

Abstract
Pseudomonas aeruginosa is an opportunistic ESKAPE pathogen that produces two lectins, LecA and LecB, as part of its large arsenal of virulence factors. Both carbohydrate‐binding proteins are central to the initial and later persistent infection processes, i. e. bacterial adhesion and biofilm formation. The biofilm matrix is a major resistance determinant and protects the bacteria against external threats such as the host immune system or antibiotic treatment. Therefore, the development of drugs against the P. aeruginosa biofilm is of particular interest to restore efficacy of antimicrobials. Carbohydrate‐based inhibitors for LecA and LecB were previously shown to efficiently reduce biofilm formations. Here, we report a new approach for inhibiting LecA with synthetic molecules bridging the established carbohydrate‐binding site and a central cavity located between two LecA protomers of the lectin tetramer. Inspired by in silico design, we synthesized various galactosidic LecA inhibitors with aromatic moieties targeting this central pocket. These compounds reached low micromolar affinities, validated in different biophysical assays. Finally, X‐ray diffraction analysis revealed the interactions of this compound class with LecA. This new mode of action paves the way to a novel route towards inhibition of P. aeruginosa biofilms.
Keywords: carbohydrates, glycoconjugates, glycomimetics, lectin, LecA
A new ligand binding pocket for additional pharmacophores in inhibitors of the bacterial lectin and virulence factor LecA from Pseudomonas aeruginosa was identified and successfully targeted, using a suite of modelling, chemical synthesis, and biophysical analyses. The crystal structure of LecA with one ligand targeting this central pocket was solved, and validated the concept.

Introduction
Pseudomonas aeruginosa belongs to the ESKAPE pathogens and is listed by the World Health Organization as the most critical bacterial pathogen. This rod‐shaped, Gram‐negative ubiquitous bacterium lives under moist conditions and thrives in hospitals, colonizing patients under ventilation or bearing catheters and cystic fibrosis patients. [1] For colonizing the host, many bacteria exploit cell surface carbohydrates, the glycocalyx, [2] through their carbohydrate‐binding proteins (lectins), adhesins or capsid proteins. [3] In case of P. aeruginosa two soluble virulence factors are involved: the d‐galactose‐binding LecA and d‐mannose‐/l‐fucose‐binding LecB. [4] These two extracellular, calcium(II)‐dependent and homotetrameric lectins are essential for establishing the biofilm matrix, a protective environment against environmental stress and antibiotic treatment and, thus, a major hurdle for therapy.[ 5 , 6 , 7 ]
To overcome this resistance mechanism, alternative strategies are currently being developed to dismantle the pathogen and restore antibiotic efficacy via inhibition of the lectins LecA and LecB.[ 8 , 9 , 10 ] Complementary to the numerous approaches for multivalent inhibition using native carbohydrates,[ 11 , 12 ] we have developed glycomimetics targeting LecA and LecB. In case of LecB, we have developed small molecule glycomimetics starting from a mannoside[ 13 , 14 , 15 ] via simple C‐glycosides [16] into orally available anti‐biofilm lead compounds[ 17 , 18 ] which are currently under further investigation. Potent monovalent glycomimetics have been proven difficult to obtain for galactophilic LecA,[ 19 , 20 , 21 ] thus requiring divalent ligands that display two galactose residues to simultaneously bind to two adjacent binding sites in the LecA tetramer and yield low nanomolar LecA inhibitors.[ 22 , 23 ] In addition, we have recently reported conceptionally new approaches for targeting LecA, i. e. covalent lectin inhibitors [24] and non‐carbohydrate glycomimetics mimicking the binding pattern of carbohydrates. [25]
For LecA, inhibitor design generally started from the monosaccharide galactose (Figure 1A) [10] which is the binding epitope from its natural ligand in human, glycosphingolipid Gb3, a host cell surface receptor mediating the engulfment of P. aeruginosa into host cells. [26] Galactose forms an extensive hydrogen bonding network with LecA using all hydroxy groups. OH3 and OH4 are further coordinating to the calcium ion of LecA. [27] OH6 is pointing into a small cavity formed by a loop from His50 to Gln53 and Val101 where it is coordinated through an extended hydrogen‐bonding network. Introduction of β‐linked aromatic aglycons led to a fivefold affinity increase compared to aliphatic analogues, which is explained by CH‐π‐interaction of the aryl aglycon with the side chain of His50. [28]
Figure 1.

Rationale for targeting the central pocket in LecA: (A) Co‐crystal structure showing the tetramer of LecA in complex with galactose (PDB: 1OKO). (B) Side view of one of the two adjacent dimers of LecA with the identified cavity (solid surface) between the two monomers defined as central pocket. The divalent ligand (PDB: 4CP9) reported by Winssinger et al. [29] is represented as sticks in electron density and is pointing towards the central pocket. (C) Structure of the divalent LecA ligand. [29] and the structural motif studied in this work highlighted in red. (D) Top view of the surface of one LecA dimer showing the cavity between the two monomers (PDB: 4LKE). The entrance of this cavity is polar due to the presence of Gln40, Lys41, Asp47, Arg48 and Glu49 and the interior is hydrophobic due to residues Trp33 and Trp42. Calcium ions in the carbohydrate binding sites are shown as green spheres.
In the present work, we localized a cavity between two monomers of LecA (Figure 1B), designated as central pocket, and developed synthetic galactosides carrying aromatic moieties directed towards this cavity.
Results and Discussion
Identification of the central pocket in LecA
23 crystallographic structures of LecA from P. aeruginosa (Uniprot reference Q05097) were retrieved from the Protein Data Bank [30] (PDB) and analyzed for the presence of the central pocket (Figure 1, Table S1). A central pocket of LecA can be identified between two adjacent monomers in the homotetrameric LecA structure. The entrance of the pocket is clearly visible (Figure 1B) and the upper part of the cavity including its entrance, formed by residues Gln40, Lys41, Asp47, Arg48 and Glu49, is highly polar. In contrast, the interior is more hydrophobic due to the presence of two tryptophan residues, Trp33 and Trp42. The central pocket‘s cavity was consistently detected in 25 out of the 31 dimers in the available crystallographic structures (Table S2).
Design and in silico analysis by molecular dynamics simulation
As a starting point for ligand design, we were inspired by a bivalent peptide‐based LecA inhibitor (K D=82 nM, Figure 1C) previously reported by Winssinger et al. [29] As often observed for multivalent ligands, the crystal structure of the complex showed electron density only for part of the molecule, i. e. the galactose residue, the aromatic aglycon and the attached triazole ring which is in proximity to the entrance of the central pocket (Figure 1B). The authors reported the crystallographically invisible phenoxy acetyl substituent adjacent to the triazole to be important for LecA binding. We hypothesized that this phenoxyacetyl moiety may bind close to or into the central pocket. To test this hypothesis, we truncated the original divalent molecule and retained the galactose residue, its aglycon and the attached phenoxyacetyl residue (shown in red in Figure 1C) and designed derivatives carrying various substituents to target the central pocket in silico.
Compound 1 (Scheme 1) was then docked to LecA, and in the resulting poses, the phenoxy acetate part reaches towards the central pocket. Subsequently, molecular dynamics simulation was performed on one selected pose obtained from docking. Visual inspection of the 240 ns MD trajectory revealed significant fluctuation between the phenoxyacetate side chain and LecA with repeated contacts of the pharmacophore and the central pocket (Figure 2). In general, the galactose moiety remained firmly bound inside the carbohydrate binding site with strong hydrogen bonds to residues His50, Gln53, Asp100 and Asn103, and hydrophobic contacts to Tyr36, Cys62 and Val101. The terminal phenyl ring was entering and leaving the central pocket repeatedly, possibly due to a suboptimal spacer length to position the phenyl group firmly within the pocket and a lack of interactions with the polar entrance of the cavity. Less frequent hydrogen bonds were also observed between the linker and Gln40 and Asp47. Last, hydrophobic contacts could be detected between the linker and Pro38, the phenoxy moiety and Trp42 (chains A and D), and the triazole ring and Pro51.
Scheme 1.

Synthesis of LecA inhibitors targeting the central pocket. The different side chains were introduced in the penultimate step via amide coupling with alkyne S9. Final assembly was achieved by coupling azide S6 with alkynes S10a–i in a copper(I)‐catalyzed cycloaddition. Reagents and conditions: (i) BF3⋅OEt2, S2, CH2Cl2, 0–25 °C, 18 h; (ii) 1. LiI, pyridine, 25 °C, 3 d; 2. S4, HOBt, EDC, DMF, 25 °C, 24 h; (iii) NaOMe, MeOH, 25 °C, 1.5 h; (iv) SOCl2, MeOH, 0–25 °C, 18 h; (v) ethanolamine, 25 °C, 18 h; (vi) various acetic acids, EDC, HOBt, DIPEA, DMF, 25 °C, 18 h; (vii) S6, CuSO4 in H2O, sodium ascorbate in H2O, DMF, 25 °C, 2 h.
Figure 2.

Molecular dynamics simulation of 1 in complex with LecA. Several snapshots of ligand 1 from the 240 ns molecular dynamic trajectory are depicted as cyan sticks. The phenoxy group of 1 is partially entering the central pocket of LecA and interacts with the Trp42 of both monomers (depicted as pink sticks). The galactosyl residue firmly coordinates the calcium ion (yellow sphere) in the carbohydrate binding site, and its β‐phenyl aglycon binds to His50. Residues involved in hydrogen bonds to the galactosyl moiety are depicted by green sticks.
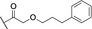
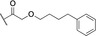
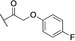
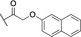
Based on these in silico considerations, we designed and synthesized derivatives 1–5 with stepwise increased linker length between the phenyl group and its ether oxygen to increase the possibility for hydrophobic contacts inside the central pocket. Furthermore, we modified the phenyl group in compounds 6–8 to assess the effect of increased lipophilicity and synthesized compound 9 carrying a hydroxy group to probe for hydrogen bonding with the cavity‘s polar entrance (see Scheme 1 for structures).
Synthesis
Galactose pentaacetate S1 was reacted with thiol S2 in presence of a Lewis acid to give β‐thioglycoside S3 in 95 % yield (Scheme 1). Then, the methyl ester in S3 was cleaved under SN2 conditions using LiI to leave the acetates unchanged. The corresponding acid intermediate was then activated with EDC/HOBt and coupled with amine S4 to give amide S5 (57 %, 2 steps). The latter was subsequently deprotected under Zemplén conditions to yield azide S6.
l‐Propargylglycin S7 was first transformed into ester S8 using thionylchloride in MeOH and then stirred in neat ethanolamine to give amide S9. In the next step, the differently substituted acetic acids bearing the central pocket‐targeting pharmacophores were introduced by amide coupling with alkynyl amine S9 into amides S10a–i (36–71 %). These alkynyl amides were then linked to azide S6 in a copper(I)‐catalyzed azide‐alkyne cycloaddition reaction resulting in the final 9 triazoles 1–9 with varying side chains in high yields (50–92 %).
Biophysical evaluation
All nine synthesized LecA ligands were then tested for LecA inhibition in a competitive binding assay based on fluorescence polarization [25] and azide S6 was included as a control devoid of the side chain targeting the central pocket. In addition, the two positive controls methyl α‐D‐galactoside (Me‐α‐D‐Gal) and para‐nitrophenyl β‐D‐galactoside (pNPG) were included (Figure 3A). Unsubstituted azide S6 (IC50=50.3±5.4 μM) showed a fourfold higher activity than Me‐α‐D‐Gal (IC50=196±7.8 μM). Phenoxy acetate 1 (IC50=49±4.5 μM), the closest derivative of the original divalent ligand, was as active as unsubstituted S6. Compounds 6 and 7 carrying electron donating or withdrawing groups in para position of the phenyl ring, as well as naphthyl 8 and phenol 9 did not show increased potency compared to 1 in this assay. Derivatives 3, 4 and 5 with increased spacer length by two to four methylene groups showed highest inhibition of LecA with IC50s between 39 and 43 μM.
Figure 3.

Analysis of synthetic inhibitor 1 interacting with LecA using (A) a competitive binding assay based on fluorescence polarization for all nine compounds including azide S6 and controls methyl α‐d‐galactoside and para‐nitrophenyl β‐d‐galactopyranoside, (B) isothermal titration calorimetry sensorgram (top panel) obtained by titration of 1 to LecA with integration of peaks and fit (bottom panel), (C) surface plasmon resonance using multi‐cycle kinetic studies (data shown for 1), top: sensorgram, bottom: affinity analysis, and (D) 19F‐protein‐observed fluorine (PrOF) NMR demonstrates the impact of binding of 1 on the NMR resonances of Trp.
To further evaluate the affinity of the ligands towards LecA in a direct binding experiment, we performed surface plasmon resonance (SPR) analysis in which LecA was immobilized and compounds 1–9 were injected. Dissociation constants at steady state were calculated from multiple cycle analysis after injection of the tested compounds in 0–200 μM range (Figure 3C). The kinetic rate constants k on and k off could not be reliably determined due to fast association and dissociation of the compounds and LecA. All compounds showed affinities towards LecA with little variation between derivatives in the μM range (6.0±0.2 to 10.3±1.3 μM, Table 1) which is comparable to previously reported aromatic galactosides. The similar affinities observed for compounds carrying the additional aryloxy pharmacophores and that of S6 suggest that the central pocket targeting pharmacophores in 1–9 either do not reach the intended site or the entropic penalties upon binding are compromising attractive interactions.
Table 1.
Analysis of LecA inhibitors S6 and 1–9 in direct binding (ITC, SPR) and competitive binding (FP) biophysical assays.
|
Structure |
Name |
ITC |
|
|
|
FP |
SPR |
|---|---|---|---|---|---|---|---|
|
|
|
K D |
ΔH |
−T ΔS |
n |
IC 50 |
K D |
|
|
|
[μM] |
[kJ/mol] |
[kJ/mol] |
|
[μM] |
[μM] |
|
|
S6 |
4.8±0.3 |
−48.9±1.6 |
18.6±0.5 |
0.9±0.0 |
50.3±5.4 |
6.7±0.3 |
|
|
1 |
9.4±4.3 |
−46.2±3.5 |
17.3±1.5 |
1.0±0.1 |
49.0±4.5 |
10.3±1.3 |
|
|
2 |
6.3±0.3 |
−50.3±1.1 |
20.7±0.3 |
1.0±0.1 |
60.7±2.5 |
6.0±0.2 |
|
|
3 |
8.3±0.3 |
−41.0±1.1 |
12.0±0.4 |
1.0±0.1 |
43.3±3.2 |
6.9±0.4 |
|
|
4 |
8.6±2.2 |
−43.9±2.6 |
14.9±0.8 |
1.0±0.0 |
41.7±3.3 |
7.2±0.2 |
|
|
5 |
6.7±2.4 |
−46.3±3.0 |
16.6±1.0 |
0.8±0.1 |
39.3±2.6 |
6.7±0.3 |
|
|
6 |
6.8±0.9 |
−45.1 ±1.7 |
15.6±0.6 |
0.8±0.1 |
60.7±6.8 |
7.7±0.4 |
|
|
7 |
8.6±0.6 |
−43.5±2.0 |
14.6±0.7 |
1.0±0.2 |
58.3±4.0 |
7.8±0.4 |
|
|
8 |
7.6±1.7 |
−38.7±2.8 |
9.4±0.8 |
1.1±0.1 |
67.7±4.5 |
6.4±0.2 |
|
|
9 |
8.8±0.7 |
−41.4±0.6 |
12.2±0.1 |
0.9±0.1 |
75.3±3.4 |
9.8±0.3 |
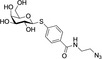
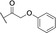
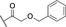
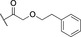

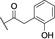
FP‐data for controls: IC50 Me‐α‐d‐Gal=196±7.8 μM; IC50 pNPG=103±6.1 μM.
Isothermal titration calorimetry was used to obtain thermodynamic data on the LecA – inhibitor interaction with both binding partners in solution. Supporting the competitive binding assay and SPR, all tested compounds showed affinities in the low micromolar range in ITC (Table 1). Surprisingly, the unsubstituted ligand S6 was the most active compound with a K d of 4.8 μM. The triazole derivatives had K d values between 6.3 and 9.4 μM, and binding enthalpies between −38.7 and −50.3 kJ/mol. Compound 2, with an increased linker length of 1 methylene group compared to 1, showed the highest affinity (K d (6)=6.3 μM) and a maximal enthalpy gain (ΔH (2)=−50.3 kJ mol−1), followed by compound 8 with the longest linker of 4 methylene groups (K d (5)=6.7 μM, ΔH (5)=−46.3 kJ mol−1). When comparing compounds 3, 4 and 5, that carry 2, 3 or 4 methylene groups respectively, a steady increase in binding enthalpy is observed which was counterbalanced by an unfavorable trend in entropy. Substitution in para/ortho position of the phenyl ring (6, 7, 9) or its replacement (8) did not lead to significant changes in binding affinity, except for tolyl 6 that was the most active (K d (6)=6.8 μM), with highest binding enthalpy (ΔH (6)=−45.1 kJ mol−1) at the cost of the highest entropic penalty (−TΔS(6)=14.6 kJ mol−1) among this subset of compounds 6–9.
Protein observed 19F (PrOF) NMR allows the detection of weak binding interactions with ligands. However, it requires the introduction of fluorine nuclei as sensitive NMR probes into the protein using 19F‐labelled amino acids or its precursors. Given the presence of tryptophans inside of the central pocket, we aimed to use PrOF NMR for detection of binders to this pocket.
To this end, all four tryptophans of LecA were simultaneously metabolically labeled at position 5 on the indole rings with fluorine and assignment was previously done by site‐directed mutagenesis. [31] Compounds 1, 2 or 8 were added to the labeled protein and PrOF NMR spectra were recorded and compared to those of the protein in absence of ligands (see Figure 3D). Trp42 is located in the carbohydrate binding site and its resonance was therefore affected by all tested ligands as observed by line broadening and chemical shift perturbation (CSP(1)=0.13 ppm, CSP(2)=0.14 ppm and CSP(8)=0.14 ppm respectively). The signals for Trp2 and Trp84 were not affected in any case while the signal intensity of the tryptophan located in the central pocket, Trp33, was shifted in presence of 1 (CSP(1)=0.08 ppm), 2 (CSP(2)=0.06 ppm) and 8 (CSP(8)=0.07 ppm) (Figure S2), supporting a contact of the arylether moieties with the central pocket of LecA.
To analyze the interaction of ligands with the central pocket at atomic resolution, we crystallized and analyzed LecA in complex with 1. The resulting structure was determined at 1.53 Å resolution in space group P212121 (Figure 1A, Table S3). The asymmetric unit consisted of a homotetramer of LecA, in agreement with previously reported LecA structures. One Ca2+‐ion and one molecule of 1 could be located in each monomer, although the completeness of electron density of the ligand beyond the triazole ring varies among the four sites (Figure 4A). The galactoside moiety of 1 interacts with LecA in the same manner as previously reported, with hydrogen bonds to residues His50, Gln53, Asp100, Thr104, Asn107 and Asn108, and coordination with the calcium ion. [27] The aglycon forms CH‐π interaction with His50 (Figure 4B) in the same way as previously reported for aromatic galactosides and LecA.[ 21 , 32 ] However, superposition of the ligand in all four sites showed that the position of the phenyl ring varied slightly (Figure 4C).
Figure 4.

Crystal structure of LecA in complex with 1. (A) The overall structure with 1 bound in all four monomers. (B) Binding pose of ligand 1 in site A with electron density displayed for the ligand. (C) Superposition of ligand conformations from all 4 binding sites. Ligand in site A is shown as sticks, whilst the others are shown as lines. (D) superposition of LecA structure in complex with 1 (protomer A) and structure in complex with peptide‐based divalent ligand (PDB: 4CP9) reported by Winssinger et al. [29] 1 is shown in white, thick stick and the visible part of the peptide‐based ligand reported by Winssinger et al. [29] in pink line.
The visible triazole rings in site A, B and D are surprisingly found in different positions and do not form productive contacts with the protein or water molecules (Figure 4B, C and Figure S4), which is in contrast to the previously reported divalent ligand with the similar phenyl aglycon and triazole design (Figure 1C), [29] where the triazole ring interacted with the side chains of His50, Tyr36 and Asp47 through a water bridge. Superposition of LecA in complex with 1 and that in complex with the peptide‐based divalent ligand reported by Winssinger et al. [29] shows that the triazole ring oriented differently among the two ligands (Figure 4D). However, the conformation of our ligand 1 in protomer A is supporting our working hypothesis, where the terminal phenyl ring reaches the targeted central pocket. This data for the shortest derivative 1 also shows that an extension of the linker as implemented in molecules 2–6 could enable a better interaction with this pocket, which is consistently supported by our SPR and FP data for the extended molecules 3–5.
Conclusions
We identified a central pocket localized between two monomers of the bacterial lectin LecA as a new target site for additional pharmacophores introduced into galactose‐based inhibitors. Inhibitors were designed to bind simultaneously to the carbohydrate binding site and the central pocket. The nine synthesized inhibitors were analyzed for their interaction with LecA in the complementary biophysical assays such as ITC, SPR and a competitive binding assay. All compounds bound to LecA in the low micromolar range. Protein NMR data supported the interaction between the phenoxyacetate moiety with the central pocket as deduced from the CSP on the central pocket localized Trp33. Finally, the crystal structure of 1 in complex with LecA revealed that the additional aromatic moiety is oriented towards the central pocket. This is further supported by the increased affinity and binding enthalpy for the extended derivative 2 and the slight increase in binding affinity for the longer spacer containing compounds 3–5 suggests their interaction with the central pocket.
One fundamental question is raised by the fact that such a galactose‐based inhibitor targeting the central pocket may block the single central pocket between two C2‐symmetry related galactose binding sites and prevent efficient binding of a second copy of the ligand bound to the other galactose site. This question may be solved by future bivalent galactosides carrying an appropriately positioned single pharmacophore addressing the central pocket.
Experimental Section
In silico crystal structure analysis: Twenty three crystallographic structures were retrieved from the Protein Data Bank [30] (PDB) (Table S1) and checked for the presence of the cavity termed here as central pocket. Cavity detection was performed using VolSite. [33] VolSite detects the shape and pharmacophore properties e. g. H‐Bond donor/acceptor, hydrophobic and aromatic points of protein cavities and gives details about its buriedness, volume and a drug‐ability score. The central pocket cavity was detected in 25 out of the 31 dimers that are available as crystallographic structures on PDB (Table S2).
Docking of 1 and molecular dynamics simulation: We used two crystal structures for docking. The structure of Winssinger's divalent LecA ligand (PDB code: 4cp9) co‐crystallized with LecA contains parts of the ligand 1 and was used to guide the docking of the whole ligand 1. It was further redocked on a LecA dimer structure with the central pocket in a more open conformation (PDB code: 4lke). Hydrogens were added to the protein structures using PROTOSS. [34] A 3D model of 1 carrying the phenoxy acetate side chain was prepared using CORINA.[ 35 , 36 , 37 ] Compound 1 was docked with the placed fragments method from Surflex. [38]
The selected docked conformation for the complex was parameterized according to the General Amber Force Field 2 (GAFF2) [39] using Ambertools 16. [40] RESP charges were fitted from the electrostatic potential and optimized at the HF/6‐31G* level of theory by using Gaussian 09. [41] The protein was prepared according to the ff14SB forcefield, [42] using tleap. Ions (K+, Cl− and Ca2+) were used to neutralize the complex. PBradii was set to mbondi3. The system was solvated with TIP3P [43] by using water molecules in an octahedral box, under periodic boundary conditions, with a distance of 12.0 Å between the protein and each face of the box. The systems were minimized in three steps by using the steepest descent, followed by a conjugate gradient after 2000 cycles: (i) position restraints for all heavy atoms of the complex (weight of 10 kcal/mol‐Å2) (ii) position restraints for the atoms of the backbone of LecA, (weight of 10 kcal/mol‐Å2); (iii) weak position restraints for the backbone atoms (weight of 5 kcal/mol‐Å2). After minimization, the system equilibration was performed on the solvent/ions in three steps: (i) the system was gradually heated from 30 to 100 K for 50 ps under the NVT ensemble with weak position restraints for the heavy atoms of LecA, by using a weight of 5 kcal/mol‐Å2; (ii) heating from 100 to 298.15 K for 250 ps under the NPT ensemble with weak position restraints for the heavy atoms of the protein using a weight of 5 kcal/mol‐Å2; (iii) MD under the NPT ensemble at constant temperature of 298.15 K for 1 ns with very weak position restraints for the protein heavy atoms, using a weight of 0.1 kcal/mol‐Å2.
The temperature was set to at 300 K using the Langevin dynamics with a collision frequency of 2 ps−1. The pressure was kept constant using the Monte Carlo barostat with a relaxation time of 2 ps. SHAKE [44] was used to control bonds length involving hydrogen atoms. The production runs, under the NPT ensemble, consisted in one replica of ∼240 ns and three independent replicas of 30 ns each, to verify the influence of sampling on ligand 1 conformations.
Visualization and hydrogen bond calculations for the trajectories were done using VMD. [45] Hydrophobic contacts were calculated using the cpptraj module available with AmberTools with the native contacts routine using a distance cutoff of 5 Å and a filter selection on non‐polar amino acids. The short trajectories and the long MD yelled similar results regarding the mentioned analyzes. Images were rendered using VMD and UCSF‐Chimera. [46]
Recombinant expression and purification of LecA: LecA expression and purification was performed as previously described. [20] The expression strain E. coli BL21(DE3) carrying the pET25pa1l [47] plasmid was grown in 4 L LB containing ampicillin (100 μg/mL) at 37 °C and 180 rpm until an OD600≈0.5 was reached. For induction of protein expression IPTG (0.25 mM) was added, and the culture was grown for additional 4 h at 30 °C. Bacterial cells were harvested by centrifugation (3000 g, 10 min, 4 °C) and the cell pellet was resuspended in TBS/Ca2+‐buffer (1 mM CaCl2, 150 mM NaCl, 20 mM TRIS, 2.5 mM KCl, pH=7.4) and PMSF (1 mM) and lysozyme (0.4 mg/mL) were added. Cell lysis was performed by 5 cycles in a homogenizer (M‐110P, Microfluidics, USA), debris was removed by centrifugation (60 min, 10000 g, 4 °C), and LecA from the supernatant was purified on a galactosylated Sepharose CL‐6B column using an Äkta start chromatography device. After washing with TBS/Ca2+‐buffer, bound LecA was eluted with 100 mM d‐galactose in buffer and then dialyzed against fresh TBS/Ca2+‐buffer every day for 7 days to remove galactose. Protein concentration was determined by UV spectroscopy (ϵ=27960 M−1 cm−1, molecular weight 12893 g mol−1, ExPASy ProtParam). LecA used for ITC was dialyzed against ddH2O for 7 days and lyophilized afterwards prior to dissolving in the ITC buffer.
Fluorescence polarization assay: This assay was performed in analogy to Joachim et al. [20] using SulfoCy5Gal, an adapted Cy5 dye. [25] 10 μL of a solution containing SulfoCy5Gal (20 nM) and LecA (40 μM) in TBS/Ca2+‐buffer were added to 10 μL of inhibitor in the same buffer at concentrations from 2000–15.6 μM and containing 2 % DMSO in a black 384‐well plate (cat no 781900, Greiner Bio‐One, Germany) in technical triplicates. Methyl α‐d‐galactopyranoside and 4‐nitrophenyl β‐d‐galactopyranoside were included as positive controls. The plate was sealed (EASYseal, cat no 676001, Greiner Bio‐One), centrifuged (1500×g, 1 min, 25 °C) and incubated in a dark chamber under shaking conditions for 16 h at r.t. The foil was removed and the fluorescence intensity was measured with a PheraStar FS microplate reader (BMG Labtech GmbH, Germany) at ex. 590 nm and em. 675 nm. Data were analyzed using MARS Data Analysis Software (BMG Labtech GmbH, Germany) after subtracting blank values (LecA in TBS/Ca2+‐buffer with 1 % DMSO) from the samples. Fluorescence polarization was calculated and the data were fitted according to the four‐parameter variable slope model. This experiment was independently repeated three times and data were averaged and visualized using GraphPad PRISM version 5.
Isothermal titration calorimetry (ITC): Galactoside (1.0 or 1.5 mM) in TBS/Ca2+‐buffer was titrated into a stirred (700 rpm) LecA solution (75–232 μM) dissolved in the same buffer using a MicroCal iTC200 (Malvern, United Kingdom) instrument at 25 °C. The reference power was set to 5 μcal s−1, the filter period to 5 s, and 20–39 injections (0.5–2 μL per injection) with an injection duration of 1 s were performed per experiment with a spacing of 240 s between each injection. For some titrations (in case of compounds 2 (1x), 4 (2x) and 8 (2x)), the syringe was refilled after the first titration ended, and the experiment was continued with the same sample cell contents to reach saturation, and the resulting files were merged using the MicroCal Concat ITC software. The first injection of each experiment was discarded, and the data were analyzed with the MicroCal Origin software using the one‐site binding model. Individual titrations are depicted in Figure S1.
Protein‐observed 19 F (PrOF) NMR: Labelled 5FW‐LecA for PrOF NMR studies was produced recombinantly as reported previously. [31] NMR experiments were conducted on a Bruker AscendTM700 (AvanceIII HD) spectrometer equipped with a 5 mm TCI700 CryoProbe in 3 mm tubes (Norell S‐3‐800‐7). PrOF NMR was recorded using 200 μM 5FW‐LecA in 20 mM Tris‐HCl pH 7.8 with 150 mM NaCl, 10 % D2O and 100 μM TFA at 310 K. Changes in the spectra in presence of DMSO and upon addition of 3 mM ligand were processed and referenced to trifluoroacetic acid (TFA) as internal reference at −75.6 ppm. We considered only changes in chemical shift perturbation (CSP) upon ligand addition being two‐fold greater than standard deviation (2x std. dev., 0.03 ppm) of the fluorine resonance.
Surface plasmon resonance (SPR): All experiments were performed on a BIACORE X100 at 25 °C. For activation and immobilization of LecA, buffer A (10 mM phosphate buffer+2.7 mM KCl, 137 mM NaCl, 0.05 % Tween 20+100 μM CaCl2) was used as a running buffer. A CM5 BIACORE chip was activated by 3 injections of 1 : 1 NHS/EDC mixture (contact time=540 s, flow rate=10 μL/min) on channel 1 and 2, followed by injections of LecA (100 μg/mL in 10 mM sodium acetate buffer pH 4.5) on channel 2 only until the final binding response reached 2939 RU. Compounds were dissolved in DMSO to the final concentration of 100 mM and subsequently diluted to 5 mM in buffer A. The compounds were then prepared to the required analytic concentrations (0.2–200 μM) in buffer A supplemented with 5 % DMSO. For multi‐cycle kinetic analysis of the compounds, buffer A supplemented with 5 % DMSO was used as a running buffer. The compounds were analyzed by injections of multiple concentrations on the immobilized LecA (contact time=30 s, dissociation time=60 s, flow rate 30 μL/min) and affinity analysis was performed using BIACORE evaluation software, which plots the binding response at the steady state against the analyzed concentrations and fits a non‐linear curve to obtain K d values. Individual experiments are depicted in Figure S3.
X‐ray crystallography: Lyophilized LecA was dissolved to saturation in PBS at pH 7.4, containing 100 μM CaCl2. 1 μL of the protein solution was mixed with 1 μL of the reservoir solution (20 % PEG6000, 1 M LiCl, 100 mM sodium acetate pH 4.2, 5 % DMSO containing 1 mM of compound 1 and the mixture was deposited on a siliconized glass circle cover slide (22 mm, Hampton research). Crystallization was performed by hanging drop vapor diffusion at 19 °C. Protein crystals were cryo‐protected in 20 % PEG6000, 1 M LiCl, 100 mM sodium acetate pH 4.2 supplemented with 20 % ethylene glycol and flash frozen in liquid nitrogen. Data collection was conducted at SOLEIL PROXIMA 1 beamline (Saint Aubin, France). The recorded data were indexed, integrated and scaled at SOLEIL using XDS, [48] and merged using AIMLESS. [49] The structure was solved by molecular replacement in PHASER [50] using 1OKO as a searching template. The model was improved by manual re‐building in COOT [51] and refinement in REFMAC5. [52] The model of compound 1 was manually built in AceDRG [53] and manually placed in the protein model. The final model was validated with MolProbity, [54] PDB‐redo [55] and wwPDB validation server (http://validate‐rcsb‐1.wwpdb.org). Structural figures were prepared using CCP4MG. [56] Data merging, phasing and model re‐building and refinement were performed through CCP4i2 [57] graphical interface.
Supporting Information
The supporting information contains the synthesis of LecA inhibitors and 1H‐ and 13C‐NMR spectra of new compounds, the tables of X‐ray structures used for the analysis and description of the carbohydrate binding site and the central pocket, ITC and PrOF NMR raw data, the sensorgrams and affinity analyses of the SPR data as well as the X‐ray structure of compound 1 with focus of each binding center of the LecA tetramer.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge the financial support of the French‐German ANR/DFG project (ANR‐AAPG‐2017) funded by the Agence Nationale de la Recherche (grant no. ANR‐17‐CE11‐0048) and Deutsche Forschungsgemeinschaft (grant no. Ti756/5‐1 and RA1944/7‐1) They also thank Glyco@Alps (ANR‐15‐IDEX‐02) for support and synchrotron SOLEIL (Saint Aubin, France) for access and technical support at beamline PROXIMA1 and the Max‐Planck Society for support. The authors also thank Emilie Gillon (CERMAV) for providing recombinant LecA for SPR and X‐ray crystallographic experiments. Open Access funding enabled and organized by Projekt DEAL.
E. Siebs, E. Shanina, S. Kuhaudomlarp, P. da Silva Figueiredo Celestino Gomes, C. Fortin, P. H. Seeberger, D. Rognan, C. Rademacher, A. Imberty, A. Titz, ChemBioChem 2022, 23, e202100563.
References
- 1. Fazzeli H., Akbari R., Moghim S., Narimani T., Arabestani M. R., Ghoddousi A. R., J. Res. Med. Sci. 2012, 17, 332–337. [PMC free article] [PubMed] [Google Scholar]
- 2. Plotkowski M. C., Chevillard M., Pierrot D., Altemayer D., Zahm J. M., Colliot E., Puchelle G., J. Clin. Invest. 1991, 87, 2018–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Imberty A., Varrot A., Curr. Opin. Struct. Biol. 2008, 18, 567–576. [DOI] [PubMed] [Google Scholar]
- 4. Gilboa-Garber N., Methods Enzymol. 1982, 83, 378–385. [DOI] [PubMed] [Google Scholar]
- 5. Diggle S. P., Stacey R. E., Dodd C., Cámara M., Williams P., Winzer K., Environ. Microbiol. 2006, 8, 1095–1104. [DOI] [PubMed] [Google Scholar]
- 6. Tielker D., Hacker S., Loris R., Strathmann M., Wingender J., Wilhelm S., Rosenau F., Jaeger K. E., Microbiology 2005, 151, 1313–1323. [DOI] [PubMed] [Google Scholar]
- 7. Davies D., Nat. Rev. Drug Discovery 2003, 2, 114–122. [DOI] [PubMed] [Google Scholar]
- 8. Meiers J., Siebs E., Zahorska E., Titz A., Curr. Opin. Chem. Biol. 2019, 53, 51–67. [DOI] [PubMed] [Google Scholar]
- 9. Calvert M. B., Jumde V. R., Titz A., Beilstein J. Org. Chem. 2018, 14, 2607–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wagner S., Sommer R., Hinsberger S., Lu C., Hartmann R. W., Empting M., Titz A., J. Med. Chem. 2016, 59, 5929–5969. [DOI] [PubMed] [Google Scholar]
- 11. Cecioni S., Imberty A., Vidal S., Chem. Rev. 2015, 115, 525–561. [DOI] [PubMed] [Google Scholar]
- 12. Bernardi A., Jiménez-Barbero J., Casnati A., De Castro C., Darbre T., Fieschi F., Finne J., Funken H., Jaeger K. E., Lahmann M., et al., Chem. Soc. Rev. 2013, 42, 4709–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hauck D., Joachim I., Frommeyer B., Varrot A., Philipp B., Möller H. M., Imberty A., Exner T. E., Titz A., ACS Chem. Biol. 2013, 8, 1775–1784. [DOI] [PubMed] [Google Scholar]
- 14. Sommer R., Hauck D., Varrot A., Wagner S., Audfray A., Prestel A., Möller H. M., Imberty A., Titz A., ChemistryOpen 2015, 4, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hofmann A., Sommer R., Hauck D., Stifel J., Göttker-Schnetmann I., Titz A., Carbohydr. Res. 2015, 412, 34–42. [DOI] [PubMed] [Google Scholar]
- 16. Sommer R., Exner T. E., Titz A., PLoS One 2014, 9, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sommer R., Rox K., Wagner S., Hauck D., Henrikus S. S., Newsad S., Arnold T., Ryckmans T., Brönstrup M., Imberty A., et al., J. Med. Chem. 2019, 62, 9201–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sommer R., Wagner S., Rox K., Varrot A., Hauck D., Wamhoff E.-C., Schreiber J., Ryckmans T., Brunner T., Rademacher C., et al., J. Am. Chem. Soc. 2018, 140, 2537–2545. [DOI] [PubMed] [Google Scholar]
- 19. Rodrigue J., Ganne G., Blanchard B., Saucier C., Giguère D., Shiao T. C., Varrot A., Imberty A., Roy R., Org. Biomol. Chem. 2013, 11, 6906. [DOI] [PubMed] [Google Scholar]
- 20. Joachim I., Rikker S., Hauck D., Ponader D., Boden S., Sommer R., Hartmann L., Titz A., Org. Biomol. Chem. 2016, 14, 7933–7948. [DOI] [PubMed] [Google Scholar]
- 21. Kadam R. U., Garg D., Schwartz J., Visini R., Sattler M., Stocker A., Darbre T., Reymond J.-L., ACS Chem. Biol. 2013, 8, 1925–1930. [DOI] [PubMed] [Google Scholar]
- 22. Pertici F., de Mol N. J., Kemmink J., Pieters R. J., Chem. Eur. J. 2013, 19, 16923–16927. [DOI] [PubMed] [Google Scholar]
- 23. Zahorska E., Kuhaudomlarp S., Minervini S., Yousaf S., Lepsik M., Kinsinger T., Hirsch A. K. H., Imberty A., Titz A., Chem. Commun. 2020, 56, 8822–8825. [DOI] [PubMed] [Google Scholar]
- 24. Wagner S., Hauck D., Hoffmann M., Sommer R., Joachim I., Müller R., Imberty A., Varrot A., Titz A., Angew. Chem. Int. Ed. 2017, 56, 16559–16564; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16786–16791. [Google Scholar]
- 25. Kuhaudomlarp S., Siebs E., Shanina E., Topin J., Joachim I., da Silva Figueiredo Celestino Gomes P., Varrot A., Rognan D., Rademacher C., Imberty A., et al., Angew. Chem. Int. Ed. 2021, 60, 8104–8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eierhoff T., Bastian B., Thuenauer R., Madl J., Audfray A., Aigal S., Juillot S., Rydell G. E., Müller S., de Bentzmann S., et al., Proc. Natl. Acad. Sci. USA 2014, 111, 12895–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cioci G., Mitchell E. P., Gautier C., Wimmerová M., Sudakevitz D., Pérez S., Gilboa-Garber N., Imberty A., FEBS Lett. 2003, 555, 297–301. [DOI] [PubMed] [Google Scholar]
- 28. Kadam R. U., Garg D., Schwartz J., Visini R., Sattler M., Stocker A., Darbre T., Reymond J.-L., ACS Chem. Biol. 2013, 8, 1925–1930. [DOI] [PubMed] [Google Scholar]
- 29. Novoa A., Eierhoff T., Topin J., Varrot A., Barluenga S., Imberty A., Römer W., Winssinger N., Angew. Chem. Int. Ed. 2014, 53, 8885–8889; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9031–9035. [Google Scholar]
- 30. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shanina E., Siebs E., Zhang H., Varón Silva D., Joachim I., Titz A., Rademacher C., Glycobiology 2021, 31, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kadam R. U., Bergmann M., Hurley M., Garg D., Cacciarini M., Swiderska M. A., Nativi C., Sattler M., Smyth A. R., Williams P., et al., Angew. Chem. Int. Ed. 2011, 50, 10631–10635; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10819–10823. [Google Scholar]
- 33. Desaphy J., Azdimousa K., Kellenberger E., Rognan D., J. Chem. Inf. Model. 2012, 52, 2287–2299. [DOI] [PubMed] [Google Scholar]
- 34. Bietz S., Urbaczek S., Schulz B., Rarey M., J. Cheminf. 2014, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.“CORINA Classic – High-Quality 3D Molecular Models|MN-AM,” can be found under https://www.mn-am.com/products/corina.
- 36. Schwab C. H., Drug Discovery Today Technol. 2010, 7, e245–e253. [Google Scholar]
- 37. Sadowski J., Gasteiger J., Klebe G., J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar]
- 38. Jain A. N., J. Med. Chem. 2003, 46, 499–511. [DOI] [PubMed] [Google Scholar]
- 39. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 40.D. A. Case, R. M. Betz, D. S. Cerutti, C. I. T. E., T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, et al., AMBER 2016, University of California, San Francisco 2016.
- 41.Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian Inc. Wallingford CT, 2009.
- 42. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C., Proteins Struct. Funct. Genet. 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jorgensen W. L., Jenson C., J. Comput. Chem. 1998, 19, 1179–1186. [Google Scholar]
- 44. Kräutler V., Van Gunsteren W. F., Hünenberger P. H., J. Comput. Chem. 2001, 22, 501–508. [Google Scholar]
- 45. Humphrey W., Andrew D., Schulten K., J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 47. Blanchard B., Nurisso A., Hollville E., Tétaud C., Wiels J., Pokorná M., Wimmerová M., Varrot A., Imberty A., J. Mol. Biol. 2008, 383, 837–853. [DOI] [PubMed] [Google Scholar]
- 48. Kabsch W., Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Evans P. R., Acta Crystallogr. Sect. D 2011, 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCoy A. J., Acta Crystallogr. Sect. D 2006, 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., Acta Crystallogr. Sect. D 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Long F., Nicholls R. A., Emsley P., Gražulis S., Merkys A., Vaitkus A., Murshudov G. N., Acta Crystallogr. Sect. D 2017, 73, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Joosten R. P., Long F., Murshudov G. N., Perrakis A., IUCrJ 2014, 1, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McNicholas S., Potterton E., Wilson K. S., Noble M. E. M., Acta Crystallogr. Sect. D 2011, 67, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Potterton L., Agirre J., Ballard C., Cowtan K., Dodson E., Evans P. R., Jenkins H. T., Keegan R., Krissinel E., Stevenson K., et al., Acta Crystallogr. Sect. D 2018, 74, 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information