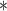Table 1.
Comparison of benchmarked deconvolution methods. Sequencing-based methods have three common criteria in the informative region selection: number of CpG, region size and read coverage. Some criteria were altered to be more suitable for the dataset we used in our analyses. For array-based methods, we specifically designated CpG sites based on methylation variance or ctDMRs. Furthermore, we described differences in cell-type composition estimation step based on three criteria again: reference-requirements, number of detectable subpopulations and estimation scope
| Method | Main data type | Informative region selection | Cell-type composition estimation | ||||
|---|---|---|---|---|---|---|---|
| # CpG | Region size (bp) | Coverage | Class | # Components | Estimation Scope | ||
| BED | RRBS | >4 |
NA
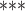
|
>20 | ref-based | 2 | local |
| ClubCpG | WGBS | >4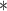
|
100 | >20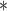
|
ref-based | 2 or more | global |
| csmFinder + coMethy | WGBS | >4 |
NA
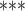
|
>10 | ref-free | 2 or more | global |
| DXM | Any kinds of BS-seq | Promoter and CpG island regions | >4 | ref-free | 2 or more | global | |
| MethylPurify | WGBS | >10 | 300/200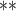
|
>10 | ref-free | 2 | local |
| Prism | RRBS | >4 |
NA
|
>20 | ref-free | 2 or more | local |
| Houseman | Methylation microarrays | CpGs overlapping with ctDMRs | ref-based | 2 or more | global | ||
| MeDeCom | Methylation microarrays | CpGs showing high methylation variance across pseudo-bulks | ref-free | 2 or more | global | ||