

Abstract
Objective
Germline loss‐of‐function mutations in DEPDC5, and in its binding partners (NPRL2/3) of the mammalian target of rapamycin (mTOR) repressor GATOR1 complex, cause focal epilepsies and increase the risk of sudden unexpected death in epilepsy (SUDEP). Here, we asked whether DEPDC5 haploinsufficiency predisposes to primary cardiac defects that could contribute to SUDEP and therefore impact the clinical management of patients at high risk of SUDEP.
Methods
Clinical cardiac investigations were performed in 16 patients with pathogenic variants in DEPDC5, NPRL2, or NPRL3. Two novel Depdc5 mouse strains, a human HA‐tagged Depdc5 strain and a Depdc5 heterozygous knockout with a neuron‐specific deletion of the second allele (Depdc5 c/−), were generated to investigate the role of Depdc5 in SUDEP and cardiac activity during seizures.
Results
Holter, echocardiographic, and electrocardiographic (ECG) examinations provided no evidence for altered clinical cardiac function in the patient cohort, of whom 3 DEPDC5 patients succumbed to SUDEP and 6 had a family history of SUDEP. There was no cardiac injury at autopsy in a postmortem DEPDC5 SUDEP case. The HA‐tagged Depdc5 mouse revealed expression of Depdc5 in the brain, heart, and lungs. Simultaneous electroencephalographic–ECG records on Depdc5 c/− mice showed that spontaneous epileptic seizures resulting in a SUDEP‐like event are not preceded by cardiac arrhythmia.
Interpretation
Mouse and human data show neither structural nor functional cardiac damage that might underlie a primary contribution to SUDEP in the spectrum of DEPDC5‐related epilepsies. ANN NEUROL 2022;91:101–116
Sudden unexpected death in epilepsy (SUDEP) is a tragic outcome that is the most common cause of epilepsy‐related death, 1 with an incidence of 1.2 per 1,000 person‐years in adults and 0.2 per 1,000 person‐years in children. 2 , 3 Although the pathophysiological mechanisms of SUDEP are still unclear, 3 explanations predominate: a cerebral shutdown associated with a generalized tonic–clonic seizure (GTCS), an autonomic dysfunction, and/or a cardiorespiratory failure. 4 , 5 , 6 , 7 Cardiac dysfunction has received much attention because SUDEP shares features with sudden cardiac death. 8 , 9 , 10 Cardiac arrhythmia can be caused by mutations in ion channel genes (SCN1A, KCNA1, KCNQ1, CACNA1A) 8 , 9 , 10 , 11 or by seizure‐induced physiological changes. 5 , 6 , 12 , 13 Discriminating between the two hypotheses would have clinical implications since genetic cardiac dysfunction leading to fatality may be preventable and indicate a need for cardiological surveillance in patients at high risk of SUDEP. 14
SUDEP was recently reported in epileptic patients with mutations in the non–ion‐channel epilepsy gene DEPDC5, as well as its binding partners NPRL2 and NPRL3, which form the GATOR1 complex. 15 , 16 , 17 , 18 First reported in two individuals from the same family, 15 DEPDC5 variants were then found in 10% of 61 SUDEP cases in an exome‐based study. 16 SUDEP was also reported in 10% of a cohort of 73 GATOR1‐related families. 17 Furthermore, sudden death is observed in rodent models of Depdc5 deficiency. 19 , 20 , 21 , 22 How DEPDC5 mutations may contribute to SUDEP is unknown. The GATOR1 complex is part of the amino‐acid–sensing branch, which inhibits complex 1 of the mammalian target of rapamycin (mTOR) pathway. 23 In this complex, DEPDC5 is the central component, which interacts directly with the Rag guanosine triphosphatases to regulate mTOR complex 1 (mTORC1) activity. 24 The mTOR signaling pathway regulates key cellular functions promoting cell growth and metabolism in response to environmental cues including growth factors, hormones, and nutrients. 25 Germline mutations in the DEPDC5 gene are the most frequent cause of genetic focal epilepsies; mutations in NPRL2 and NPRL3 are also encountered, although more rarely. 17 , 18 , 26 , 27 , 28 , 29 GATOR1 mutations act typically as a loss of function leading to constitutive activation of the mTOR pathway, as observed in resected brain tissues from operated patients with refractory epilepsy and mouse models. 18 , 19 , 20 , 21 , 22 , 30 Recently, DEPDC5 brain somatic second‐hit mutations occurring on top of germline DEPDC5 heterozygous mutations have been shown to cause focal cortical dysplasia (FCD) type 2, a localized cortical malformation manifesting with intractable epilepsy. 19 , 30 , 31 , 32 , 33
Preventive strategies to allay the risk of SUDEP in individuals with GATOR1 mutations require the identification of biomarkers to predict SUDEP susceptibility and understanding of how seizures lead to death. Here, to test the hypothesis that mutations in DEPDC5 (or in NPRL2 and NPRL3) may induce primary cardiac alterations predisposing to SUDEP, we conducted cardiac investigations in a cohort of 16 GATOR1 patients and assessed by simultaneous electroencephalographic (EEG)–electrocardiographic (ECG) records cardiac‐related mechanisms in a novel mouse model of Depdc5 deficiency presenting with fatal spontaneous seizures, resembling SUDEP‐like events.
Materials and Methods
Study Approval
Written informed consent was obtained from all participants (French cohort: C14‐56 GENEPI; UK cohort: 11/LO/2016). Mouse studies were approved by the French Ministry of Research (no. APAFIS#3506).
Cardiac Investigation in Patients
We enrolled a cohort of 16 patients with pathogenic mutations in DEPDC5 (n = 14), NPRL2 (n = 1), or NPRL3 (n = 1), among which 6 were previously reported. 17 , 18 Genetic testing was performed antemortem from genomic blood DNA. Cardiac and neurological characteristics including patient history, physical examination, 12‐lead ECGs, and in some cases Holter monitor and transthoracic echocardiographic (TTE) records were collected retrospectively.
Brain and Heart Postmortem Examination of a SUDEP Case
Postmortem examination of heart and brain tissue from a patient with a DEPDC5 mutation was conducted by an expert pathologist (H.A.‐B). Samples of different regions from whole fixed brain and heart were assessed using a comprehensive gross and histologic examination as previously performed. 34
Generation of Depdc5 Strain Mice
Two mouse strains were generated by genOway (Lyon, France) on a C57BL6/J background: a mouse carrying a human HA‐tagged Depdc5 flanked by LoxP sequences at the endogenous locus of Depdc5 and a heterozygous deletion of Depdc5 termed Depdc5 +/−. The universal Cre‐driver synapsin1‐Cre mouse (B6.Cg‐Tg(Syn1‐cre)671Jxm/J) was imported from Jackson Laboratory (Bar Harbor, ME).
Generation of Depdc5 HA‐Tagged and Floxed Mice
HA‐Depdc5 flox/flox mice were generated by flanking exons 1–3 with LoxP sites and an HA tag in the N‐terminal region. An HA tag in the N‐terminal part of Depdc5 was previously reported not to affect the protein interactions, and preserve the role of Depdc5 as a repressor of mTORC1. 23 HA‐Depdc5 flox/flox mice are viable and show no gross physical differences from wild‐type littermates. To simplify, theses mice are termed "HA‐Depdc5."
Generation of Depdc5 Conditional Knockout Mice
Depdc5 conditional knockout (KO) mice were generated by breeding Depdc5 +/− males and Syn1‐Cre+ females to obtain Depdc5 +/−;Syn1‐Cre+ mice. Depdc5 +/−;Syn1‐Cre+ females were then crossed with Depdc5 flox/flox males to generate 4 genotypes, among which 3 were used: Depdc5 c/− conditional KO (Depdc5 flox/−;Syn1‐Cre+), Depdc5 +/− constitutive heterozygous (Depdc5 flox/−;Syn1‐Cre−), and Depdc5 +/+ control wild type (Depdc5 flox/+;Syn1‐Cre−). Genomic DNA was extracted from tail biopsies. All mice were genotyped by polymerase chain reaction at the iGenSeq sequencing facility of the Paris Brain Institute (ICM). Three primer pairs were used to genotype Depdc5 alleles: 2 forwards (CCTTTCAGCCGAACAACCACCAGT and GCACCAGAAGTCAGATCTCATTATGG) and one reverse (GCTCTCATTTCCAACCATCCCTG). All breeding couples were previously back‐crossed for >10 generations into the C57BL/6J background.
Mice were housed in groups of 2 to 6 in standard plastic cages on a 12‐hour light/dark cycle under pathogen‐free conditions at the ICM animal core facility. Animals implanted for EEG and ECG records were housed singly. Water and food were provided ad libitum. Animals were treated according to the guidelines of the European Community. All efforts were made to minimize the number and the suffering of animals. Both males and females were used in the study.
Western Blots
Mouse organs were processed as previously described. 35 The following primary antibodies were used: rat anti‐HA (1/1,000, 3F10; Sigma‐Aldrich, St Louis, MO), rabbit anti‐Depdc5 (1/250, ab185565; Abcam, Cambridge, UK), rabbit anti–phospho‐S6 ribosomal protein (1/500, 5,364; Cell Signaling Technology, Danvers, MA), and rabbit anti–α‐actin antibody (1/1,000, A2066, Sigma‐Aldrich); secondary antibodies were antirat horseradish peroxidase linked (1/2,000, 7,077, Cell Signaling Technology) and antirabbit peroxidase stabilized (1/2,000, 32,460; Invitrogen, Carlsbad, CA). Protein expression was quantified by densitometry using Fiji software and normalized against actin.
Immunohistochemistry
Adult (postnatal day 85 [P85]) mouse brains were collected after perfusion with 4% paraformaldehyde (PFA), as previously described. 35 All immunostainings were performed on sections from the somatosensory cortex, in duplicate in 3 sections per animal (n = 2 mice per genotype). Primary antibodies used were rabbit anti‐HA (1/1,500, C29F4, Cell Signaling Technology), mouse anti‐NeuN (1/200, MAB377; Millipore, Billerica, MA), mouse anti‐Map2 (1/500, ab11267, Abcam), mouse anti‐CamkII (1/500, ab22609, Abcam), rat anti‐LAMP1 (1/500, 1D4B; DSHB, Iowa City, IA), goat anti‐Gad67 (1/400, MAB5406, Millipore), mouse antiparvalbumin (anti‐PV; 1/1,000, P3088, Sigma), mouse anti‐Gfap (1/300, MA5‐15086; Thermo Fisher Scientific, Waltham, MA), rat anti‐Plp (1/10, DSHB), mouse anti‐Olig2 (1/400, ab236540, Abcam), and goat anti‐Iba1 (1/500, 011‐27991; Wako, Osaka, Japan). Secondary antibodies were donkey antirabbit Alexa Fluor 555, antirat Alexa Fluor 488, antigoat Alexa Fluor 488, and antimouse Alexa Fluor 488 (1/1,000; Invitrogen A32794, A‐21208, A32814, and A32766, respectively). Images were acquired using Zen software with an upright, widefield, apotome Zeiss (Oberkochen, Germany) microscope and a confocal Leica (Wetzlar, Germany) SP8 microscope. Quantification of colocalization was performed using the JaCoP plugin in ImageJ software. The fraction of cell types expressing Depdc5 was assessed using ImageJ software (using automatic particle counting).
Adult (P85) mouse hearts were collected before seizure onset or after SUDEP and were fixed in PFA 4%. Transverse sections (20μm) were cut and stained with hematoxylin and eosin (HE; n = 3 Depdc5 +/+ hearts, 3 Depdc5 c/− hearts before seizures, and 4 Depdc5 c/− after SUDEP).
Simultaneous EEG‐ECG Recordings
Two groups of mice were used: one for EEG recordings (n = 10 Depdc5 c/−, n = 7 Depdc5 +/+, and n = 3 Depdc5 +/− mice) and one for simultaneous EEG‐ECG recordings (n = 23 Depdc5 c/− and n = 4 Depdc5 +/+ mice). Mice were anesthetized (2–4% isoflurane) under analgesia (0.1mg/kg buprecare for 48 hours). EEG recording electrodes were implanted into mice placed in a stereotaxic frame. Homemade enamel‐coated stainless‐steel electrodes (#791400; A‐M Systems, Sequim, WA) were placed in primary motor cortex (bregma: anteroposterior [AP], 2.2mm; mediodorsal [MD], 2.2mm) and the lateral parietal association cortex (bregma: AP, −1.8mm; MD, −1.2mm). A bipolar electrode was placed in the left hippocampus (bregma: AP, −1.8mm; MD, −1.8mm; dorsoventral, −1.8mm) and a reference electrode on the cerebellum. Coordinates were derived and adjusted from the Franklin and Paxinos atlas. 36 ECG records were obtained from 2 electrodes placed by tunneling subcutaneously, one in the right scapular region and the other in the left abdominal muscle. 37 After a 1‐week recovery period, implanted mice were permitted free movement, connected to an analog to digital converter amplifier (Brainbox EEG‐1166) as part of a video‐EEG acquisition system (DELTAMED, Natus Medical Incorporated, Pleasanton, CA). EEG signals were acquired at 2kHz and pass band filtered (0.5–70Hz). Video was recorded at 25 frames/s, synchronized to electrophysiological signals. For the EEG group, recordings were made 24/24 over 4 to 6 consecutive days from P21 until P117, or until a fatal seizure; for the EEG‐ECG group, recordings were made 24/24, 7/7 from P80 until a fatal seizure. Seizures were detected electrically as EEG spikes causing a rapid increase in signal amplitude and power analyzed over a sliding window of 20 seconds. Signal analysis for representative images was made using MATLAB (MathWorks, Natick, MA). Morlet wavelets were computed from 0.5 to 45Hz at a resolution of 0.1Hz, over a sliding time window of 1 second, updated each 0.1 second. Events were detected on both time and frequency criteria. RR intervals were analyzed with Spike2 software, and heart rate was calculated as number of RRs in 2 seconds (Spike2 v7.06; Cambridge Electronic Design, Cambridge, UK).
Heart rate variability (HRV) was assessed by different parameters: standard deviation of all RRs (SDRR), square root of mean of successive square differences between RRs (rMSSD), and using Poincaré plots. 38 , 39 This latter method graphs the time between one pair of QRS complexes on an ECG (RR) versus the time between the next pair of QRS complexes (RR’). The SD1 and SD2 refer to short‐term and long‐term adaptation, respectively. For all Poincaré plots, more than 500 pairs of RR intervals were plotted and analyzed. Recordings during animal movements were excluded to eliminate artifacts.
ECG Recordings in Anesthetized Mice
Mice (n = 18) were used for this study (n = 9 Depdc5 c/− and n = 9 Depdc5 +/+ mice) aged P80. The QRS component of the ECG was recorded from anesthetized mice (2–4% isoflurane) kept warm at 35–37°C with a heating pad. A specific ECG recording system was used (ElectroMyography; Neurosoft, Athens, Greece) to amplify records made with a needle electrode (TE/B50600‐001; Technomed, Maastricht, the Netherlands) inserted subcutaneously near the heart. Ground signals were obtained from a needle electrode (N3512P150; Spes Medica, Genoa, Italy) inserted into the back of the animal. Signals were band pass filtered (0.02–10kHz) and analyzed with Spike2 software. RR interval length and PR, QT, and QRS lengths were measured, and SDRR and rMSSD were calculated.
Statistical Analysis
The number of mice per group was as determined from the resource equation method to sample size and is indicated in each figure legend. 40 Normality distributions of data were tested with the Shapiro–Wilk normality test. Unpaired 2‐tailed t tests or Mann–Whitney tests were used to compare sets of data obtained from independent groups of animals (Depdc5 +/+ vs Depdc5 c/−, or fatal vs nonfatal groups). For survival, log‐rank test was performed. Two‐way analysis of variance test was performed for comparisons that included >2 groups. All statistical analyses were performed with Prism 7 (GraphPad Software, San Diego, CA), as described in figure legends. Statistical significance was set at α < 0.05.
Results
Cardiac Investigations in the GATOR1 Patient Cohort
We explored cardiac function in subjects carrying mutations in GATOR1‐encoding genes and suffering recurrent seizures. Table 1 presents the cohort that comprised patients with germline pathogenic nonsense variants in DEPDC5 (n = 14), NPRL2 (n = 1), or NPRL3 (n = 1). All patients were diagnosed with focal epilepsy (Table 2). Age at epilepsy onset ranged from 1 to 27 years (mean = 8 years and 3 months). A family history of SUDEP was documented in 6 patients. Two females with DEPDC5 mutations aged 18 and 50 years (Patients 1 and 2) and 1 male patient aged 20 years (Patient 16) had definite (1/3) and probable (2/3) SUDEP after they were recruited into the genetic study. Notably, 2 of these 3 patients (Patients 1 and 2) were seizure‐free for 8 months and 9 years before SUDEP.
TABLE 1.
Genetic Features and SUDEP History of the Patient Cohort
| Case | Gender | Gene: Variant | SUDEP | Family History of SUDEP | Ref |
|---|---|---|---|---|---|
| 1 | F | DEPDC5: p.Arg874* | Definite SUDEP during night at 18 yr | Paternal granduncle died of probable SUDEP at 59 yr after a diurnal sz | 18 |
| 2 | F | DEPDC5: p.Glu1385* | Probable SUDEP during night at 50 yr | No | 18 |
| 3 | M | DEPDC5: p.Glu1421Argfs* | No | No | 17 |
| 4 | M | DEPDC5: p.Met152fs | No | No | 17 |
| 5 | F | NPRL2: p.Ile23Asnfs*6 | No | Uncle died of probable SUDEP at 22 yr | 17 |
| 6 | F | NPRL3: p.Arg424* | No | Half‐sister died of probable SUDEP at 25 yr, and two cousins had near‐SUDEP at 30 yr | 17 |
| 7 | M | DEPDC5: p.Arg243* | No | No | |
| 8 | F | DEPDC5: p.Tyr1082* | No | Father died of probable SUDEP | |
| 9 | F | DEPDC5: p.Gln981* | No | No | |
| 10 | F | DEPDC5: p.Arg422* | No | No | |
| 11 | F | DEPDC5: p.Arg264GlufsTer9 | No | Father died of possible SUDEP at 40 yr | |
| 12 | M | DEPDC5: c.3564‐3C > G | No | No | |
| 13 | F | DEPDC5: p.E448* | No | Brother died of definite SUDEP at 21 yr | |
| 14 | M | DEPDC5: p.R1087* | No | No | |
| 15 | M | DEPDC5:c.280‐1G > A | No | No | |
| 16 | M | DEPDC5: c.523_526del:p.I175Rfs*2 | Probable SUDEP during night at 20 yr | No |
DEPDC5 (Refseq NM_001242896; NP_001229825), NPRL2 (Refseq NM_006545; NP_006536), and NPRL3 (Refseq NM_001077350; NP_001070818).
F = female; M = male; Ref = reference; SUDEP = sudden unexpected death in epilepsy; sz = seizure.
TABLE 2.
Epilepsy Features of the Patient Cohort
| Case | Epilepsy Type; Age at Onset | Seizure Frequency | Antiseizure Medication; Outcome | Sodium Channel Blocker | Brain MRI | EEG/sEEG |
|---|---|---|---|---|---|---|
| 1 a | SHE; 7 yr | NA | Low dosage of CBZ (recurrence after withdrawal); good outcome | Yes | Negative (1.5T) | Interictal EEG: normal |
| 2 a | SHE; 12 yr | 4–10 sz/yr | TPM + LTG, CBZ added at 44, sz‐free for 8 mo before SUDEP | Yes | Negative | EEG: bifrontal epileptic activity |
| 3 | SHE; 5 yr | Several sz/night several days per week | CBZ, LEV | Yes | Negative (1.5T) | Interictal EEG: normal; sEEG: right insula onset |
| 4 | SHE; 4 yr | 1 nocturnal convulsive sz/month | CLB, OXC, PER; drug‐resistant | Yes | Negative | Ictal EEG: right frontocentral seizure |
| 5 | FLE (operated at 18 yr: FCD 1); 3 yr | 1 nocturnal convulsive sz/month | LAC, CLB; drug‐resistant | Yes | Negative (1.5T) | sEEG: right frontobasal and insular onset |
| 6 | FLE; 8 yr | Monthly to weekly nocturnal focal frontal sz | OXC, LTG, VPA, PER; drug‐resistant | Yes | Negative (1.5T) | EEG: frontal epileptic activity |
| 7 | FLE (left frontal lobectomy, sz recurrence afterward); 11 yr | 2 clusters of 2 focal to bilateral tonic–clonic sz/month | VPA, LTG, PHE, rescue protocol with midazolam after the first sz; drug‐resistant | Yes | Negative (3T) | EEG: left frontal lobe epilepsy; sEEG: left orbitofrontal lobe epilepsy |
| 8 | Focal epilepsy, localization unclear, 7 yr | 2–4 focal to bilateral tonic–clonic sz/month, almost all arising from sleep | LTG, TPM | Yes | Negative (3T) | EEG (3 days VT): focal epilepsy, localization unclear (most likely left frontotemporal) |
| 9 | Focal epilepsy, left neocortical temporal onset; 4 yr | 1–2 focal sz with or without impaired awareness every 2 weeks, rare focal to bilateral tonic–clonic sz | LEV, VPA, clobazam | No | Negative (3T) | EEG (VT): neocortical focal epilepsy, possible left lateral temporal region |
| 10 | FTE, lateralization unclear; 1 yr | Brief blank spells 2–3 times per week; no focal to bilateral tonic–clonic sz | VPA, LAC | Yes | Negative (3T) | EEG (VT): interictal epileptiform discharges from both temporal regions |
| 11 | FTE; 7 yr | No clear sz, but frequent syncopal episodes (most likely of autonomic origin) | LTG | Yes | Mild cerebellar atrophy, small right parafalcine meningioma (3T) | EEG: bitemporal interictal epileptiform activity, more prominent on the left |
| 12 | Focal epilepsy, left parietal onset; surgically resected FCD type 2; 7 yr | sz‐free after lesionectomy | LAC, PER, TPM, clobazam | Yes | Left parietal FCD (3T) | Presurgical VT: ictal bradycardia down to 42 bpm |
| 13 | Focal epilepsy, left hemispheric onset; 11 yr | 3–7 sz/week (30–40% focal to bilateral tonic–clonic sz; others: focal impaired awareness sz) | OXC, TPM, rescue protocol with lorazepam and CLB | Yes | Negative (3T) | VT: focal epilepsy, left hemispheric onset, localization unclear |
| 14 | Focal epilepsy; 10 yr | sz‐free for 3 yr | LEV, LTG, CBZ | Yes | Negative (3T) | 24‐h ambulatory EEG (23 yr): normal |
| 15 | FTE, 27 yr | sz‐free for the past 2 yr | BRV, ZNS, pregabalin | No | Negative (3T) | 24‐h ambulatory EEG: left temporal slow, no clear interictal epileptiform abnormalities |
| 16 a | Focal epilepsy; 7 yr | 1 unprovoked nocturnal convulsive sz every 2–3 mo (but issues with medication compliance) | OXC | Yes | Negative | 24 h ambulatory EEG: bihemispheric cortical dysfunction, suggestive of multifocal irritative regions |
Succumbed to SUDEP.
bpm = beats per minute; BRV = brivaracetam; CBZ = carbamazepine; CLB = clonazepam; EEG = electroencephalogram; FCD = focal cortical dysplasia; FLE = frontal lobe epilepsy; FTE = focal temporal epilepsy; LAC = lacosamide; LEV = levetiracetam; LTG = lamotrigine; MRI, magnetic resonance imaging; NA = not available; OXC = oxcarbazepine; PER = perampanel; PHE = phenytoin; sEEG = stereotaxic EEG; SHE = sleep‐related hypermotor epilepsy; SUDEP = sudden unexpected death in epilepsy; sz = seizure(s); TPM = topiramate; VPA = valproate; VT = video telemetry; ZNS = zonisamide.
Age at cardiac investigation ranged from 16 to 60 years (mean = 39 years and 9 months; Table 3). Patient 11 (with family history of SUDEP) had a history of recurrent syncopal episodes, unrelated to epilepsy and most likely of autonomic origin, as orthostatic intolerance was diagnosed. In the cohort, no abnormal heart sounds were detected in cardiac auscultations. Twelve‐lead ECG monitoring revealed PR, QRS, and QTc intervals that fell within normal ranges in 14 of 16 patients (87.5%); the exceptions were 1 NPRL3 subject (Patient 6) and 1 DEPDC5 subject (Patient 10), who presented with an isolated incomplete right bundle branch block. No ST segment changes were evident, indicating normal ventricular repolarization. One patient (Patient 12) showed borderline left axis deviation and subtle T abnormalities, with normal QTc and PR intervals. In the context of presurgical evaluation of Patient 12, the video‐EEG‐ECG telemetric system captured 4 episodes of ictal bradycardia with heart rate down to 42 beats per minute (bpm), which were followed with postictal heart rate normalization. Holter monitoring for 24 hours to 8 continuous days revealed no major cardiac arrhythmias in 4 of 4 patients, including 3 (Patients 5, 6, and 13) with a family history of SUDEP. Two‐dimensional TTE excluded any major structural or functional heart disease in 6 of 6 patients. Most patients (14/16) received sodium channel blockers as antiepileptic medication (see Table 2), among whom were the 3 patients who died from SUDEP. Overall, these data provide no evidence that baseline cardiac parameters can accurately predict sudden death in patients with GATOR1 pathogenic variants, including in high‐risk patients who have a family history of SUDEP.
TABLE 3.
Clinical Cardiac Features of the Patient Cohort
| Case | Age at Cardiac Exam | 12‐Lead ECG | Holter | TTE |
|---|---|---|---|---|
| 1 a | 16 yr | Normal (HR = 68 bpm, PR < 200 ms, QRS < 120 ms, QTc < 460 ms) | NA | Normal |
| 2 a | 43 yr | Normal (HR = 75 bpm, PR = 160 ms, QRS = 60 ms, QTc = 390 ms) | NA | NA |
| 3 | 36 yr | Normal including an optimal cardiac stress test (HR = 75 bpm, PR = 114 ms, QRS = 78 ms, QTc = 389 ms) | Normal (24‐h monitoring with rare SVES) | Normal |
| 4 | 32 yr | Normal (HR = 79 bpm, PR = 140 ms, QRS < 80 ms, QTc < 440 ms) | Normal (24‐h monitoring with rare SVES) | Normal |
| 5 | 29 yr | Normal (HR = 80 bpm, PR = 180 ms, QRS = 60 ms, QTc = 362 ms) | Normal (24‐h monitoring with rare SVES and 1 VES) | Normal (isolated subtle type I mitral regurgitation) |
| 6 | 47 yr | Subnormal; isolated incomplete right bundle branch block (HR = 62 bpm, PR = 180 ms, QRS = 100 ms, QTc = 360 ms) | Normal (8‐day monitoring with rare VES) | Normal (isolated ventricular septal aneurysm) |
| 7 | 28 yr | Normal (HR = 78 bpm, PR = 144 ms, QRS = 106 ms, QTc = 424 ms) | NA | NA |
| 8 | 47 yr | Normal (HR = 61 bpm, PR = 142 ms, QRS = 80 ms, QTc = 422 ms) | NA | NA |
| 9 | 51 yr, 60 yr | 51 yr: normal (HR = 60 bpm); 60 yr: junctional rhythm (HR = 52 bpm, QRS = 86 ms, QTc = 407 ms) | NA | NA |
| 10 | 41 yr | Normal with incomplete right bundle branch block (HR = 66 bpm, PR = 164 ms, QRS = 100 ms, QTc = 448 ms) | NA | NA |
| 11 | 59 yr | Normal (HR = 71 bpm, PR = 152 ms, QRS = 76 ms, QTc = 434 ms); 24‐h monitoring: HR = 80 bpm; positive tilt testing: mild sympathetic failure and orthostatic intolerance | NA | NA |
| 12 | 46 yr | Normal (HR = 70 bpm, borderline left axis deviation, PR = 170 ms, borderline T‐wave abnormalities, QTc = 413 ms) | NA | NA |
| 13 | 35 yr, 45 yr | 35 yr: normal (HR = 68 bpm, PR = 176 ms, QRS = 88 ms) | Normal | 45 yr: normal |
| 14 | 23 yr | Normal (HR = 64 bpm, PR = 140 ms, QRS = 90 ms, QTc = 418 ms) | NA | NA |
| 15 | 50 yr | Normal (HR = 70 bpm, PR = 154 ms, QRS = 86 ms, QTc = 403 ms); cardiac computed tomography: angiogram for chest pain, normal | NA | NA |
| 16 a | 18 yr | Normal (HR = 50 bpm, PR = 136 ms, QRS < 120 ms, QTc < 450 ms) | NA | NA |
Succumbed to sudden unexpected death in epilepsy.
bpm = beats per minute; ECG = electrocardiogram; HR = heart rate; NA = not assessed; QTc = corrected QT; SVES = supraventricular extrasystole; TTE = Transthoracic echocardiography; VES = ventricular extrasystole.
Postmortem Cardiac Examination of a SUDEP DEPDC5 Case
Patient 1 had nonlesional sleep‐related hypermotor epilepsy since the age of 7 years; seizures responded well to carbamazepine from the age of 9 years. She carried a paternally inherited germline pathogenic DEPDC5: c.2620C>T/p.Arg874* variant. SUDEP was suspected in the sudden death of her paternal granduncle after a diurnal seizure. No specific anomalies were detected in the ECG and TTE examinations that were performed at the age of 16 years in the context of a possible familial SUDEP history (see Tab1, 3). At the age of 18 years, Patient 1 died during sleep. A probable seizure that night was identified on questioning the family (noise of convulsive movements), although the patient had been seizure‐free on carbamazepine for 9 years.
Possible medical causes of unnatural death, including illicit drug use, were excluded. Definite SUDEP was confirmed following autopsy, which included the heart and brain examination, and did not reveal any abnormalities. The heart weighed 279g. Histological examination of mapped, labeled blocks of myocardium from representative transverse slices of both ventricles was performed (Fig 1). No lesions were identified in the coronary arteries, the myocardium, or the cardiac valves. No ischemic sequelae, thrombus, fibrosis, or scars were evident. Moreover, we did not detect contraction band necrosis, a common finding in neurocardiac death (see Fig 1C–E). 41 Brain autopsy was also performed. The brain weighed 1,630g, which is 2.5 standard deviations above the average for age and gender, suggesting macrocephaly. Macroscopic examination excluded contributions of any other major cerebral injury to the SUDEP.
FIGURE 1.
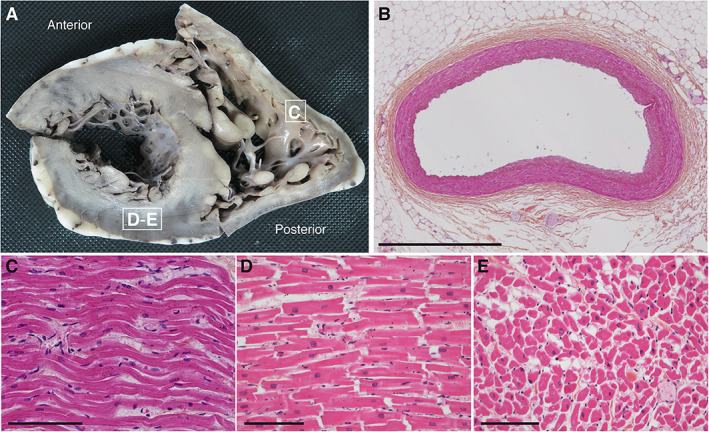
Macroscopic and microscopic pictures of the heart autopsy of Patient 1. (A) Short axis view of a transverse section of the right and left ventricles. Morphological cardiovascular examination including valves and coronary arteries inspection revealed no gross abnormalities. White squares indicate sites of sections used for histological analysis in C–E. (B–E) Hematoxylin and eosin–stained transversal sections of the anterior interventricular artery (B), longitudinal sections of the right ventricle myocardium (C), the posterior wall of the left ventricle myocardium (D), and cross‐sectional section of the left ventricle (E). No contraction band necrosis was detected. Scale bars represent 1mm (B) or 100μm (C–E).
There was no evidence for primary cardiac dysfunction leading to SUDEP in this DEPDC5 case. To further investigate the role of DEPDC5 in SUDEP pathomechanism, we generated mice models of Depdc5 haploinsufficiency.
Depdc5 Mouse Spatiotemporal Expression Pattern
Although interest in the DEPDC5 epilepsy gene has tremendously increased, the pattern of expression of Depdc5 protein remains unclear, because no specific antibody is available for immunohistochemistry. To provide this missing information, we generated a mouse strain expressing an endogenous version of Depdc5 HA‐tagged at the N‐terminus. Western blot with an anti‐HA tag antibodies (Fig 2A–C) detected specific high expression levels of the HA‐Depdc5 in the brain, and moderate levels in the heart, lungs, muscle, skin, and adipose tissue, but there was little or no expression in liver or kidneys. During development, brain HA‐Depdc5 expression was apparent from embryonic day 10, and levels increased until they reached a stable expression from P21 to adulthood. In adult mice, HA‐Depdc5 was detected in all brain regions examined, including cortex, hippocampus, striatum, septum, olfactory bulb, thalamus–hypothalamus, cerebellum, and brainstem. We next assessed which cortical cell subtypes express Depdc5 (Fig 2D–H). HA staining was specific when comparing HA‐Depdc5 cortical slices to wild‐type untagged. All NeuN‐positive neurons expressed HA‐Depdc5, particularly in the soma, where it colocalizes with the lysosomal marker Lamp1 (Pearson coefficient = 0.49, corresponding to a moderate/high colocalization). HA‐Depdc5 was also detected in both proximal and distal MAP2‐positive dendrites, as shown in mouse primary cortical neuron cultures. HA‐Depdc5 was detected in both excitatory (CaMKII‐positive) and inhibitory (Gad67‐ and PV‐positive) cortical neurons but not in glial cells (identified by Gfap immunostaining for astrocytes, Olig2 and Plp for oligodendrocytes, and Iba1 for microglia).
FIGURE 2.
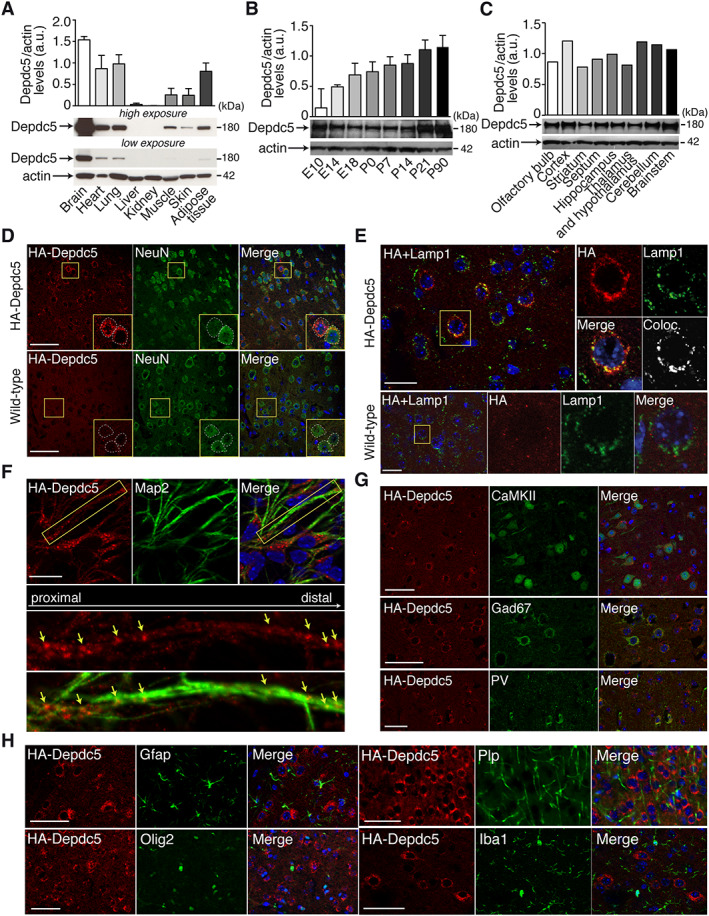
Depdc5 expression in the HA‐tagged Depdc5 mouse. (A–C) Quantified representative Western blots show HA‐Depdc5 expression in (A) various mouse organ lysates, (B) mouse brain lysates at developmental stages from embryonic day 10 (E10) to postnatal day 90 (P90), and (C) different brain regions of adult mouse (n = 1–3). Actin was used as the loading control. (D–H) Depdc5 expression in the somatosensory cortex. (D) Immunofluorescent colabeling of HA‐Depdc5 (red) with NeuN (green) showing specific expression of Depdc5 (compared to wild‐type untagged cortex, bottom) in neuronal soma (see insets, corresponding to the yellow squares). (E) Immunofluorescent colabeling of HA‐Depdc5 (red) with Lamp1 (green) showing specific enriched expression in lysosomes (compared to wild‐type untagged cortex, bottom). On the right of insets (corresponding to the yellow square) are the colocalization (Coloc.) between HA and Lamp1 (Pearson correlation coefficient for HA‐Depdc5 mouse = 0.49). (F) Immunofluorescent colabeling of HA‐Depdc5 (red) with Map2 (green) showing expression of Depdc5 in neurites. Bottom images are the insets (corresponding to the yellow square) showing HA (red) distribution along the MAP2‐positive (green) neurite. Arrows show aggregation of HA‐Depdc5 staining. (G) Immunofluorescent colabeling of HA‐Depdc5 (red) and CaMKII (excitatory neurons), Gad67 (inhibitory neurons) or parvalbumin (PV; PV interneurons) shows Depdc5 is expressed in excitatory and inhibitory neurons. (H) Colabeling of HA‐Depdc5 (red) and Gfap, Olig2, Plp, and Iba1, shows no coexpression in glial and microglial cells. Three sections were done in duplicate. Scale bars represent 50μm (D, G, H), 25μm (E), or 20μm (F).
In summary, Depdc5 protein is most highly expressed in the brain, and moderately in the heart and the lungs. To assess cardiac activity during seizures, we next generated a novel mouse model of Depdc5 deficiency.
Spontaneous Seizures and SUDEP in Depdc5 c/− Mice
To model the human genetic condition, we generated a heterozygous Depdc5 +/− mouse and deleted the second Depdc5 allele specifically in neurons (using a synapsin1‐driven Cre recombinase in Depdc5 floxed strain), termed Depdc5 c/− (see Fig 3 and Materials and Methods for details). We confirmed both a significant reduction of brain Depdc5 expression and a robust increase in mTOR signaling in Depdc5 c/− mice, measured with the phosphorylation level of the downstream target ribosomal protein S6 (pS6) by Western blot. Depdc5 c/− mice were born in the expected Mendelian ratio, with no obvious morphological differences from wild‐type littermates (Depdc5 +/+). However, the lifespan of Depdc5 c/− mice was much reduced compared to Depdc5 +/+ littermates. No Depdc5 c/− mouse survived after 140 days (median = 103 days), whereas wild‐type Depdc5 +/+ mice all survived beyond 200 days of age. Spontaneous behavioral seizures were often observed in adult Depdc5 c/− mice. Eventually, all Depdc5 c/− mice were found dead in a tonic posture, presumably after an epileptic seizure, suggesting a SUDEP‐like event. To confirm this hypothesis, cortical and hippocampal EEG electrodes were implanted in Depdc5 c/− (n = 10), Depdc5 +/+ (n = 7), and Depdc5 +/− (n = 3) mice to record brain activity by continuous video‐EEG in free‐moving animals. All 10 Depdc5 c/− mice died suddenly in a tonic posture; we were able to record 6 fatal seizures (the other 4 were not connected to the recording device at time of death), confirming spontaneous fatal electroclinical seizures. In 4 of 6 Depdc5 c/− mice, a single seizure was immediately followed by sudden death (age at death = 83–116 days). In 2 of 6 Depdc5 c/− mice, recurrent spontaneous seizures occurred at a mean frequency of 1.14 seizures/day over a week before a terminal seizure (age at death = 106–117 days).
FIGURE 3.
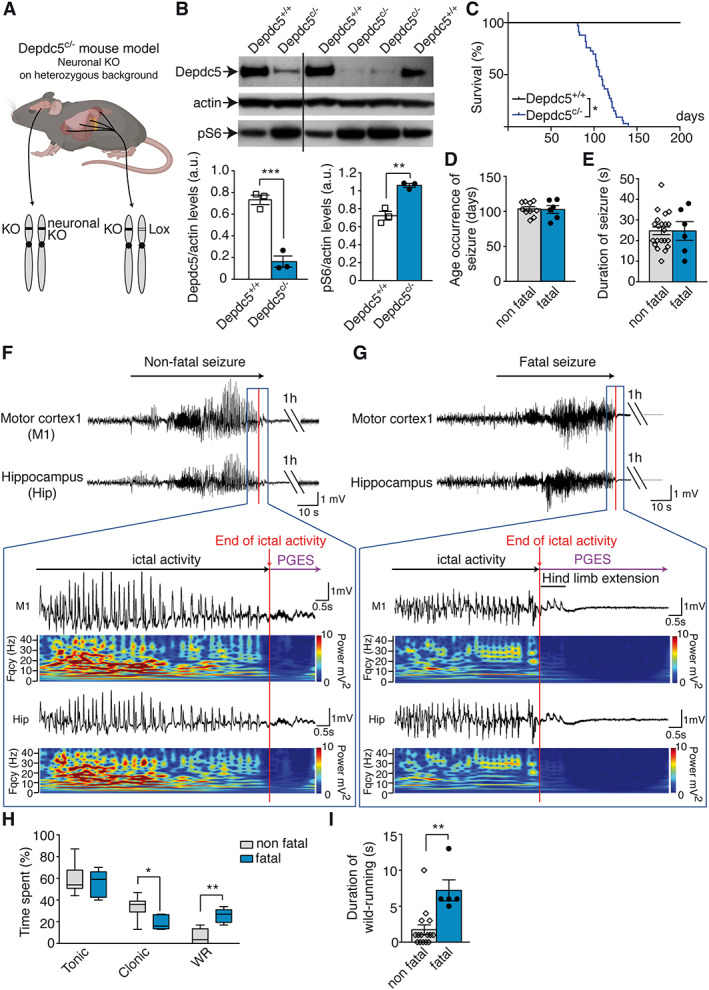
Spontaneous seizures in Depdc5 c/− mice. (A) Illustration of the Depdc5 c/− mouse model generated. KO = knockout. (B) Representative Western blot of brain lysates from Depdc5 c/− and control wild type (Depdc5 +/+) immunostained for Depdc5, actin, and pS6 (top) and quantification (bottom) of Depdc5 levels (left, unpaired t test, t 4 = 8.4, p = 0.0006) and pS6 levels (right, unpaired t test, t 4 = 6.4, p = 0.0031). (C) Survival of Depdc5 c/− mice (n = 33, log‐rank test, p = 0.047). (D) Age at onset of nonfatal (n = 6) and fatal seizures (n = 10, unpaired t test, t 14 = 0.17, p = 0.87). (E) Duration of nonfatal (n = 6) and fatal seizures (n = 21, unpaired t test, t 25 = 0.011, p = 0.99). (F) Representative electroencephalographic (EEG) recordings and fast Fourier transform (FFT) power spectrum of nonfatal seizure in a Depdc5 c/− mouse, followed with a postictal generalized EEG suppression (PGES) of 3 minutes 34 seconds. The color‐coded FFT power spectrum shows EEG amplitude and frequency changes from motor cortex (M1) and hippocampus (Hip). (G) Representative EEG recordings and FFT of a fatal seizure in a Depdc5 c/− mouse terminating with hind limb extension and prolonged PGES. (H) Duration of tonic, clonic, and wild running (WR) phases during nonfatal and fatal seizures in Depdc5 c/− mice (n = 5–12; 2‐way analysis of variance revealed main effect of behavior type, F 2,45 = 62.85, p < 0.0003; and main effect of interaction, F 2,45 = 9.98, p = 0.0003; Bonferroni post hoc tests). (I) Duration of wild running in nonfatal and fatal seizures (n = 5–21, unpaired t test, t 24 = 3.83, p = 0.0012). Results are given as mean ± standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001 versus Depdc5 +/+.
Attempting to define predictive biomarkers for SUDEP, we asked whether fatal and nonfatal seizures could be distinguished in terms of age onset, seizure duration, and behavior. The ages of occurrence of fatal and nonfatal seizures were similar (mean = 103.5 ± 3.1 vs 102.5 ± 5.5 days, respectively; see Fig 3). The duration of fatal and nonfatal seizures was comparable (average = 25 seconds, range = 10–35 seconds, n = 27 seizures). Fatal seizures occurred during both sleep and waking phases. The frequency of nonfatal seizures did not increase before the terminal seizure as reported in other mouse models of SUDEP. 42 , 43 EEG signal patterns were not strikingly different in nonfatal and fatal seizures: generalized high‐amplitude spikes and polyspikes at 10 to 15Hz, with no detectable focal onset. Figure 3F, G shows typical patterns and fast Fourier transform power spectra, which were similar for fatal and nonfatal seizures. Nonfatal seizures ended with a postictal generalized EEG suppression (PGES) of 4 minutes 30.6 ± 25.3 seconds before normal EEG activity was re‐established, whereas for fatal seizures the PGES was prolonged indefinitely, coinciding with death of the animal. Duration of PGES associated with nonfatal seizures did not change over the days preceding the final SUDEP seizure and therefore could not predict the fatal seizure. Seizures consisted of a stereotyped sequence of behaviors; behavioral tonic arrest with short periods of forelimb clonus (Racine stage 3) was followed by rearing and falling to the side (Racine stage 4), a characteristic of a GTCS (videos shown in Supplementary Videos S1 and S2, corresponding to nonfatal and fatal seizure, respectively). Whereas the duration of the tonic phase was similar between fatal and nonfatal seizures, duration of the clonic phase was shorter in fatal seizures, in which wild running was longer. Specifically, wild running (Racine stage 5) was observed in all fatal seizures, but only in 50% of nonfatal seizures, and episodes were shorter for nonfatal than for fatal seizures (duration = 1.7 ± 0.7 vs 7.2 ± 1.4 seconds). Fatal seizures ended with hind limb extension. Control Depdc5 +/+ and heterozygous Depdc5 +/− mice did not exhibit seizures or die prematurely.
In summary, all Depdc5 c/− mice exhibited spontaneous GTCSs. Fatal and nonfatal seizures had similar durations and frequencies and began at comparable ages. None of the parameters measured from EEG traces could be reliably linked with SUDEP‐like events. Longer lasting wild running behaviors were more likely to precede sudden death. We next asked whether alterations in cardiac activity could predispose Depdc5 c/− mice to SUDEP and predict fatal seizures.
Cardiac Investigation in Depdc5 c/− Mice
We first assessed baseline cardiac function in adult anesthetized mice (n = 9 Depdc5 c/− and n = 9 Depdc5 +/+ mice). A representative ECG trace is shown in Figure 4A. There were no significant differences in baseline heart rate, assessed with the RR length (144.5 ± 8.9 milliseconds for Depdc5 c/− vs 141 ± 5.5 milliseconds for Depdc5 +/+, p = 0.75), PR length (46.9 ± 1.5 vs 47.9 ± 2.1 milliseconds, p = 0.7), QT interval (36.7 ± 1.4 vs 33.4 ± 1.7 milliseconds, p = 0.17), and QRS length (7.2 ± 0.3 vs 6.8 ± 0.3 milliseconds, p = 0.35) between Depdc5 c/− and Depdc5 +/+ mice. To complete the characterization of baseline cardiac function, we assessed the HRV with the SDRR parameter (0.054 ± 0.018 vs 0.039 ± 0.002, p > 0.99), and the rMSSD parameter (0.62 ± 0.1 vs 0.42 ± 0.07, p = 0.33); both indicated no significant differences between Depdc5 c/− mice and controls.
FIGURE 4.
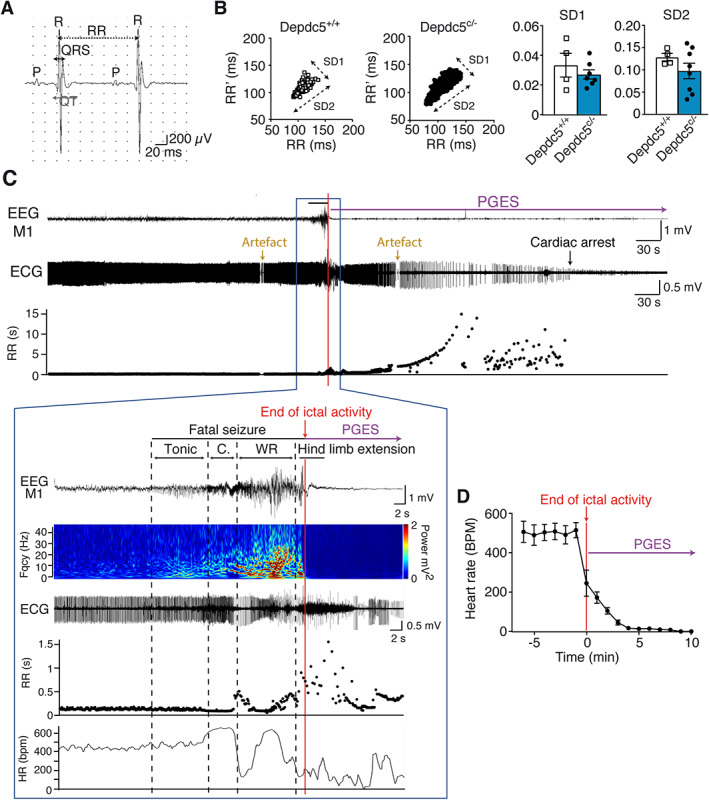
Electroencephalographic (EEG) and electrocardiographic (ECG) records from Depdc5 c/− mice at rest and during a spontaneous fatal seizure. (A) Example of ECG records in anesthetized mice. (B) In vivo recordings of heart rate variability are represented with Poincaré plots in Depdc5 c/− and Depdc5 +/+ mice, and quantified by SD1 (Mann–Whitney, U = 7, p = 0.53) and SD2 (Mann–Whitney, U = 11, p = 0.46) parameters (n = 4–8 mice, n > 500 RR measures per animal). (C) Representative simultaneous EEG‐ECG records and RR plot before, during, and after a fatal seizure in a Depdc5 c/− mouse, terminating with prolonged postictal generalized EEG suppression (PGES). Fast Fourier transform power spectrum shows EEG amplitude and frequency changes from motor cortex (M1). Dashed lines indicate the different behavioral phases (Tonic, C = Clonic, and WR = Wild Running). RR length and heart rate (HR; mean number of RR in 2 seconds) changed after the beginning of the seizure, during the clonic phase. The ECG during the seizure and after the hind limb extension phase is partially obscured by electrical activity of muscle contraction. Artifacted RR measures were excluded. (D) Average heart rate changes during and after the seizure in 6 Depdc5 c/− mice. Results are mean ± standard error of the mean.
Second, we performed simultaneous video‐EEG‐ECG recordings to assess cardiac activity during fatal seizures in Depdc5 c/− (n = 23) compared to Depdc5 +/+ (n = 4) conscious unrestrained mice. During the period preceding a seizure, the baseline heart rate was in the normal range (mean RR length = 95.33 ± 4.9 milliseconds for Depdc5 c/− vs 94.7 ± 3.2 milliseconds for Depdc5 +/+, p = 0.57). The HRV, assessed with SDRR (0.051 ± 0.009 vs 0.065 ± 0.005, p = 0.46), rMSSD (1.52 ± 1.2 vs 1.77 ± 1.2, p = 0.57), and the short‐term HRV (SD1) and long‐term HRV (SD2) parameters of the Poincaré plots, 38 , 39 showed no cardiac arrhythmia in Depdc5 c/− mice (see Fig 4B).
In the ictal period, we were able to record SUDEP events by simultaneous EEG‐ECG signals in 6 of 23 Depdc5 c/− mice at the age of P98 ± 9. Representative EEG and ECG traces before, during, and after a fatal seizure are shown in Figure 4C. Electroclinical fatal seizures displayed a succession of tonic, clonic, wild running phases, terminated by a tonic posturing with a hind limb extension (see Supplementary Videos S2 and S3). During the tonic phase, cardiac activity assessed by RR intervals and heart rate was normal; a moderate heart rate increase (tachycardia) occurred during the clonic phase. During the wild running phase, the RR intervals increased rapidly, reflecting a variable and progressive slowing of the ventricular rate. After hind limb extension, during the PGES, the heart rate markedly decreased until cardiac arrest. Qualitative examination of videos shows that a respiratory arrest occurred after the fatal seizure (see Supplementary Videos S2 and S3). All 6 recorded fatal seizures presented the same pattern of EEG‐ECG. On average, heart rate was normal (540 ± 20 bpm) before seizure and decreased to 250 ± 50 bpm at the end of the seizure (see Fig 4D), indicating that bradycardia followed by heart arrest 4 minutes later was consecutive with the fatal seizure, and not causal. We conclude that Depdc5 c/− mice did not present primary cardiac abnormalities during the preictal period of fatal seizures.
Finally, we investigated heart histology in Depdc5 c/− mice, before and after SUDEP‐like event. Compared to Depdc5 +/+ control, H&E‐stained transverse heart sections did not show structural differences before the seizure occurrence or after SUDEP‐like events (Fig 5). Notably, no contraction band necrosis or fibrosis was observed in Depdc5 c/− mice after SUDEP‐like event (see Fig 5C).
FIGURE 5.
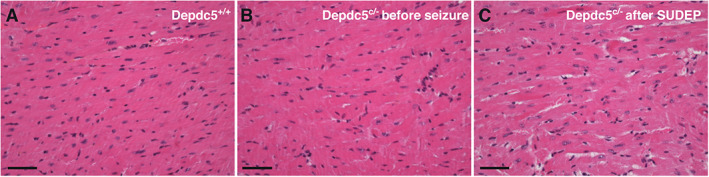
Mouse heart histology before and after sudden unexpected death in epilepsy (SUDEP)‐like event. Hematoxylin and eosin–stained transversal sections of the anterior interventricular artery are shown in a control Depdc5 +/+ (A), Depdc5 c/− before seizure (B), and Depdc5 c/− after SUDEP‐like event (C). No contraction band necrosis or fibrosis was detected in Depdc5 c/− heart after SUDEP. Scale bars represent 100μm.
Discussion
Mutations in either DEPDC5, NPRL2, or NPRL3 are commonly encountered among subjects with childhood onset focal seizures either with a normal magnetic resonance imaging or associated with FCD type 2. 17 Several clinical observations suggest that mutations in DEPDC5, NPRL2, and NPRL3, the components of the GATOR1 complex of the mTOR pathway, may confer a higher risk to SUDEP. 15 , 16 , 17 , 18 Mutations in GATOR1 genes cause a loss of function leading to a haploinsufficiency and hyperactivation of the mTOR signaling pathway. 18 , 27 , 28 , 44 Pathophysiological mechanisms that induce sudden death in GATOR1‐related epilepsy remain unknown.
Epilepsy genes associated with SUDEP include cardiac and neuronal ion channel genes regulating excitability in both heart and brain, thus generating cardiac arrhythmias and seizures. 8 , 9 , 10 , 11 In this study, we asked whether DEPDC5‐related SUDEP may have a primary cardiac etiology, and we aimed to identify possible predictive cardiac biomarkers. Whether DEPDC5 has a direct impact on cardiac activity and could lead to fatal seizures is a crucial question. This study shows that Depdc5 protein is expressed in the brain and the heart. It is noteworthy that in a previous study, 45 invalidation of Depdc5 in mouse skeletal muscle led to muscle cell hypertrophy and alterations of mitochondrial respiratory capacity, suggesting that Depdc5 deficiency could contribute to cardiac dysfunction.
For this purpose, we present here an unprecedented cohort of rare cases of 16 patients with GATOR1 mutations who underwent cardiac evaluation because of risk for SUDEP, among whom 6 of 16 were at high risk because of family history of SUDEP, and 3 of 16 died of SUDEP. There was no evidence for cardiac arrhythmia or structural abnormality on resting 12‐lead ECG, Holter‐ECG monitoring, and TTE in any of the patients, suggesting that seizures did not have long‐term interictal consequences for cardiac activity. Notably, no clinical cardiac anomalies were detected in 3 of 3 patients carrying pathogenic DEPDC5 variants who succumbed to SUDEP at 2, 3, and 7 years after cardiac assessment. All 3 SUDEP patients had received a sodium channel blocker antiseizure drug. Although this treatment may increase the risk for sudden cardiac death, 46 a recent large case–control study did not show any clear association between antiseizure medication and SUDEP; rather, it showed an association between nonadherence and SUDEP. 47 Noticeably, SUDEP Patient 16 had issues with medication compliance. The 2 other SUDEP patients (Patients 1 and 2) had been seizure‐free for at least 8 months before SUDEP, suggesting it is unlikely that a poor control of seizures could contribute to sudden cardiac death. We also present the first record of an autopsy on heart tissue in a DEPDC5 patient, which revealed no structural pathology that might affect the integrity or electrical function of the heart. Myocardial fibrosis, previously reported at autopsy in some SUDEP cases, 48 and contraction band necrosis, associated with sudden cardiac death, 41 were absent. Altogether, our human data indicate that DEPDC5‐related SUDEP is unlikely to result from primary fatal cardiac arrhythmia due to repetitive seizures or as a direct consequence of GATOR1 mutations affecting cardiac activity. This hitherto unique study suggests a distinct SUDEP mechanism between patients with DEPDC5 mutations and patients with epilepsy ion channel gene mutations. 10
The clinical evaluation of patients has several limitations because of the lack of (1) high‐resolution TTE capable of identifying subtle changes such as cardiac stiffness in people with epilepsy, 49 although such stiffness is unlikely to have been present as fibrosis, a marker of cardiac stiffness, 49 was not observed in the postmortem heart; (2) prolonged ECG recordings (only 4/16 patients had Holter‐ECG monitoring, ranging from 24 hours to 8 days); (3) ictal ECG recordings (only available in 1 patient during presurgical evaluation), which may reveal transient cardiac arrhythmias as previously reported. 4 In addition, the patient cohort size is relatively small, and we cannot completely exclude additional possibilities, such as dynamic alterations in cardiac electrophysiology, as seen in ATP1A3‐related disease, another genetic disorder characterized by paroxysmal neurological events, including seizures. 50
The limits of both antemortem and postmortem human clinical investigations prompted us to explore cardiac activity during seizures in a mouse model of SUDEP linked to Depdc5 deficiency. Using a tagged HA‐Depdc5 mouse, we show that Depdc5 protein is highly expressed in the cortex, and in both excitatory and inhibitory neurons, but not in glial cells, in accord with mouse and human single‐cell RNA sequencing data. 51 As expected for an mTOR repressor, 24 Depdc5 colocalized with lysosomes. During neurodevelopment, Depdc5 expression increased to a peak in adulthood, correlating with mTOR protein expression. 52 A recent report suggested that Depdc5 knockdown in a mixed culture of excitatory and inhibitory cortical neurons enhances excitatory neurotransmission, with presumed proepileptic effects, 53 which could be the cause of seizures related to DEPDC5 deficiency.
We next generated a mouse model with a Depdc5 neuronal deletion in synapsin1‐cre–expressing neurons on a Depdc5 heterozygous background to mimic brain second‐hit events reported in patients with FCD type 2. 18 , 19 , 20 , 21 , 22 , 30 All Depdc5 c/− mice exhibited spontaneous seizures with a fatal outcome at ~3 months of age. Whereas few Depdc5 c/− mice presented nonfatal seizures preceding the fatal one, most of them had a single seizure that proved fatal. This notable observation has its parallel in the histories of 2 patients (Patients 1 and 2) who had been seizure‐free for prolonged intervals (9 years and 8 months, respectively) before succumbing to SUDEP. The established clinical feature most strongly associated with SUDEP risk is the uncontrolled tonic–clonic seizures. 3 , 4 Neither of these patients were reported to have such seizures, and both died after prolonged periods of seizure freedom, raising questions about our current ideas on the mechanisms of SUDEP.
Cardiac activity of Depdc5 c/− mice showed no significant alterations, nor different HRV compared to controls, which could have predicted or predisposed to a fatal seizure. HRV, a marker of autonomic tone, 39 , 54 was not impaired, suggesting autonomic function is not altered. ECG recordings during fatal seizures revealed a short tachycardia, followed by a progressive bradycardia, which are cardiac features that can be associated with typical epileptic seizures. 55 Therefore, no obvious cardiological defects were present in the Depdc5 c/− mice, nor autonomic dysfunction that could impact arrhythmogenesis, as mentioned in sudden cardiac death. 54
Cerebral biomarkers of SUDEP are still missing, and so far, GTCSs represent the greatest risk factor for SUDEP in humans. 4 , 5 , 7 , 56 Comparison of fatal and nonfatal seizures in Depdc5 c/− mice showed fatal seizures were accompanied by a longer episode of wild running, and GTCS electrographic features were similar. This wild running behavior is reported during audiogenic seizures, where the brainstem may be involved. 57 Brainstem nuclei control visceral functions including cardiac and respiratory pacemakers. Brainstem dysfunction has been linked to SUDEP, where postictal apnea and bradycardia precede asystole and death. 5 Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in both genetic and induced mouse SUDEP models. 39 , 58 Our data suggest that a SUDEP‐like event due to Depdc5 haploinsufficiency might be a consequence of brainstem failure, rather than a predisposition to cardiac arrhythmia.
In conclusion, we report a non–ion‐channel SUDEP mouse model, and our data do not support primary cardiac etiology as the proximate cause of death. Canonical cardiac biomarkers such as QT interval or heart rate are poor predictors of sudden death in DEPDC5‐mutated patients, in contrast to SUDEP cases linked to ion channel gene mutations. 10 Effective preventive strategies in high‐risk patients will rely on the understanding of the mechanisms that lead from seizures to death.
Author Contributions
A.B. and S.B. contributed to the conception and design of the study; A.B., D.R., T.B., S.Z., M.M., T.R., H.A.‐B., C.M., M.J., I.A., and F.P. contributed to the acquisition and analysis of data; A.B., D.R., T.B., V.N., S.M.S., and S.B. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Table S1. List of the Genomics England Research Consortium contributors
Video S1. Video of a nonfatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 3F
Video S2. Video of a fatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 3G
Video S3. Video of a fatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 4C
Acknowledgments
We thank the families that took part in this study, Dr J. Devendeville for sharing the autopsy material, Dr R. Miles for proofreading the article, the clinicians who performed cardiac examinations, and the Paris Brain Institute's core facilities: PHENO‐ICMice, ICM.Quant, Histomics, iGenSeq, CELIS, and DNA and Cell Bank. All animal work was conducted at the PHENO‐ICMice facility (supported by ANR‐10‐ IAIHU‐06, ANR‐11‐INBS‐0011‐NeurATRIS, and FRM). This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. This work was funded by the European Research Council (682345 to S.B.), the program “Investissements d'avenir” (ANR‐10‐IAIHU‐06), and the Fondation pour la Recherche Médicale (FDT201904008269 to T.R.). Part of this work was undertaken at University College London Hospitals, which received a proportion of funding from the National Institute for Health Research Biomedical Research Centres funding scheme. Additional funding was provided by the Epilepsy Society. The 100,000 Genomes Project is funded by the National Institute for Health Research and National Health Service England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure.
Contributor Information
Stéphanie Baulac, Email: stephanie.baulac@icm-institute.org, @BaulacS.
Genomics England Research Consortium:
J. C. Ambrose, R. Jackson, M. Ryten, P. Arumugam, L. J. Jones, K. Savage, E. L. Baple, D. Kasperaviciute, K. Sawant, M. Bleda, M. Kayikci, R. H. Scott, F. Boardman‐Pretty, L. Lahnstein, A. Siddiq, J. M. Boissiere, K. Lawson, A. Sieghart, C. R. Boustred, S. E. A. Leigh, D. Smedley, H. Brittain, I. U. S. Leong, K. R. Smith, M. J. Caulfield, F. J. Lopez, A. Sosinsky, G. C. Chan, F. Maleady‐Crowe, W. Spooner, C. E. H. Craig, J. Mason, H. E. Stevens, L. C. Daugherty, E. M. McDonagh, A. Stuckey, A. de Burca, L. Moutsianas, R. Sultana, A. Devereau, M. Mueller, E. R. A. Thomas, G. Elgar, N. Murugaesu, S. R. Thompson, R. E. Foulger, A. C. Need, C. Tregidgo, T. Fowler, C. A. Odhams, A. Tucci, P. Furió‐Tarí, C. Patch, E. Walsh, J. M. Hackett, D. Perez‐Gil, S. A. Watters, D. Halai, D. Polychronopoulos, M. J. Welland, A. Hamblin, J. Pullinger, E. Williams, S. Henderson, T. Rahim, K. Witkowska, J. E. Holman, A. Rendon, S. M. Wood, T. J. P. Hubbard, P. Riesgo‐Ferreiro, M. Zarowiecki, K. Ibáñez, and T. Rogers
References
- 1. Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 2012;53:227–233. [DOI] [PubMed] [Google Scholar]
- 2. Harden C, Tomson T, Gloss D, et al. Practice guideline summary: sudden unexpected death in epilepsy incidence rates and risk factors: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Epilepsy Curr 2017;17:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whitney R, Donner EJ. Risk factors for sudden unexpected death in epilepsy (SUDEP) and their mitigation. Curr Treat Options Neurol 2019;21:7. [DOI] [PubMed] [Google Scholar]
- 4. Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 2013;12:966–977. [DOI] [PubMed] [Google Scholar]
- 5. Devinsky O, Hesdorffer DC, Thurman DJ, et al. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 2016;15:1075–1088. [DOI] [PubMed] [Google Scholar]
- 6. Lacuey N, Zonjy B, Hampson JP, et al. The incidence and significance of periictal apnea in epileptic seizures. Epilepsia 2018;59:573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pensel MC, Nass RD, Taubøll E, et al. Prevention of sudden unexpected death in epilepsy: current status and future perspectives. Expert Rev Neurother 2020;20:497–508. [DOI] [PubMed] [Google Scholar]
- 8. Friedman D, Kannan K, Faustin A, et al. Cardiac arrhythmia and neuroexcitability gene variants in resected brain tissue from patients with sudden unexpected death in epilepsy (SUDEP). NPJ Genom Med 2018;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chahal CAA, Salloum MN, Alahdab F, et al. Systematic review of the genetics of sudden unexpected death in epilepsy: potential overlap with sudden cardiac death and arrhythmia‐related genes. J Am Heart Assoc 2020;9:e012264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bleakley LE, Soh MS, Bagnall RD, et al. Are variants causing cardiac arrhythmia risk factors in sudden unexpected death in epilepsy? Front Neurol 2020;11:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bagnall RD, Crompton DE, Semsarian C. Genetic basis of sudden unexpected death in epilepsy. Front Neurol 2017;8:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zijlmans M, Flanagan D, Gotman J. Heart rate changes and ECG abnormalities during epileptic seizures: prevalence and definition of an objective clinical sign. Epilepsia 2002;43:847–854. [DOI] [PubMed] [Google Scholar]
- 13. Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol 2014;10:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwartz PJ, Ackerman MJ, George AL, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013;62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nascimento FA, Borlot F, Cossette P, et al. Two definite cases of sudden unexpected death in epilepsy in a family with a DEPDC5 mutation. Neurol Genet 2015;1:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bagnall RD, Crompton DE, Petrovski S, et al. Exome‐based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol 2016;79:522–534. [DOI] [PubMed] [Google Scholar]
- 17. Baldassari S, Picard F, Verbeek NE, et al. The landscape of epilepsy‐related GATOR1 variants. Genet Med 2019;21:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weckhuysen S, Marsan E, Lambrecq V, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 2016;57:994–1003. [DOI] [PubMed] [Google Scholar]
- 19. Ribierre T, Deleuze C, Bacq A, et al. Second‐hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia‐associated epilepsy. J Clin Invest 2018;128:2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuskaitis CJ, Jones BM, Wolfson RL, et al. A mouse model of DEPDC5‐related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis 2018;111:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuskaitis CJ, Rossitto L‐A, Gurnani S, et al. Chronic mTORC1 inhibition rescues behavioral and biochemical deficits resulting from neuronal Depdc5 loss in mice. Hum Mol Genet 2019;28:2952–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klofas LK, Short BP, Zhou C, Carson RP. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum Mol Genet 2020;29:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bar‐Peled L, Chantranupong L, Cherniack AD, et al. A tumor suppressor complex with GAP activity for the rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340:1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen K, Huang RK, Brignole EJ, et al. Architecture of the human GATOR1 and GATOR1‐rag GTPases complexes. Nature 2018;556:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 2020;21:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546–551. [DOI] [PubMed] [Google Scholar]
- 27. Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45:552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology 2014;82:2101–2106. [DOI] [PubMed] [Google Scholar]
- 29. Ricos MG, Hodgson BL, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol 2016;79:120–131. [DOI] [PubMed] [Google Scholar]
- 30. Baldassari S, Ribierre T, Marsan E, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol 2019;138:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol 2015;77:675–683. [DOI] [PubMed] [Google Scholar]
- 32. Lee WS, Stephenson SEM, Howell KB, et al. Second‐hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol 2019;6:1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sim NS, Ko A, Kim WK, et al. Precise detection of low‐level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol 2019;138:901–912. [DOI] [PubMed] [Google Scholar]
- 34. Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Archiv 2017;471:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marsan E, Ishida S, Schramm A, et al. Depdc5 knockout rat: a novel model of mTORopathy. Neurobiol Dis 2016;89:180–189. [DOI] [PubMed] [Google Scholar]
- 36. Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates, compact. 3rd ed. Waltham, MA: Academic Press, 2008. [Google Scholar]
- 37. Mishra V, Gautier NM, Glasscock E. Simultaneous video‐EEG‐ECG monitoring to identify neurocardiac dysfunction in mouse models of epilepsy. J Vis Exp 2018;131:57300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kamen PW, Krum H, Tonkin AM. Poincaré plot of heart rate variability allows quantitative display of parasympathetic nervous activity in humans. Clin Sci 1996;91:201–208. [DOI] [PubMed] [Google Scholar]
- 39. Kim Y, Bravo E, Thirnbeck CK, et al. Severe peri‐ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 2018;128:1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Charan J, Kantharia ND. How to calculate sample size in animal studies? J Pharmacol Pharmacother 2013;4:303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hopster DJ, Milroy CM, Burns J, Roberts NB. Necropsy study of the association between sudden cardiac death, cardiac isoenzymes and contraction band necrosis. J Clin Pathol 1996;49:403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ren Y, Chang J, Li C, et al. The effects of ketogenic diet treatment in Kcna1‐null mouse, a model of sudden unexpected death in epilepsy. Front Neurol 2019;10:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teran FA, Kim Y, Crotts MS, et al. Time of day and a ketogenic diet influence susceptibility to SUDEP in Scn1aR1407X/+ mice. Front Neurol 2019;10:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sim JC, Scerri T, Fanjul‐Fernández M, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol 2016;79:132–137. [DOI] [PubMed] [Google Scholar]
- 45. Graber TG, Fry CS, Brightwell CR, et al. Skeletal muscle‐specific knockout of DEP domain containing 5 protein increases mTORC1 signaling, muscle cell hypertrophy, and mitochondrial respiration. J Biol Chem 2019;294:4091–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bardai A, Blom MT, van Noord C, et al. Sudden cardiac death is associated both with epilepsy and with use of antiepileptic medications. Heart 2015;101:17–22. [DOI] [PubMed] [Google Scholar]
- 47. Sveinsson O, Andersson T, Mattsson P, et al. Pharmacologic treatment and SUDEP risk: a nationwide, population‐based, case‐control study. Neurology 2020;95:e2509–e2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. P‐Codrea Tigaran S, Dalager‐Pedersen S, Baandrup U, et al. Sudden unexpected death in epilepsy: is death by seizures a cardiac disease? Am J Forensic Med Pathol 2005;26:99–105. [PubMed] [Google Scholar]
- 49. Fialho GL, Pagani AG, Wolf P, et al. Echocardiographic risk markers of sudden death in patients with temporal lobe epilepsy. Epilepsy Res 2018;140:192–197. [DOI] [PubMed] [Google Scholar]
- 50. Balestrini S, Mikati MA, Álvarez‐García‐Rovés R, et al. Cardiac phenotype in ATP1A3‐related syndromes: a multicenter cohort study. Neurology 2020;95:e2866–e2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hodge RD, Bakken TE, Miller JA, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019;573:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Talos DM, Sun H, Zhou X, et al. The interaction between early life epilepsy and autistic‐like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One 2012;7:e35885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Fusco A, Cerullo MS, Marte A, et al. Acute knockdown of Depdc5 leads to synaptic defects in mTOR‐related epileptogenesis. Neurobiol Dis 2020;139:104822. [DOI] [PubMed] [Google Scholar]
- 54. Verrier RL, Pang TD, Nearing BD, Schachter SC. Epileptic heart: a clinical syndromic approach. Epilepsia 2021;62:1780–1789. [DOI] [PubMed] [Google Scholar]
- 55. Jefferys JGR, Ashby‐Lumsden A, Lovick TA. Cardiac effects of repeated focal seizures in rats induced by intrahippocampal tetanus toxin: bradyarrhythmias, tachycardias, and prolonged interictal QT interval. Epilepsia 2020;61:798–809. [DOI] [PubMed] [Google Scholar]
- 56. King‐Stephens D. Biomarkers for SUDEP: are we there yet? Epilepsy Curr 2019;19:231–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Faingold CL. Brainstem networks: reticulo‐cortical synchronization in generalized convulsive seizures. In: Noebels JL, Avoli M, Rogawski MA, et al., eds. Jasper's basic mechanisms of the epilepsies. 4th ed. Bethesda, MD: National Center for Biotechnology Information, 2012;1–22. [PubMed] [Google Scholar]
- 58. Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med 2015;7:282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the Genomics England Research Consortium contributors
Video S1. Video of a nonfatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 3F
Video S2. Video of a fatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 3G
Video S3. Video of a fatal seizure in Depdc5 c/‐ mouse corresponding to Fig. 4C