recommended for treated patients to ensure baseline compliance, reduce study dropout, and help standardize data collection.( 21 ) The most common design is to randomize treatment‐experienced patients to the trial drug or the patient’s current treatment. Although this pragmatic choice is supported by the author panel, certain possible biases must be taken into account. Because using a run‐in period and requiring clinical stability (see section on Stability below) likely ensure that current treatment is optimized at inclusion, the trial drug may be held to a higher standard than if only treatment‐naïve patients were studied. At the same time, double blinding may be difficult and more costly; however, certain outcome measures can be obtained in a single‐blinded fashion.
Inclusion Criteria
Diagnosis
The diagnosis of WD should rest on standardized, validated diagnostic criteria such as the Leipzig score.( 22 , 23 ) A liver biopsy is not a requisite for WD diagnosis or for inclusion in a clinical trial unless study endpoints include hepatic copper content or histology.
Stability
Inclusion criteria may require clinical and biochemical stability; however, a generally accepted definition is lacking. After 3 years of uninterrupted treatment, further symptom regression is unlikely and clinical condition, treatment dosing, and measures of copper metabolism are usually stable. For patients with <3 years of treatment, the definition of stability should leave room for possible symptom regression. Some patients will never be stable despite treatment (“treatment failures”) and RCTs with that specific focus are needed.
Exclusion Criteria Specific to WD
ALF
Patients with ALF or at high risk of ALF should be excluded from pharmacological trials. Use of the new Wilson’s index for predicting mortality, developed for WD children presenting with liver failure,( 24 ) can help identify these persons. Given that a score of ≥11 predicted death, we recommend excluding WD patients with a score >10 despite limited data in adults.( 25 )
End‐Stage Liver Disease
Patients with clinical instability attributable to refractory ascites, overt HE, or gastroesophageal variceal bleeding within 6 months should be excluded unless treated and stabilized. HCC and cholangiocarcinoma should also exclude enrollment. patients with compensated cirrhosis may be included. Listed Patients with a waiting time for transplant >1 year can be enrolled. Liver transplantation should be an exclusion criterion.
Neurological End‐Stage Disease
Patients with marked disabilities may improve and be included in an RCT. Those with severe neurological deficits (bedridden, fixed dystonia or parkinsonism, and severe cognitive impairment) nonresponsive to treatment for >12 months should be excluded from treatment trials.
Withdrawal Criteria From Trials
Patients should be withdrawn from treatment trials if they experience drug injury (alanine aminotransferase [ALT] increases >5‐ to 10‐fold normal or hyperbilirubinemia >2‐fold normal); worsening of cirrhosis (new onset of ascites, encephalopathy, variceal bleeding, and/or jaundice); neurological deterioration (i.e., by a predefined increase in the United Wilson’s Disease Rating Scale [UWDRS]); or significant psychiatric disease, such as onset of psychosis, severe depression, or behavioral changes.
Paradoxical neurological deterioration has been described as rapid neurological worsening within the 6 months of the start of an initial or secondary treatment.( 13 ) If this is not defined as a treatment failure per protocol, the protocol should provide concise instructions about dose reduction and a subsequent reduced rate of dose escalation.
Endpoints
Clinically important or Surrogate Endpoints
The primary endpoint should define the effectiveness of treatment. It is needed for power calculations to determine the number of patients needed for the trial. In a phase 3 trial, the EMA states that “ideally a ´hard´ and clinically relevant endpoint” is used as the primary endpoint variable.( 26 ) We define “a clinically important” endpoint as a clinical effect of treatment on how the patient feels, functions, and survives.( 27 , 28 ) This endpoint should be objectively measurable, reflect important aspects of clinical disease progression, and have a meaningful relation to patients’ quality of life.( 27 , 28 )
Surrogate endpoints must be validated to ensure that they adequately reflect the clinically important outcome. Their use as a primary endpoint may shorten the study duration and reduce the sample size. As discussed below, surrogate endpoints meeting these criteria are lacking for WD. Identifying surrogate endpoints should be prioritized in future work.
Surrogate markers are chosen because of their relation to the pathophysiology and disease natural history( 26 ); however, they are insufficient to verify long‐term patient benefit.
Exploratory endpoints are included to better estimate the efficacy and confirm the mechanism of action of treatments.
Composite endpoints combining different endpoints are necessary when a single meaningful primary endpoint cannot be defined. Use of multiple simultaneous endpoints, clinical or biochemical, may be necessary despite a less‐clear interpretation.( 26 )
Hepatic Endpoints
Endpoints should relate to the goals of treatment. On treatment, patients with near‐normal histology or minimal steatosis should remain stable, whereas those with inflammation, fibrosis, or cirrhosis should improve or at least remain stable (Fig. 1). Markers of treatment failure include fibrosis progression, cirrhotic decompensation, or liver failure requiring transplantation or causing death (Table 2).
TABLE 2.
Endpoints in Trials for Patients With WD
| Hepatic Endpoints |
|
| Identified areas of research |
| High priority |
|
| Others |
|
| Neurological endpoints |
|
| Identified areas of research |
| High priority |
|
| Others |
|
| Endpoints related to assessment of copper metabolism |
|
| Identified areas of research |
| High priority |
|
| Others |
|
Abbreviations: ARFI, acoustic radiation force impulse; MRE, magnetic resonance elastography.
Routine Laboratory Parameters
ALT and aspartate aminotransferase (AST) are markers of hepatocellular necrosis and should be included as secondary or exploratory endpoints and measured for monitoring treatment safety. Biomarkers of liver protein synthesis (albumin, international normalized ratio, and pseudocholinesterase) and excretion (bilirubin) should be included as estimates for liver function. These parameters form part of the scoring systems for those patients with cirrhosis, such as the Model for End‐Stage Liver Disease (MELD) score and Child‐Pugh score.
Development of Fibrosis
Change in hepatic fibrosis is a potentially useful endpoint and can be included as a secondary or exploratory endpoint or as part of a composite endpoint.
The best way to assess hepatic fibrosis is uncertain, but includes histological grading, elastography (sound wave or obtained by MRI), and biochemical methods. In WD, liver biopsy may be less useful because histological findings did not clearly differentiate between progressors and nonprogressors in past trials,( 29 , 30 ) and some patients hesitate to undergo biopsy. Transient elastography is a potential surrogate endpoint (Supporting Information S.1.1), but prospective studies of the rate of fibrosis progression/regression in WD are needed. Until then, it is recommended as a surrogate marker. Noninvasive biochemical markers of fibrosis, such as AST to Platelet Ratio Index (APRI), Fibrosis‐4 (FIB‐4) index, and Enhanced Liver Fibrosis (ELF) index, are less sensitive than elastography and may be included as exploratory endpoints (Supporting Information S1.1).
Further developments of MR methodology assessing hepatic fibroinflammation, steatosis, and iron content may be of interest as exploratory endpoints (Supporting Information S.1.1).
Progression to Cirrhosis and Its Complications
Development of complications of cirrhosis evolve slowly, but are clinically important as endpoints in studies with long‐term duration. Ideally, on treatment they may improve (i.e., disappearance of ascites or esophageal varices), but worsening may lead to study withdrawal (see Withdrawal section).
For WD patients with cirrhosis, validated prognostic information can be obtained using the Child‐Pugh,( 31 ) MELD,( 32 ) and MELD‐sodium scores.( 33 ) These scores could be included as surrogate markers of liver disease progression or regression on treatment; however, there are no supportive data for their use in WD patients without cirrhosis.
Other Possible Surrogate Hepatic Markers
One treatment target is the reduction or prevention of inflammation. Biomarkers for hepatic inflammation need to be developed given that ALT alone is insufficient (Supporting Information S.1.2).
The new Wilson’s index for predicting mortality( 24 ) discussed above (see Exclusion Criteria) may be a useful endpoint for safety because a rising score may portend severe liver injury given that the score captures elements of systemic inflammatory response syndrome and acute‐phase injury.
The potential use of quantitative dynamic liver function tests described in Supporting Information S.1.3) as surrogate endpoints should be evaluated.
Neurological Endpoints
Neurological manifestations of WD can be classified into syndrome types based on predominant signs, such as tremor, ataxia, bradykinesia (parkinsonism‐like), and dystonia. The choice of neurological endpoint should encompass this wide variability. This consideration led to the development of scoring systems for assessment of neurological status in clinical trials.( 3 , 34 , 35 , 36 , 37 )
The UWDRS
The UWDRS is a widely used scoring system for WD.( 35 , 36 , 37 ) Part I of the UWDRS assesses consciousness, Part II is a patient‐reported evaluation of disability, and Part III a rater‐determined neurological examination (Supporting Information S.2.1). Use of the UWDRS can be blinded if the assessor is unaware of the treatment. Video recordings may allow a centralized evaluation. Interobserver agreement is sufficient to permit the use of single‐observer assessments (see Supporting Information S.2.1). A possible limitation of Part III is that if the total score is used to estimate disease severity, a positive change in one item (i.e., handwriting) can neutralize a negative change in another (i.e., speech), which may not be equivalent for the patient. Analysis of elements of the UWDRS is indicated to determine which are most relevant to patient functionality.
The use of a common neurological rating scale will facilitate comparison between studies, and presently we recommend use of the UWDRS. However, less complex and time‐consuming measurements of patients’ neurological functional status are desirable. UWDRS Part II may be of interest given that it is less time‐consuming (patient reported) and correlated with UWDRS Part III.( 37 ) The modified Rankin score( 38 ) deserves further evaluation given that it correlated with the UWDRS after liver transplantation for neurological WD.( 39 )
MRI
To be a useful surrogate endpoint, MRI findings require objective and reproducible evaluation parameters and longitudinal studies demonstrating that MRI changes correlate with clinical findings in individual patients. Until such data are available, the use of MRI in clinical trials is exploratory. Validation of MRI is in progress (Supporting Information S.2.2). Importantly, it would allow for blinded, centralized evaluation.
Other Possible Clinical Neurological Endpoints
Small interesting reports suggest a possible value of evoked potentials (Supporting Information S.2.3), but further studies are needed.
Psychiatric and Other Endpoints
Psychiatric manifestations of WD are relevant as study endpoint(s) given that they affect quality of life.( 40 ) At the present time, with no validated instruments specific for WD available, we recommend the use of a simple standardized questionnaires, such as the Patient Health Questionnaire‐9. Any treatment of psychiatric disorders should be monitored during trials. Psychometric test batteries may be useful to detect subtle changes in cognition and/or psychomotor performance, but need validation in WD (Supporting Information S.2.3).
Other less‐common symptoms of WD, such as arthropathy, female reproductive abnormalities, and renal and skin disturbances, may be considered as tertiary or exploratory endpoints.
Copper Metabolism and Study Endpoints
For treatments modifying copper metabolism, their impact on copper metabolism should be a focus of phase 1 and 2 trials whereas phase 3 trials should focus on the impact of the treatment on clinical outcomes. At present, none of the measures of copper metabolism discussed below are validated as surrogate endpoints given that there still is a need to demonstrate that with treatment they have a positive correlation with good clinical outcome.( 20 )
Measurements of Bioavailable Copper
Determination of “free” bioavailable copper concentration has been proposed as a possible surrogate marker. This copper fraction is considered biologically active and is the target of treatment to prevent the extrahepatic uptake of copper. There are several approaches to measure free copper.
Non‐ceruloplasmin‐bound copper (NCC) is estimated by subtracting ceruloplasmin‐bound copper from the total serum copper concentration.( 1 , 2 ) A weakness of the methodology is biologically implausible negative values in some patients (Supporting Information S.3.1). Reports on the correlation between NCC normalization and clinical outcome are conflicting,( 41 , 42 , 43 ) but in a recent phase 2 study, the NCC estimate correlated with clinical outcome during treatment with bis‐choline tetrathiomolybdate.( 44 )
Measurement of exchangeable copper (CuEXC) is obtained by the incubation of serum with EDTA to remove loosely bound copper and subsequent removal of ceruloplasmin‐bound copper by ultrafiltration.( 49 ) The method does not depend on the measurement of ceruloplasmin. Correlation between CuEXC and organ damage was observed in an animal study.( 45 ) CuEXC was related to patient compliance,( 46 ) but longitudinal data in patients have not been reported.
For further discussion of the measurement of bioavailable (free) copper, see Supporting Information S.3.1. At the present stage, neither NCC nor CuEXC has been validated as a surrogate endpoint. The data do not allow a conclusion as to which is more valuable for treatment monitoring. At least one of these should be included as an exploratory endpoint.
Newly reported mass spectrometry–based methods directly measure ceruloplasmin copper and total copper.( 47 ) This method suggests weaknesses with both the estimation of NCC and CuEXC methodologies.
Twenty‐Four‐Hour Urinary Copper Excretion
Twenty‐four‐hour urinary copper excretion is used for diagnosis and treatment monitoring of WD.( 1 , 2 , 23 , 43 ) Symptomatic patients treated with cupriuretic chelators have initial increases in copper excretion that decrease with time, but remain above the normal range.( 20 , 43 , 48 ) The intraindividual variation is pronounced, but lower than with NCC.( 43 ) Because 24‐hour urine copper excretion is dose dependent and reflects dietary intake and total body copper content,( 48 ) it is useful to monitor the treatment of a given patient assuming relatively consistent dietary copper intake.
With zinc therapy (no cupriuretic effect), the pattern is different, and in those with elevated urinary copper excretion, there is a slow decrease in copper excretion that takes months to years to reach the normal range.( 42 )
Measurement of 24‐hour urinary copper excretion after a 48‐hour “drug holiday” might overcome the problems of interpretation during chelator therapy( 1 , 48 , 49 ) and may reflect whole‐body copper in these persons. In compliant patients, and for an individual patient, urinary copper excretion after a 48‐hour drug holiday reflects whole‐body copper content and not differences in dosing or drug absorption, facilitating treatment comparisons.( 43 , 50 )
Twenty‐four‐hour urinary copper excretion should be included in a clinical study as a surrogate marker. In studies including chelating agents, collection after a 48‐hour drug holiday may be preferred to facilitate comparison between treatments.
Other Possible Exploratory Endpoints Related to Copper Metabolism
For long‐term treatments, changes in organ copper content may be ideal but very hard to obtain. One noninvasive approach is the quantification of Kayser‐Fleischer (KF) rings intensity by anterior segment optical coherence tomography (Supporting Information S.3.2). Measurement of copper in cerebrospinal fluid is more invasive, but may reflect cerebral copper burden (Supporting Information S.3.2). Hepatic copper concentration in liver biopsy samples is not useful for evaluating therapy because it may vary within the liver( 51 ) and remains elevated despite clinical improvement.( 30 , 52 )
Patient‐Reported Outcomes
Quality of life and functional status are important efficacy measures of long‐term therapy,( 28 ) but cannot be used as primary endpoints because their relation to long‐term disease progression is unknown. The “minimal UWDRS” transformed into a patient‐reported outcome included nine items related to activities of daily living that correlated with UWDRS scores.( 37 ) It is recommended that a specific quality‐of‐life index for WD be developed and used as a secondary outcome measure until validation.
Choosing Endpoints in Clinical Trials in WD
Although copper parameters are useful primary endpoints in phase 2 studies, none have been validated to be used as the primary surrogate endpoint in phase 3 studies. In these studies, which include various clinical phenotypes, no single clinical endpoint would cover all situations (Fig. 2). Therefore, the primary endpoint must be a composite, including assessment of the most relevant clinical and biochemical features. The simplest form will include definitions of progression, regression, or no change of disease. A more advanced composite endpoint would be a “WD severity score” including more parameters with weighting according to their impact on disease severity and patient functionality. Such a score may be a more sensitive composite primary endpoint in future RCTs in WD and would also be useful for the validation of specific measures of copper metabolism for use as surrogate outcome measures.
FIG. 2.
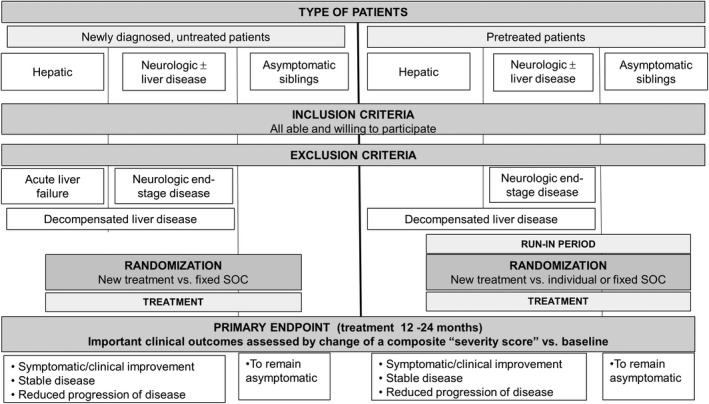
Proposed design for prospective, randomized phase 2 and phase 3 studies in WD. Given that currently there is no single endpoint describing all possible features of WD, we propose to develop a composite score (“severity score”) that includes and weights several clinical and laboratory parameters. Until then, a combination of changes of single parameters from baseline can be described as improved, unchanged, or worse.
Trial Design Considerations
Choice of Study Drug and Comparator for WD Treatment in Trials
With the currently available treatments for WD, survival is near normal.( 4 , 6 , 7 ) Given that clinical deterioration can develop within months after treatment discontinuation,( 53 , 54 ) placebo monotherapy as the comparator in an RCT is impossible. In current studies, standard of care (SOC) is used as the comparator. New treatments can be directly compared to SOC or as an add‐on treatment to SOC alone. Ideally, SOC should be standardized for all study participants. In current trials, SOC varies according to local traditions, economics, and differences in regulatory approval of medications.
Sample Size
In any RCT, the necessary sample size depends on the minimally relevant difference in treatment outcomes. Regulatory authorities( 26 ) and the scientific community( 55 , 56 ) recognize the need for innovative solutions to design and analyze clinical trials with as few participants as possible, especially for rare disorders. To increase the number of eligible patients, stratification should only be used if an impact on the outcome is expected.( 26 , 57 ) Also, sample size may be reduced if longitudinal evaluation of endpoint variables is applied using all available data rather than baseline to end‐of‐study comparisons.( 58 )
Length of Trial
The optimal trial duration is derived from knowledge of the natural history of the disease. When patients with WD were treated with chelators or zinc, partial normalization of ALT, albumin, and prothrombin time was observed after 6 months, and most patients reached values close to or within the normal range after 12‐24 months.( 30 , 42 ) Histological normalization may take 6‐10 years in adults,( 30 , 59 ) possibly shorter in children.( 60 ) Neurological symptoms will typically stabilize and start improving after 2 months of treatment, but improvements after 3 years are possible.( 14 ) Thus, studies with clinical endpoints may need trial durations of ≥1‐2 years. The use of surrogate endpoints will help to shorten trial length.
Specific Designs
Depending on the specific aim of the study, the choice of study design will be influenced by the rarity of the disease and the availability of suitable study subjects.
Crossover trial designs reduce variability and thus the necessary sample size. The crossover design requires that the disease should not progress between periods and there should be no residual treatment effects. This may be difficult to fulfil in long‐term studies in WD, but the design may be applicable to short‐term studies.
Sequential designs have been developed for use in superiority studies and will often reduce sample size. The trial continues until superiority is demonstrated in one arm or until there is a certain number of included patients. Outcomes must be available shortly after the individual patient’s trial termination.
Adaptive designs have specific advantages in rare diseases.( 55 ) With adaptive designs, the trial is separated into two or more independent phases in which an analysis described in the protocol can lead to protocol changes, such as stop for futility or efficacy, or changes in sample size, endpoints, inclusion criteria, or even removal or addition of new arms of active treatment.( 56 , 61 ) With the responsive‐adaptive randomization designs, the randomization ratios change during the trial according to the observed responses.( 26 ) A flexible adaptive enrichment design allows the trial to start with a large population of “straightforward” patients. Based on the experience of the first part of the trial, more specific subpopulations are assessed in the second phase.
Even more advanced solutions are under development,( 56 ) including multiarm sequential designs and the use of external data for the analysis of phase 3 trials.( 62 ) In the latter case, it is important that the data obtained are of similar quality (i.e., according to the recommendations in this article).
Statistical Methods
Small sample‐size studies require more complex statistical analysis than larger studies. Methods are being developed to deal with multiple endpoints, sensitivity analyses, adjustment for baseline variables, and stratification and the evaluation of repeated measurements.
Bayesian methods may further increase the information that can be extracted from an RCT, although regulatory authorities will require validation of the earlier beliefs that will be included in the analysis. These methods may be most valuable for sample‐size estimations.( 63 )
Monitoring
For monitoring the clinical and biological improvement, adherence, and safety of a new treatment, the frequency of visits must take into account disease phase and severity.( 20 ) During the initial phase of treatment after diagnosis, symptomatic patients should be assessed every 2‐4 weeks for 2 months and then at 2‐ to 3‐month intervals until the end of the first year. If a treatment‐naïve patient is randomized to receive SOC, dose modifications may be needed according to the drug selected. In the late phase of a trial, follow‐up should be twice‐yearly, even in asymptomatic or stable patients.( 2 ) More frequent monitoring may be needed after treatment modifications or based on clinical indication.
Monitoring should also detect signs of overtreatment, such as neutropenia, sideroblastic anemia, hyperferritinemia, and, possibly, hepatic iron accumulation. Overtreated patients are also expected to present with low serum copper and low NCC and CuEXC. They will also have low 24‐hour urinary copper relative to the treatment used. Urinary excretion after a 48‐hour drug holiday may be helpful in monitoring patients on D‐penicillamine or trientine. In case of overtreatment, therapy should temporarily be discontinued and, rarely, copper replaced.
Conclusion
With the ongoing development of new therapies for WD, we recommend that newly initiated clinical trials follow the above consensus guidance to improve the impact of the individual studies and facilitate their comparison. In order to achieve adequate recruitment, the trial population should include all clinical phenotypes and treatment‐experienced and ‐naïve patients. The most important clinical hepatic and neurological endpoints were discussed. The primary study endpoint should be clinical or a composite endpoint, given that no surrogate endpoints are validated. Centers around the world are urged to provide this validation given that the use of surrogate endpoints would shorten trial duration and speed the development of therapies for WD.
Author contributions
P.O., T.D.S., M.L.S., and P.F. conceived the article. All authors contributed to the draft and critical revision of the article. All authors approved the final version.
Supporting information
Supplementary Material
Acknowledgment
This work was supported by The Memorial Foundation for Manufacturer Vilhelm Pedersen & Wife. Ralf‐Dieter Hilgers received funding from the European Union’s 7th Framework Programme for research, technological development, and demonstration under the IDEAL Grant Agreement no. 602552, the European Union’s Horizon 2020 research and innovation programme under the EJP RD COFUND‐EJP no. 825575, and ERICA under Grant Agreement no. 964908.
Supported by an unrestricted grant from The Memorial Foundation of Manufacturer Vilhelm Pedersen & Wife. The foundation played no role in the planning or any other phase of the study.
Potential conflict of interest: Dr. Ala advises for, is on the speakers’ bureau for, and received grants from GMPO and Univar. He advises for and received grants from Alexion. Dr. Ferenci advises for Univar, Vivet, Ambys, Ultragenix, and Gilead. He received grants from Alexion. Dr. Ott is on the speakers’ bureau for GMPO. Dr. Askari received grants from Alexion, Ultragenix, and Vivet. Dr. Sandahl advises for Alexion. Dr. Weiss consults for Univar, Alexion, Vivet, Pfizer, and Orphalan. Dr. Schilsky received grants from Alexion, Orphalan, and Vivet.
References
Author names in bold designate shared co‐first authorship.
- 1. EASL . EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol 2012;56:671‐685. [DOI] [PubMed] [Google Scholar]
- 2. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008;47:2089‐2111. [DOI] [PubMed] [Google Scholar]
- 3. Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, et al. Wilson disease. Nat Rev Dis Primers 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beinhardt S, Leiss W, Stättermayer AF, Graziadei I, Zoller H, Stauber R, et al. Long‐term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol 2014;12:683‐689. [DOI] [PubMed] [Google Scholar]
- 5. Bruha R, Marecek Z, Pospisilova L, Nevsimalova S, Vitek L, Martasek P, et al. Long‐term follow‐up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int 2011;31:83‐91. [DOI] [PubMed] [Google Scholar]
- 6. Dziezyc K, Karlinski M, Litwin T, Czlonkowska A. Compliant treatment with anti‐copper agents prevents clinically overt Wilson’s disease in pre‐symptomatic patients. Eur J Neurol 2014;21:332‐337. [DOI] [PubMed] [Google Scholar]
- 7. Czlonkowska A, Tarnacka B, Litwin T, Gajda J, Rodo M. Wilson’s disease‐cause of mortality in 164 patients during 1992‐2003 observation period. J Neurol 2005;252:698‐703. [DOI] [PubMed] [Google Scholar]
- 8. Maselbas W, Czlonkowska A, Litwin T, Niewada M. Persistence with treatment for Wilson disease: a retrospective study. BMC Neurol 2019;19:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss KH, Thurik F, Gotthardt DN, Schäfer M, Teufel U, Wiegand F, et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol 2013;11:1028‐1035.e1‐2. [DOI] [PubMed] [Google Scholar]
- 10. Litwin T, Dziezyc K, Czlonkowska A. Wilson disease—treatment perspectives. Ann Transl Med 2019;7(Suppl. 2):S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Appenzeller‐Herzog C, Mathes T, Heeres MLS, Weiss KH, Houwen RHJ, Ewald H. Comparative effectiveness of common therapies for Wilson disease: a systematic review and meta‐analysis of controlled studies. Liver Int 2019;39:2136‐2152. [DOI] [PubMed] [Google Scholar]
- 12. Czlonkowska A, Litwin T, Karlinski M, Dziezyc K, Chabik G, Czerska M. D‐penicillamine versus zinc sulfate as first‐line therapy for Wilson’s disease. Eur J Neurol 2014;21:599‐606. [DOI] [PubMed] [Google Scholar]
- 13. Litwin T, Dziezyc K, Karlinski M, Chabik G, Czepiel W, Czlonkowska A. Early neurological worsening in patients with Wilson’s disease. J Neurol Sci 2015;355:162‐167. [DOI] [PubMed] [Google Scholar]
- 14. Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double‐blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol 2006;63:521‐527. [DOI] [PubMed] [Google Scholar]
- 15. Ferenci P, Stremmel W, Członkowska A, Szalay F, Viveiros A, Stättermayer AF, et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 2019;69:1464‐1476. [DOI] [PubMed] [Google Scholar]
- 16. Valentino PL, Roberts EA, Beer S, Miloh T, Arnon R, Vittorio JM, et al. Management of Wilson disease diagnosed in infancy: an appraisal of available experience to generate discussion. J Pediatr Gastroenterol Nutr 2020;70:547‐554. [DOI] [PubMed] [Google Scholar]
- 17. The Human Gene Mutation Database (HGMD) . The Human Gene Mutation Database. At the Institute of Medical Genetics in Cardiff. 2020. http://www.hgmd.cf.ac.uk/ac/index.php. Accessed August 18, 2021.
- 18. Ferenci P, Roberts EA. Defining Wilson disease phenotypes: from the patient to the bench and back again. Gastroenterology 2012;142:692‐696. [DOI] [PubMed] [Google Scholar]
- 19. Czlonkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord 2009;24:1066‐1069. [DOI] [PubMed] [Google Scholar]
- 20. Woimant F, Poujois A. Monitoring of medical therapy and copper endpoints. In: Weiss KH, Schilsky M, eds. Wilson Disease Pathogenesis, Molecular Mechanisms, Diagnosis, Treatment and Monitoring. Amsterdam: Academic; 2019:223‐232. [Google Scholar]
- 21. Huo X, Armitage J. Use of run‐in periods in randomized trials. JAMA 2020;324:188‐189. [DOI] [PubMed] [Google Scholar]
- 22. Ferenci P, Caca K, Loudianos G, Mieli‐Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003;23:139‐142. [DOI] [PubMed] [Google Scholar]
- 23. Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re‐evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology 2010;52:1948‐1956. [DOI] [PubMed] [Google Scholar]
- 24. Dhawan A, Taylor RM, Cheeseman P, De Silva P, Katsiyiannakis L, Mieli‐Vergani G. Wilson's disease in children: 37‐year experience and revised King’s score for liver transplantation. Liver Transpl 2005;11:441‐448. [DOI] [PubMed] [Google Scholar]
- 25. Petrasek J, Jirsa M, Sperl J, Kozak L, Taimr P, Spicak J, et al. Revised King’s College score for liver transplantation in adult patients with Wilson’s disease. Liver Transpl 2007;13:55‐61. [DOI] [PubMed] [Google Scholar]
- 26. EMEA . Guideline on clinical trials in small populations. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐clinical‐trials‐small‐populations_en.pdf. 2006. Accessed October 31, 2020.
- 27. Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69:89‐95. [DOI] [PubMed] [Google Scholar]
- 28. International Rare Diseases Research Consortium . Patient‐Centered Outcome Measures. Initiatives in the Field of Rare Diseases. https://www.irdirc.org/wp‐content/uploads/2017/12/PCOM_Post‐Workshop_Report_Final.pdf. 2016. Accessed October 31, 2020.
- 29. Sini M, Sorbello O, Sanna F, Battolu F, Civolani A, Fanni D, et al. Histologic evolution and long‐term outcome of Wilson’s disease: results of a single‐center experience. Eur J Gastroenterol Hepatol 2013;25:111‐117. [DOI] [PubMed] [Google Scholar]
- 30. Schilsky ML, Scheinberg IH, Sternlieb I. Prognosis of Wilsonian chronic active hepatitis. Gastroenterology 1991;100:762‐767. [DOI] [PubMed] [Google Scholar]
- 31. Peng Y, Qi X, Guo X. Child‐Pugh versus MELD score for the assessment of prognosis in liver cirrhosis: a systematic review and meta‐analysis of observational studies. Medicine (Baltimore) 2016;95:e2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end‐stage liver disease. Hepatology 2001;33:464‐470. [DOI] [PubMed] [Google Scholar]
- 33. Kim WR, Biggins SW, Kremers WK, Wiesner RH, Kamath PS, Benson JT, et al. Hyponatremia and mortality among patients on the liver‐transplant waiting list. N Engl J Med 2008;359:1018‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aggarwal A, Aggarwal N, Nagral A, Jankharia G, Bhatt M. A novel Global Assessment Scale for Wilson’s Disease (GAS for WD). Mov Disord 2009;24:509‐518. [DOI] [PubMed] [Google Scholar]
- 35. Czlonkowska A, Tarnacka B, Möller JC, Leinweber B, Bandmann O, Woimant F, Oertel WH. Unified Wilson’s Disease Rating Scale—a proposal for the neurological scoring of Wilson’s disease patients. Neurol Neurochir Pol 2007;41:1‐12. [PubMed] [Google Scholar]
- 36. Leinweber B, Möller JC, Scherag A, Reuner U, Günther P, Lang CJG, et al. Evaluation of the Unified Wilson’s Disease Rating Scale (UWDRS) in German patients with treated Wilson’s disease. Mov Disord 2008;23:54‐62. [DOI] [PubMed] [Google Scholar]
- 37. Volpert HM, Pfeiffenberger J, Gröner JB, Stremmel W, Gotthardt DN, Schäfer M, et al. Comparative assessment of clinical rating scales in Wilson’s disease. BMC Neurol 2017;17:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Haan R, Limburg M, Bossuyt P, van der Meulen J, Aaronson N. The clinical meaning of Rankin ‘handicap’ grades after stroke. Stroke 1995;26:2027‐2030. [DOI] [PubMed] [Google Scholar]
- 39. Poujois A, Sobesky R, Meissner WG, Brunet AS, Broussolle E, Laurencin C, et al. Liver transplantation as a rescue therapy for severe neurologic forms of Wilson disease. Neurology 2020;94:e2189‐e2202. [DOI] [PubMed] [Google Scholar]
- 40. Dening TR, Berrios GE. Wilson’s disease. Psychiatric symptoms in 195 cases. Arch Gen Psychiatry 1989;46:1126‐1134. [DOI] [PubMed] [Google Scholar]
- 41. Brewer GJ, Askari F, Dick RB, Sitterly J, Fink JK, Carlson M, et al. Treatment of Wilson’s disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res 2009;154:70‐77. [DOI] [PubMed] [Google Scholar]
- 42. Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson’s disease with zinc: XV long‐term follow‐up studies. J Lab Clin Med 1998;132:264‐278. [DOI] [PubMed] [Google Scholar]
- 43. Pfeiffenberger J, Lohse CM, Gotthardt D, Rupp C, Weiler M, Teufel U, et al. Long‐term evaluation of urinary copper excretion and non‐caeruloplasmin associated copper in Wilson disease patients under medical treatment. J Inherit Metab Dis 2019;42:371‐380. [DOI] [PubMed] [Google Scholar]
- 44. Weiss KH, Askari FK, Czlonkowska A, Ferenci P, Bronstein JM, Bega D, et al. Bis‐choline tetrathiomolybdate in patients with Wilson’s disease: an open‐label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol 2017;2:869‐876. [DOI] [PubMed] [Google Scholar]
- 45. Schmitt F, Podevin G, Poupon J, Roux J, Legras P, Trocello JM, et al. Evolution of exchangeable copper and relative exchangeable copper through the course of Wilson’s disease in the Long Evans Cinnamon rat. PLoS One 2013;8:e82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillaud O, Brunet AS, Mallet I, Dumortier J, Pelosse M, Heissat S, et al. Relative exchangeable copper: a valuable tool for the diagnosis of Wilson disease. Liver Int 2018;38:350‐357. [DOI] [PubMed] [Google Scholar]
- 47. Solovyev N, Ala A, Schilsky M, Mills C, Willis K, Harrington CF. Biomedical copper speciation in relation to Wilson’s disease using strong anion exchange chromatography coupled to triple quadrupole inductively coupled plasma mass spectrometry. Anal Chim Acta 2020;1098:27‐36. [DOI] [PubMed] [Google Scholar]
- 48. Walshe JM. The pattern of urinary copper excretion and its response to treatment in patients with Wilson’s disease. QJM 2011;104:775‐778. [DOI] [PubMed] [Google Scholar]
- 49. Walshe JM. Monitoring copper in Wilson’s disease. Adv Clin Chem 2010;50:151‐163. [DOI] [PubMed] [Google Scholar]
- 50. Dzieżyc K, Litwin T, Chabik G, Czlonkowska A. Measurement of urinary copper excretion after 48‐h d‐penicillamine cessation as a compliance assessment in Wilson’s disease. Funct Neurol 2015;30:264‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liggi M, Mais C, Demurtas M, Sorbello O, Demelia E, Civolani A, et al. Uneven distribution of hepatic copper concentration and diagnostic value of double‐sample biopsy in Wilson’s disease. Scand J Gastroenterol 2013;48:1452‐1458. [DOI] [PubMed] [Google Scholar]
- 52. Ferenci P, Steindl‐Munda P, Vogel W, Jessner W, Gschwantler M, Stauber R, et al. Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s Disease. Clin Gastroenterol Hepatol 2005;3:811‐818. [DOI] [PubMed] [Google Scholar]
- 53. Ping CC, Hassan Y, Aziz NA, Ghazali R, Awaisu A. Discontinuation of penicillamine in the absence of alternative orphan drugs (trientine‐zinc): a case of decompensated liver cirrhosis in Wilson’s disease. J Clin Pharm Ther 2007;32:101‐107. [DOI] [PubMed] [Google Scholar]
- 54. Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med 1987;317:209‐213. [DOI] [PubMed] [Google Scholar]
- 55. Day S, Jonker AH, Lau LPL, Hilgers RD, Irony I, Larsson K, et al. Recommendations for the design of small population clinical trials. Orphanet J Rare Dis 2018;13:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hilgers RD, Bogdan M, Burman CF, Dette H, Karlsson M, König F, et al. Lessons learned from IDeAl—33 recommendations from the IDeAl‐net about design and analysis of small population clinical trials. Orphanet J Rare Dis 2018;13:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hilgers RD, Manolov M, Heussen N, Rosenberger WF. Design and analysis of stratified clinical trials in the presence of bias. Stat Methods Med Res 2020;29:1715‐1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mohlenberghs G, Verbeke G. An Introduction to Generalized (Non)linear Mixed Models. In: de Boeck PW, Wilson M, eds. Explanatory Item Response Models. A Generalized Linear and Nonlinear Approach. New York, NY: Springer‐Verlag New York; 2004:111‐153. [Google Scholar]
- 59. Cope‐Yokoyama S, Finegold MJ, Sturniolo GC, Kim K, Mescoli C, Rugge M, et al. Wilson disease: histopathological correlations with treatment on follow‐up liver biopsies. World J Gastroenterol 2010;16:1487‐1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arnon R, Calderon JF, Schilsky M, Emre S, Shneider BL. Wilson disease in children: serum aminotransferases and urinary copper on triethylene tetramine dihydrochloride (trientine) treatment. J Pediatr Gastroenterol Nutr 2007;44:596‐602. [DOI] [PubMed] [Google Scholar]
- 61. Bauer P, Brannath W. The advantages and disadvantages of adaptive designs for clinical trials. Drug Discov Today 2004;9:351‐357. [DOI] [PubMed] [Google Scholar]
- 62. Eichler HG, Bloechl‐Daum B, Bauer P, Bretz F, Brown J, Hampson LV, et al. “Threshold‐crossing”: a useful way to establish the counterfactual in clinical trials? Clin Pharmacol Ther 2016;100:699‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brakenhoff TB, Roes K, Nikolakopoulos S. Bayesian sample size re‐estimation using power priors. Stat Methods Med Res 2019;28:1664‐1675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material