
Figure 1.
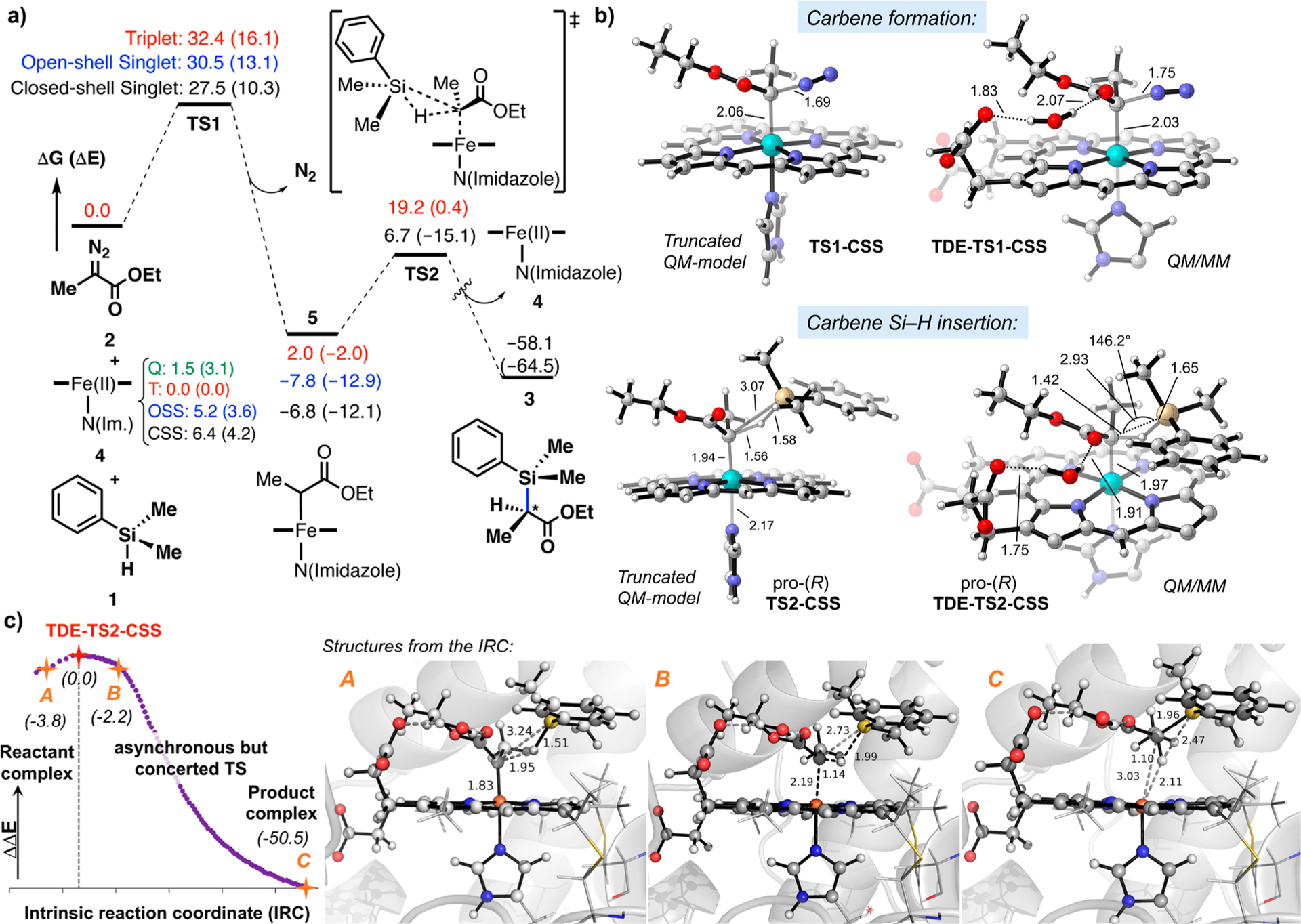
(a) Computed reaction pathway for carbene Si–H insertion reaction between PhMe2SiH 1 and Me-EDA 2 catalyzed by a model imidazole-ligated iron porphyrin (truncated DFT model). Results with three iron electronic states are given. The calculated intrinsic electronic and Gibbs free energies are given in kcal·mol−1. Gibbs free energies do not consider the formation of substrate–substrate or enzyme–substrate reactant complexes, thus neglecting the stabilizing interactions that overcome the unfavorable −TΔS term of a bimolecular reaction (usually about 12 kcal· mol−1 in the free energies). (b) Optimized geometries for the lowest in energy TS1-CSS and TS2-CSS obtained from truncated QM and QM/MM models (only atoms included in the QM region are shown for clarity). (c) Intrinsic reaction coordinate (IRC) calculation for the lowest energy Si–H carbene insertion transition state (TDE-TS2-CSS) calculated using QM/MM (atoms included in the QM region are shown in ball and stick format). Relative electronic energies are given in kcal·mol−1. Key distances and angles are shown in Å and degrees.