
Figure 3.
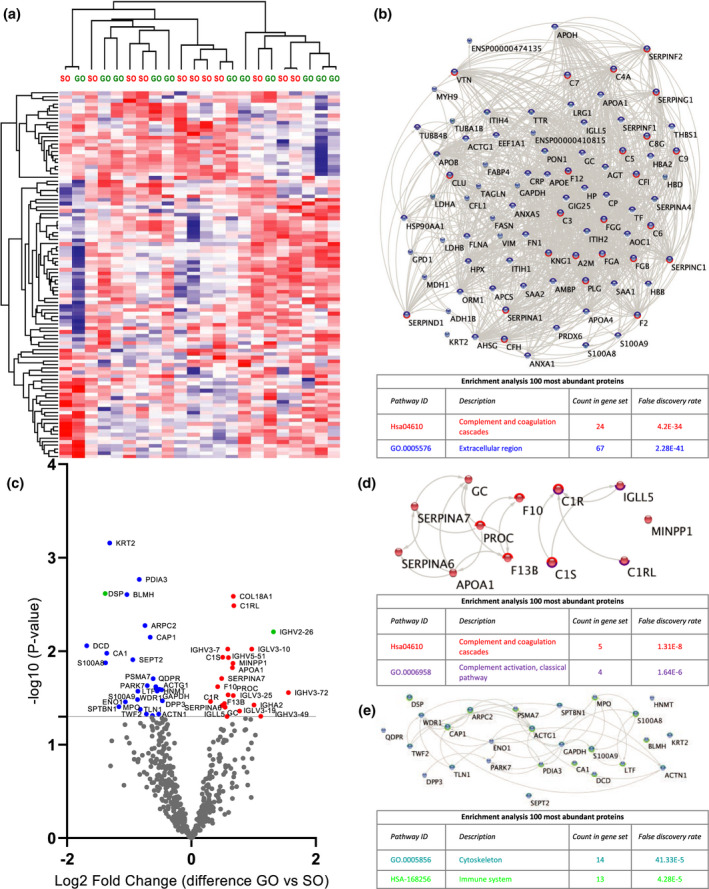
Quantitative proteomic analysis of perfusate samples taken before the termination of HMP (T2). (a) Heat map and hierarchical clustering of 100 most abundant proteins at T2. (b) STRING analysis of 100 most abundant proteins at T2. Nodes circled with blue represent proteins located in extracellular regions (FDR: 2.28E‐41) and red represent functional enrichment of the KEGG complement and coagulation pathway (FDR: 4.2E‐34). (c) Volcano plot showing differential protein expression of 498 identified proteins at T2 between good outcome (GO) and suboptimal outcome (SO) at 1‐year post‐transplantation. X‐axis demonstrates protein level difference indicated by log2 fold change; Y‐axis demonstrates statistical significance indicated by ‐log10 (P‐value). A ‐log10 (P‐value) of > 1.3, and a fold change of > 0 was considered significant. Blue dots represent significant downregulated proteins. Red dots represent significantly upregulated proteins. Green dots represent proteins identified using prediction models. (d) STRING pathway analysis of significantly upregulated proteins at T2. Nodes circled with red represent functional enrichment of the KEGG complement and coagulation pathway (FDR 1.21e‐8) and purple represents proteins involved in complement activation of the classical pathway (FDR: 1.6E‐6). (e) STRING analysis of significantly downregulated proteins at T2. Nodes circled with turquoise represent functional enrichment GO component cytoskeleton (FDR 1.33e‐5) and green represents proteins affiliated with the immune system (FDR: 4.28E‐5). FDR, false discovery rate.