
Fig. 1 |. SPTLC1 variants in patients with childhood-onset ALS.
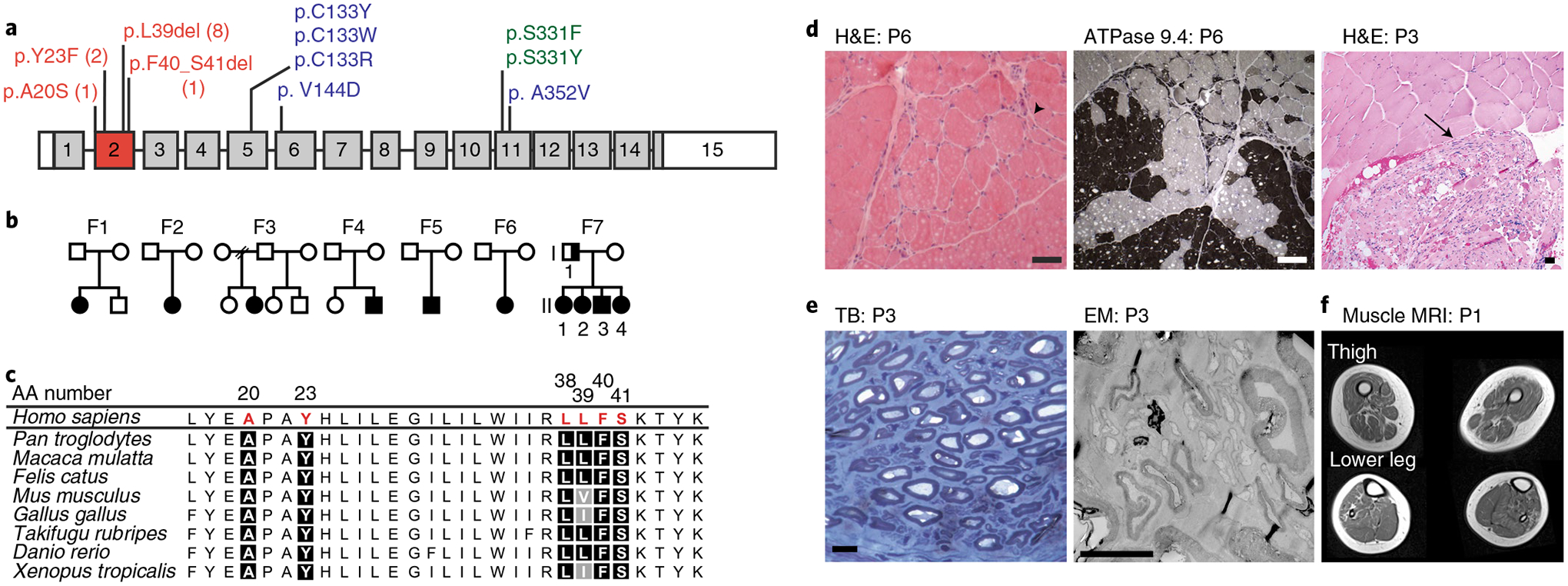
a, SPTLC1 gene with ALS variants (red) in exon 2, compared with classic (blue) and atypical (green) HSAN1 variants. The numbers for ALS variants indicate the number of individuals in this report with that variant. b, Pedigrees of families with ALS and SPTLC1 variants. Solid symbols indicate ALS/motor neuron disease; a half-solid symbol indicates sensorimotor neuropathy. Empty symbols indicate unaffected family members. F, family c, Evolutionary conservation of altered SPTLC1 amino acids associated with ALS. Due to the repeat sequence, distinction of the deletion of L38 versus L39 residues in humans is not possible. However, the presence of both residues in series is highly evolutionarily conserved. AA number, amino acid number of human SPTLC1 protein. d, Muscle biopsies showing angular atrophic fibers (arrowhead) and fiber type grouping (ATPase stain) highlighting acute and chronic neurogenic atrophy, respectively. Pyknotic nuclear clumps and grouped, fascicular atrophy (arrow) both also suggest chronic neurogenic atrophy. Scale bars, 50 μm; H&E, hematoxylin and eosin; P, patient. e, Biopsy of sural nerve (a sensory nerve). Semithin toluidine blue (TB) stain (left) and electron microscopy (EM) photograph (right) showing normal density and morphology of myelinated and unmyelinated axons. There are no degenerating axonal profiles or empty Remak Schwann cells. Scale bars, 5 μm. f, T1 axial MRI images of the lower extremities in patient 1 showing global skeletal muscle atrophy and patchy, heterogeneous T1 hyperintensity consistent with denervation-related changes in the muscle.