

Abstract
Bottom-up proteomic strategies rely on efficient digestion of proteins into peptides for mass spectrometry analysis. In-solution and filter-based strategies are commonly used for proteomic analysis. In recent years, filter-aided sample preparation (FASP) has become the dominant filter-based method due to its ability to remove SDS prior to mass spectrometry analysis. However, the time-consuming nature of FASP protocols have led to the development of new filter-based strategies. Suspension traps (S-Traps) were recently reported as an alternative to FASP and in-solution strategies as they allow for high concentrations of SDS in a fraction of the time of a typical FASP protocol. In this study, we compare the yields from in-solution, FASP, and S-Trap based digestions of proteins extracted in SDS and urea-based lysis buffers. We performed label-free quantification to analyze the differences in the portions of the proteome identified using each method. Overall, our results show that each digestion method had a high degree of reproducibility within the method type. However, S-Traps outperformed FASP and in-solution digestions by providing the most efficient digestion with the greatest number of unique protein identifications. This is the first work to provide a direct quantitative comparison of two filter-based digestion methods and a traditional in-solution approach to provide information regarding the most efficient proteomic preparation.
Keywords: sample preparation techniques, bottom-up proteomics, suspension trap, filter-aided sample preparation, sodium dodecyl sulfate, label-free quantification, digestion comparison, tandem mass spectrometry, quantitative proteomics
Graphical Abstract
INTRODUCTION
Global identification and quantification of proteins and peptides has become a critical tool for the analysis of biological systems.1–3 Mass spectrometry-based studies using ultra performance liquid chromatography (UPLC)4–6 or capillary zone electrophoresis (CZE)7–9 coupled to a mass spectrometer are often performed to identify thousands of proteins in a sample of interest. Large-scale proteomic studies often rely on digestion of proteins into peptides using the enzyme trypsin, and the corresponding peptide ion intensities can be exploited for quantification.10 Efficient digestion of proteins into their corresponding peptides is critical for the success of bottom-up protein quantification. The depth of coverage of the proteome is largely dependent on the chosen sample preparation and separation techniques. Sample preparation methods upstream of mass spectrometry can vary greatly depending on sample type, lysis conditions, digestion, and offline fractionation methods, with each method having distinct advantages and drawbacks.
A number of previous studies have compared lysis conditions,11,12 fractionation methods,13,14 and digestion conditions10,15,16 to derive the optimal proteomic preparation protocol. Most studies emphasize the importance of detergents to be able to detect hydrophobic membrane proteins that perform many important functions in the cell. The most commonly used detergent to achieve this result is sodium dodecyl sulfate (SDS), which readily solubilizes proteins in biological matrices. However, SDS removal is critical before mass spectrometry analysis due to its ability to contaminate liquid chromatography systems and dominate mass spectra. The most popular method for detergent removal and digestion in recent years has been filter-aided sample preparation (FASP),17 which was designed based on previous methods from Manza et al. using spin filters.18 In this preparation, SDS-containing protein samples are applied to a filter and washed with 8 M urea to disrupt SDS micelles. Filters are then washed with various buffers to remove excess urea before digestion. Digestion is performed on the filter and peptides are eluted the following day. Although this method has been widely successful for many applications,19–21 the tedious nature of the protocol and batch-to-batch variation often hinders its use for high-throughput proteomic studies. For these reasons, new technologies have been developed to aid SDS-based proteomic preparations.
Recently, a suspension trapping (S-Trap) method was described by Zougman et al., which allows for preparation of SDS-containing protein lysates in a fraction of the time needed for FASP protocols.22 In this method, proteins are lysed in 5% SDS and a fine protein particulate suspension is created through the addition of phosphoric acid and a methanolic buffer solution. The suspension is trapped in a stack of filtration material and residual SDS is washed away in one short wash step. Proteins are digested in the filter using a protease of choice before analysis via LC–MS/MS. The S-Trap possesses the advantages of FASP and other filter-based methods while decreasing the sample handling steps and time prior to mass spectrometry analysis.
In this study, we examined the quantitative and qualitative differences in proteins from the SW480 colon cancer cell line that were lysed and digested under various conditions. We compare commonly used SDS and urea-based lysis buffers, and digest these lysates using a traditional in-solution approach, a FASP method, and an S-Trap method. We used label-free quantification in MaxQuant23 to determine quantitative differences that occur due to the various lysis and digestion strategies. The results of this work show that each method of digestion preferentially enriches different parts of the proteome and yields different total numbers of protein identifications. Furthermore, we found that filter-based methods were more consistent across experimental replicates when compared to in-solution digests. It is critical to assess the efficacy and robustness of these proteomic methods to determine their use in bottom-up proteomic studies with clinical and biological consequences.
MATERIALS AND METHODS
Reagents
Cell culture reagents and phosphate buffered saline were purchased from Invitrogen (Gaithersburg, MD). Fetal bovine serum (FBS) was obtained from Hyclone. Sodium dodecyl sulfate (SDS), triethylammonium bicarbonate (TEAB), urea, iodoacetamide (IAA), dithiothreitol (DTT), sodium orthovanadate, were purchased from Sigma-Aldrich (St. Louis, MO). TPCK-treated trypsin was obtained in-house. Mass spectrometry solvents were obtained from Burdick and Jackson (Muskegon, MI). Suspension-traps were purchased from Protifi (Huntington, NY), while FASP filters were obtained from Millipore (Burlington, MA).
Cell Culture and Protein Harvest
Colorectal cancer SW480 cells were purchased from the American Type Culture collection (ATCC) and cultured in RPMI medium supplemented with 10% FBS and l-glutamine. Cells were maintained at 37 °C with 5% CO2 and were verified by STR sequencing in the summer of 2016. Cells were grown to 80% confluency and harvested with one of two lysis buffers. The first lysis buffer contained 8 M urea, 75 mM NaCl, 50 mM Tris-HCl (pH 8), 1 mM NaF, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM PMSF, and 1 tablet of EDTA-free protease inhibitor cocktail. The second lysis buffer contained 3% SDS in 50 mM Tris-HCl (pH 8), 1 mM NaF, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, and 1 tablet of EDTA-free protease inhibitor cocktail. Cells were harvested in 2 mL of lysis buffer and sonicated on ice for three 1 min rounds at 15% amplitude. SDS lysates were heated to 90 °C for 10 min. Lysates were then clarified at 15 000 rpm for 10 min. Total protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific Pierce, Rockford, IL). 100 μg aliquots of sample were used for all subsequent steps.
In-Solution Sample Preparation
Only proteins harvested with the 8 M urea buffer were prepared using in-solution methods. 100 μg of proteins was reduced with 5 mM DTT for 1 h at 37 °C. Proteins were then alkylated with 14 mM IAA for 30 min in the dark. The alkylation reaction was quenched with an additional 5 mM DTT at room temperature for 1 h. Samples were diluted with 50 mM Tris-HCl so that the final concentration of urea was 1.5 M. Trypsin was added 1:50 (enzyme:protein) and digested overnight at 37 °C. Digestion reactions were quenched with the addition of 10% formic acid until the pH < 3. Peptides were then vacuum centrifuged to dryness.
Suspension-Trapping (S-Trap) Preparation
Proteins were reduced and alkylated as described above. After quenching the alkylation reaction, additional SDS was added so that the final concentration was 5%. Samples were then prepared according to manufacturer’s instructions with slight modifications. Trypsin was added to a 100 mM TEAB solution and spun through the S-Trap prior to digestion. The flow-through trypsin was reloaded to the top of the column and allowed to digest overnight at 37 °C. Peptides were eluted according to the manufacturer’s protocol and vacuum centrifuged to dryness.
Filter-Aided Sample Preparation (FASP)
Proteins were reduced and alkylated as described above. After quenching the alkylation reaction, SDS-containing samples were combined 1:1 with 8 M urea in 100 mM TEAB (pH 8). FASP filters were activated with 100 μL of 100 mM TEAB and centrifuged for 15 min at 13 000g. Samples were then added to the membrane and centrifuged 40 min at 14 000g. The membrane was washed 3× with 200 μL of 8 M urea to remove SDS by centrifuging 15 min at 13 000g. The membrane was then washed twice with 200 μL of 100 mM TEAB to remove urea. 100 μL of 100 mM TEAB containing 2 μg of trypsin was added to the membrane and digested overnight at 37 °C. The next day, peptides were eluted from the membrane by centrifuging for 15 min at 13 000g. 100 μL of water was used for a second elution. Samples were dried using a vacuum centrifuge.
High-pH Reversed-Phase Fractionation
Dried peptides were resuspended in 500 μL of 20 mM TEAB at pH 9. A 50 mg C18 Sep-Pak (Waters) was used for peptide fractionation. The column was conditioned with 2 mL of ACN and equilibrated with 3 mL of 10 mM TEAB (pH 9). Sample was then applied to the column and washed with 3 mL of 10 mM TEAB (pH 9). Peptides were eluted with 8%, 15%, 22%, 30% and 50% ACN in 10 mM TEAB (pH 9). All fractions were dried completely and resuspended in 0.1% formic acid in water.
Mass Spectrometry and Data Analysis
All fractions were analyzed using a Waters NanoAcquity liquid chromatography (LC) system coupled to a Q-Exactive mass spectrometer (Thermo). The LC system was equipped with a BEH C18 column (Waters, 10 cm × 100 mm, 1.7 μm particle size) on which peptides were separated using a binary solvent system over a 90 min gradient. Solvent A consisted of 0.1% formic acid in water, while solvent B consisted of 0.1% formic acid in acetonitrile. The following linear gradient was used for all samples: 2% B for 0–8 min, 2–6% B from 8 to 10 min, 6–30% B from 10 to 60 min, 30–50% B from 60 to 70 min, 50–80% B at 71 min, 80% B until 80 min, and re-equilibration at 2% B from 80 to 90 min. The mass spectrometer was operated in Top 12 data-dependent mode with automated switching between MS and MS/MS. The ion source was operated in positive ion mode at 2.0 kV, and the ion transfer tube was maintained at 280 °C. Full MS scans were acquired from 350 to 2000 m/z at a resolution of 70 000, with an AGC target of 1 × 106 ions and a fill time of 250 ms. MS2 scans were performed from 100 to 1500 m/z at a resolution of 17 500 and a maximum fill time of 120 ms. The AGC target was set at 5 × 105 ions with an underfill ratio of 0.6%. An isolation window of 2 m/z was used for fragmentation with a normalized collision energy of 28. Dynamic exclusion was set at 40 s. Ions with a charge of +1 or greater than +7 were excluded from fragmentation. All samples were run in triplicate.
Raw files were searched using MaxQuant software (version 1.6.0.16) equipped with the Andromeda search engine. The first search peptide mass tolerance was set to 20 ppm, and the main search peptide mass tolerance was 4.5 ppm. Product ions were searched with a tolerance of 20 ppm. Trypsin was set as the digestion enzyme with a maximum of two missed cleavages. Carbamidomethylation was set as a fixed modification, while oxidation (M), acetylation (protein N-term), and deamidation (NQ) were set as variable modifications. Spectra were searched against the Uniprot human database (88 266 sequences) concatenated with common contaminants. The “match between runs” feature was used with a matching time window of 0.7 min and an alignment time window of 20 min. Target decoy analysis was performed by searching a reverse database with an overall FDR of 1% at the protein and peptide levels. Label-free quantification was performed using the MaxLFQ feature included in MaxQuant according to default LFQ parameters. The data output from MaxQuant was analyzed using Microsoft Excel, Perseus, and ProteoSign software.
For statistical quantitative analysis, the “evidence” and “proteingroups” files from MaxQuant were input into the web-based Proteosign software24 (http://bioinformatics.med.uoc.gr/ProteoSign/). The software is specifically designed to analyze quantitative proteomic experiments. Protein quantification analysis was performed using the default settings for label-free quantification. The output from Proteosign was analyzed using R and Matlab statistical software. Isoelectric point of peptides and protein GRAVY scores were obtained using ExPasy.25 Cellular compartment data for proteins was obtained from Uniprot. Functional analysis was performed using DAVID Bioinformatics Resources 6.826 (https://david.ncifcrf.gov/tools.jsp).
RESULTS AND DISCUSSION
Bottom-up proteomic protocols rely on the solubilization of proteins from samples of interest, digestion of these proteins into peptides, and identification of peptides using mass spectrometry.4 Often, detergents including SDS are used to obtain both the hydrophobic and hydrophilic proteins contained in the sample. However, detergent removal is critical before mass spectrometry analysis. In this study, we analyzed the various available digestion filters that allow for the use of high concentrations of SDS, including Suspension-Traps22 and Filter-Aided Sample Preparation,17 against a traditional in-solution digestion approach. For our analysis, 100 μg of SW480 human cell lysate in 8 M urea buffer or 5% SDS buffer was digested using all compatible methods. Proteins lysed with 8 M urea were digested using in-solution, FASP and S-Trap methods, while SDS-lysed proteins were digested only using FASP and S-Traps. Peptides were separated using high-pH reversed-phase chromatography and analyzed using a Q-Exactive mass spectrometer (Figure 1). Peptide and protein quantification was performed using maxLFQ in MaxQuant statistical software.23,27
Figure 1.
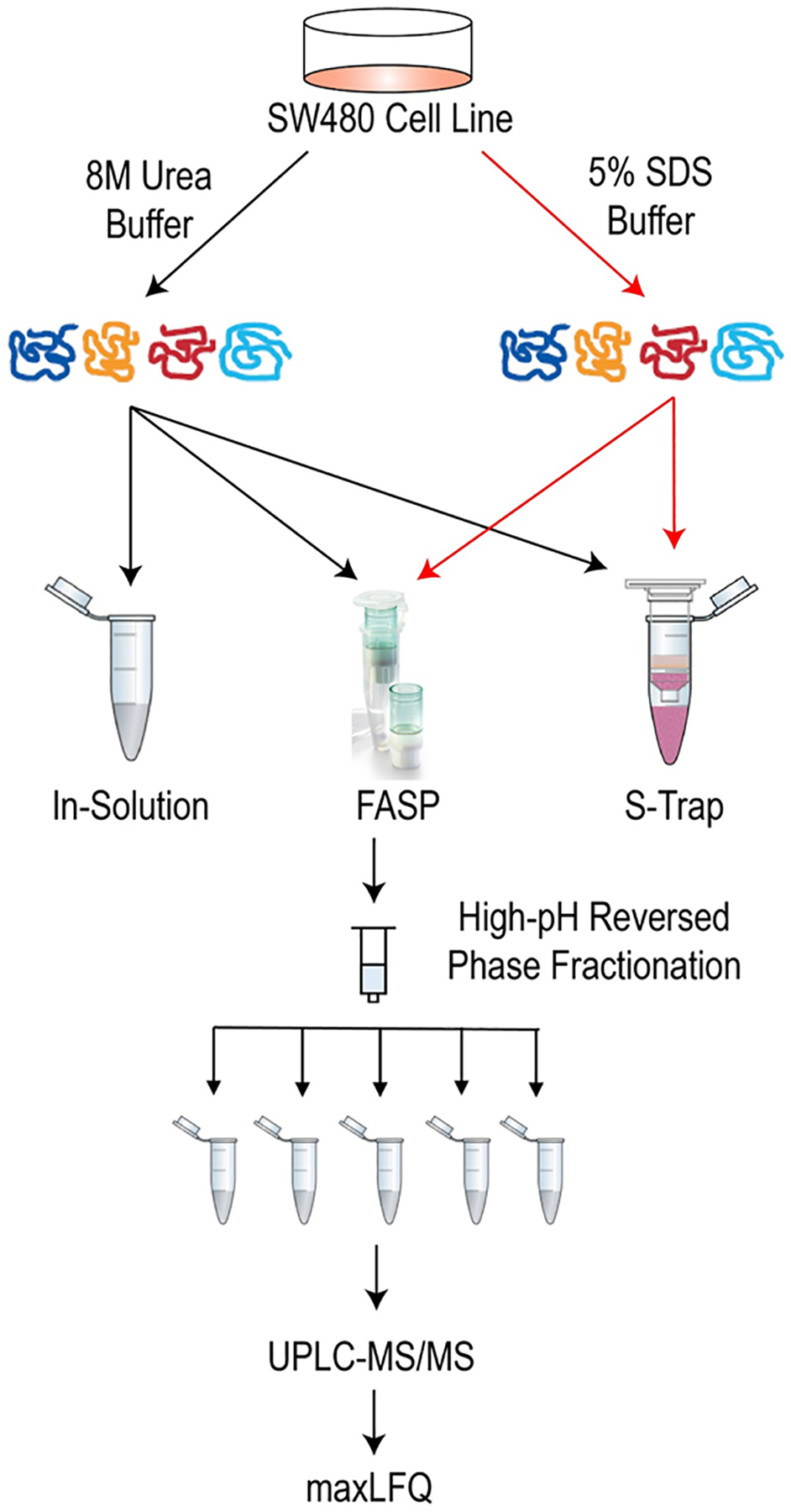
Workflow for experiments. SW480 cells were harvested in lysis buffers containing 8 M urea or 5% SDS. Lysates in 8 M urea were digested using in-solution, FASP and S-Trap methods. SDS lysates were digested using FASP and S-Trap methods. All digests were separated into five fractions using high-pH reversed phase separations on a C18 Sep-Pak. Fractions were analyzed via UPLC–MS/MS and searched using MaxQuant software.
Protein and Peptide Identifications
In this study, we compared three bottom-up proteomic sample preparation methods. We first analyzed a traditional in-solution digestion approach, and compared it to the popular filter-aided sample preparation (FASP) method. In addition, we analyzed the utility of Suspension-Traps (S-Traps), which have not yet been widely adopted in the bottom-up proteomics community. Five different digestion conditions, as indicated in Figure 1, were analyzed in experimental duplicate. Each condition resulted in different combined protein identifications (Figure 2A,B), ranging from 3757 to 4662 (Figure 2C). Overall, the S-Trap digested proteins that were harvested in urea-containing buffer yielded the greatest number of protein identifications (4662), while the SDS-lysed cells digested using the FASP method resulted in the fewest identifications (3757). There was significant overlap in the protein identifications among all methods (Figure 2A). The urea S-Trap and in-solution digest samples resulted in the greatest number of unique protein identifications. For the urea S-Trap, this finding is likely because this method yielded the greatest number of proteins, and therefore would have a large contingent of proteins not seen by other methods. However, it is somewhat surprising for the in-solution digest to contain an equal number of unique proteins. This finding indicates that filter-based methods and in-solution based methods preferentially isolate different portions of the proteome.
Figure 2.
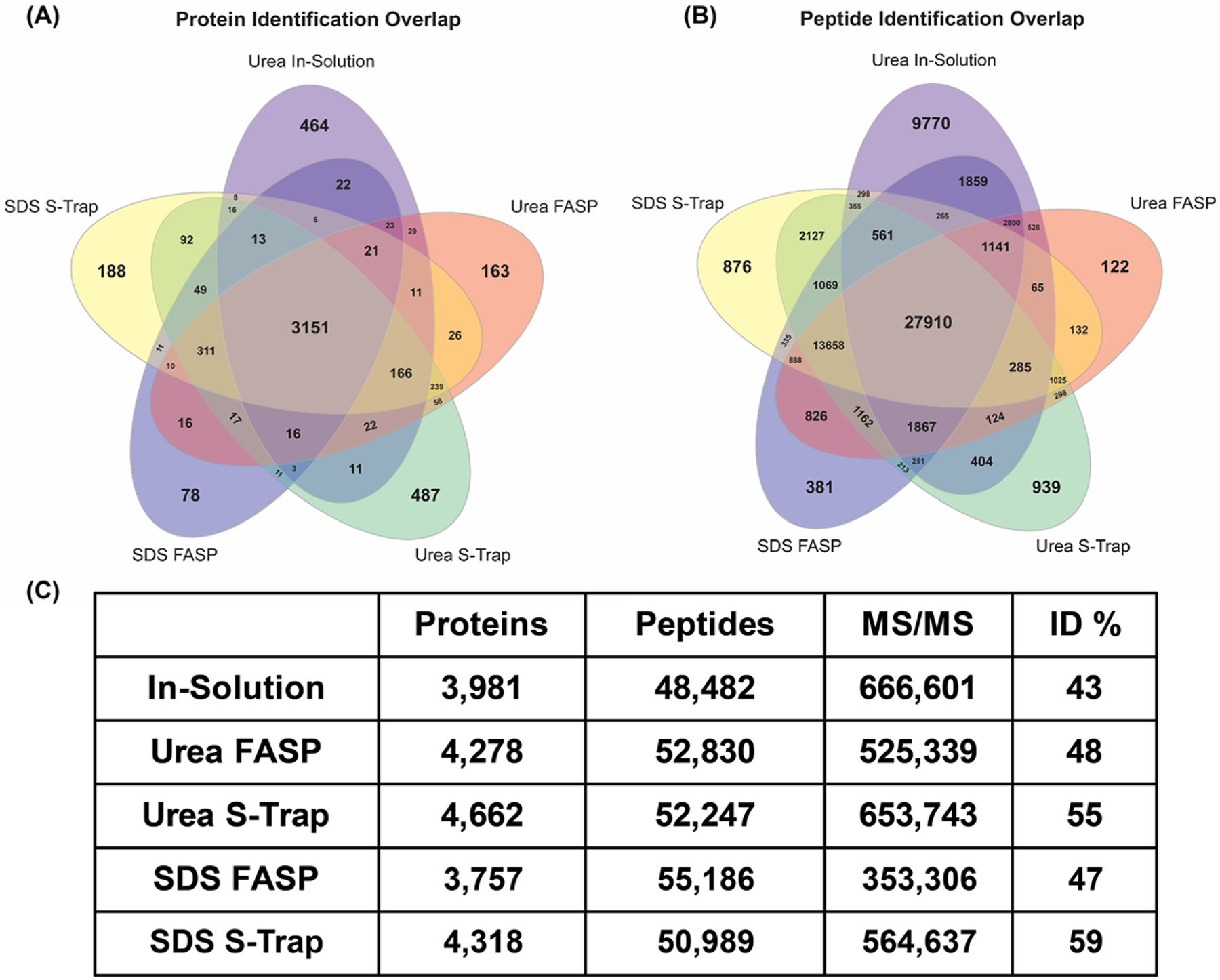
Protein and peptide identifications. (A) Overlap in unique protein identifications from two combined experimental replicates for each digestion condition. (B) Overlap in unique peptide identifications from two combined experimental replicates in each digestion condition. (C) Overall numbers of unique protein identifications, peptide identifications, MS/MS spectra, and percentage of identified spectra for two experimental replicates of each digestion condition.
The number of peptide identifications for each method was next compared (Figure 2B). The total number of unique peptides identified in each experiment ranged from 48 482 to 55 186 with an overlap among all methods of 27 910 peptides. Both the SDS S-Trap and Urea S-Trap digests resulted in larger numbers of unique peptides than the SDS or Urea-based FASP digests. However, the in-solution digest resulted in the greatest number of unique peptides, with a total of 9770. Upon further investigation, it was determined that this value is an artifact of the large proportion of missed tryptic cleavages in the in-solution digest. Of the 9770 unique peptides, 9318 peptides contained at least one missed cleavage. Therefore, these unique peptide identifications did not result in an increased number of protein identifications.
We further explored this trend in missed cleavages by examining the trypsin efficiency in all digestion conditions (Figure 3). All filter-based digestion methods showed superior trypsin efficiency compared to in-solution digestion. Between 61 and 69% of peptides identified using FASP or S-Traps had no missed cleavages, while only 43% of peptides digested in-solution contained no missed cleavages. Peptides identified after S-Trap digestion had slightly higher percentages with no missed cleavages than FASP-digested peptides. These trends continued when comparing peptides containing one or two missed cleavages. One in five peptides identified by in-solution digestion contained two missed tryptic cleavages, while only 5–8% of peptides from filter-based methods contained two missed cleavages. This substantial difference is likely due to the high trypsin concentrations used when samples are digested on a filter. In-solution digestion using urea requires a large dilution resulting in urea concentrations below 1.5 M, which dilutes trypsin and vastly decreases its efficiency. For this reason, filter-based methods are superior to in-solution digestion for targeted studies that rely on quantification of specific peptides, and also provide more fully tryptic peptides for global proteomic analysis.
Figure 3.
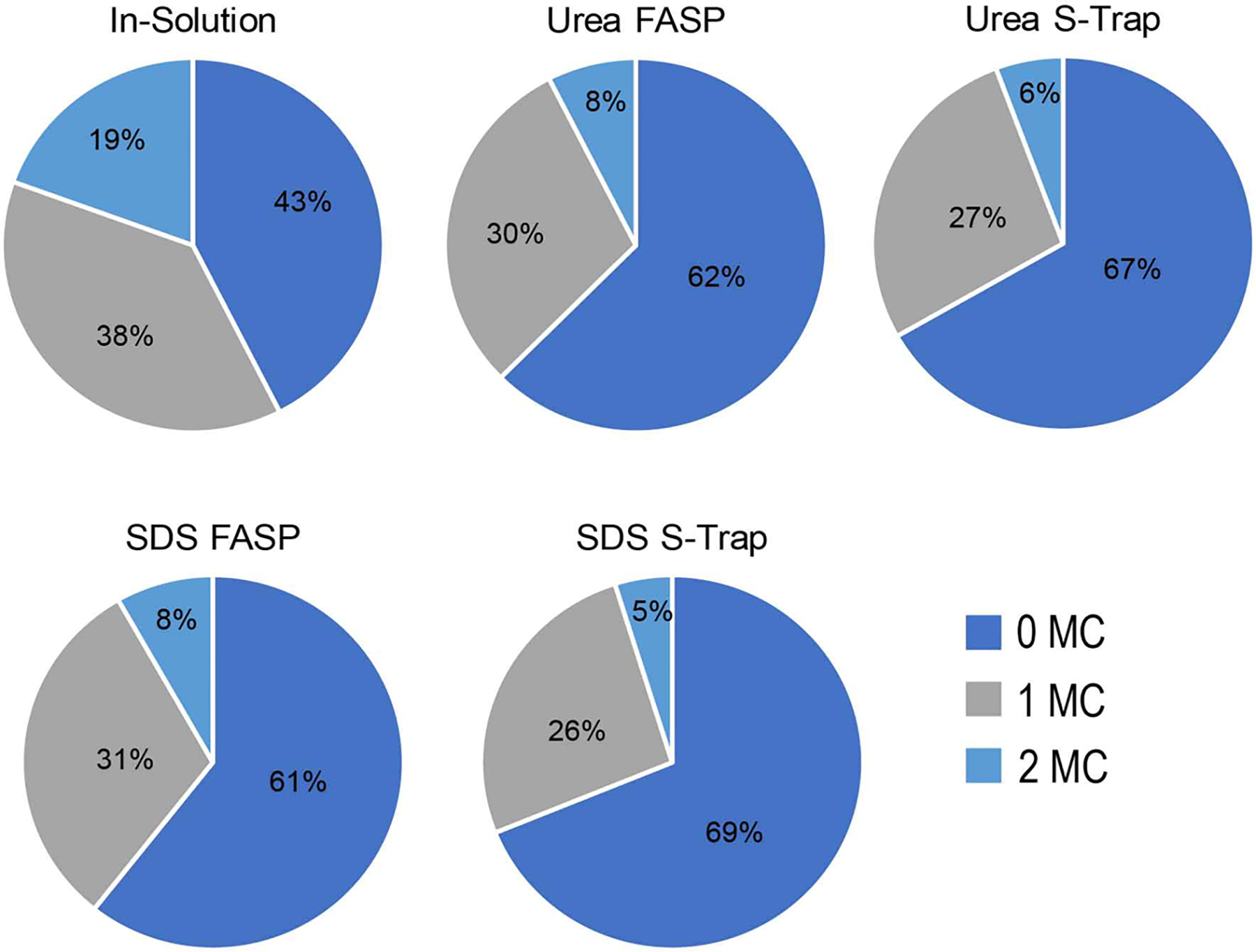
Trypsin efficiency across experiments. The percentage of identified peptides containing zero, one, or two missed tryptic cleavages is shown for each digestion condition. Percentages shown are an average of two experimental replicates.
The proteins identified by each method were further analyzed to discern any specific trends in their physical characteristics. All filter-based methods showed a similar distribution of peptide identifications based on their isoelectric points (Supplemental Figure S1A). The greatest number of peptides were identified with a pI between 4 and 4.5 for all digestion methods. Interestingly, the in-solution digested samples showed a cumulative distribution at slightly higher pI values than filter-based methods. Furthermore, tryptic peptide lengths were examined for each digestion mode (Supplemental Figure S1B). Between 72 and 78% of peptides were 20 amino acids or fewer in length for all methods. However, the in-solution digested samples showed a distinct shift toward larger peptides in their cumulative distribution. This difference is likely a result of the decreased trypsin efficiency in in-solution digested samples. No distinct trends regarding the molecular weight of the identified proteins were found for any of the different digestion methods (Supplemental Figure S1C).
Lysis buffers containing SDS are often preferred for proteomic analysis due to their ability to extract greater amounts of protein, particularly from cell membranes.12,11 We assessed our protein identification data from samples lysed in SDS or urea-based buffers to identify global trends regarding protein hydrophobicity. Overall, all data sets showed a bias toward hydrophilic proteins, with an average GRAVY score below zero (Supplemental Figure S2). In addition, neither buffer showed a bias toward proteins from a particular cellular compartment (data not shown). The overall sequence coverage of proteins was also comparable among the different methods and lysis buffers (Supplemental Figure S3), indicating that the largest qualitative differences in these methods is the efficiency of trypsin and overall numbers of protein identifications.
Functional analysis was performed using DAVID bioinformatics software. The cellular component and biological processes for the proteins found in each data set were considered. Data sets were filtered for overrepresented cellular components and biological process with a Bonferroni-corrected p-value less than 0.05. The unique cellular components overrepresented in each data set are shown in Supplemental Table S4. The urea S-Trap digested sample had the greatest number of unique cellular components, including transcription factor complexes and extrinsic components of membranes. In-solution digested samples were enriched for proteins from vesicle membranes and mitochondrial intermembrane space proteins. Furthermore, a number of biological processes were found to be unique to each method (Supplemental Table S5). Apoptotic proteins, as well as proteins involved in mitosis and spindle formation were enriched in in-solution digested samples. Urea S-Trap digested samples contained proteins involved in chaperone-mediated protein folding, EGFR signaling, histone deacetylation, and positive regulation of cell growth. Urea FASP-digested proteins were enriched for miroautophagy and mitochondrial changes, while SDS FASP-digested proteins were enriched for oxidative phosphorylation and mRNA cleavage. SDS S-Trap-digested proteins were enriched for 2-oxoglutarate metabolic processes, protein targeting to the endoplasmic reticulum, and negative regulation of the release of cytochrome c. The complete list of enriched cellular components and biological processes are provided in the Supporting Information, and may be of use for researchers targeting specific biological pathways through bottom-up proteomics.
Label-Free Quantification Analysis
MaxLFQ,27 a feature included in MaxQuant statistical software,23 was used for label-free quantification of SW480 proteins that were lysed in SDS or urea and digested with the various methods described. All samples were fractionated to increase proteome coverage for a more extensive analysis. Because the proteins analyzed all originated from the same homogenized biological sample, any quantification differences are not due to true biological changes. Up or down-regulation of proteins is a reflection of biases introduced by each bottom-up experimental preparation.
Each digest was performed in experimental duplicate to analyze the reproducibility of the preparation and quantification methods. The overlap in protein identifications between replicates can be seen in Figure 4. At the protein level, between 3085 and 3807 protein groups (76.4–88.2% of protein identifications) were found in both experimental replicates. The greatest overlap was observed in the SDS S-Trap digested samples, while the least overlap was seen in-solution. Quantitative reproducibility was assessed based on the maxLFQ-calculated intensities of proteins between replicates. The correlations between replicates showed a strong linear trend for all filter-based methods, with R squared values ranging from 0.94 to 0.98. The correlation between the in-solution replicates displayed a significantly weaker correlation, with an R squared value of 0.74, indicating a lower degree of reproducibility for in-solution digests. These trends were further explored at the peptide level (Figure 5). The SDS S-Trap digested samples showed the greatest peptide identification overlap (79.9%) and the strongest quantitative correlation (R2 = 0.87). The urea S-Trap and SDS FASP samples showed similarly strong correlations with R2 values of 0.83 and 0.84, respectively.
Figure 4.
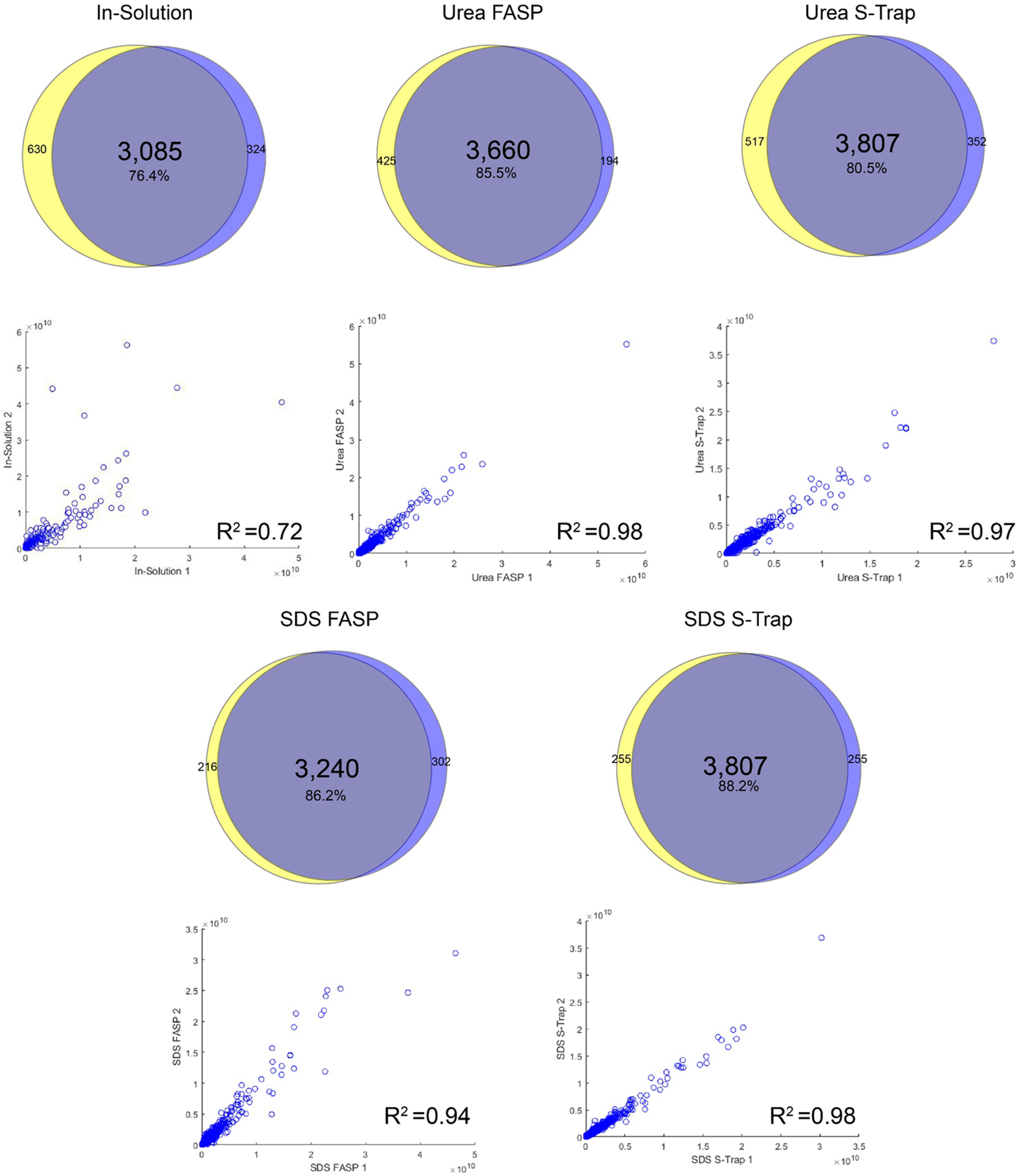
Experimental repeatability on the protein level. Venn diagrams display the overlap in protein identifications across two experimental replicates of each digestion condition. The scatter plots display the LFQ intensities of proteins detected in both replicates and their corresponding R2 values.
Figure 5.
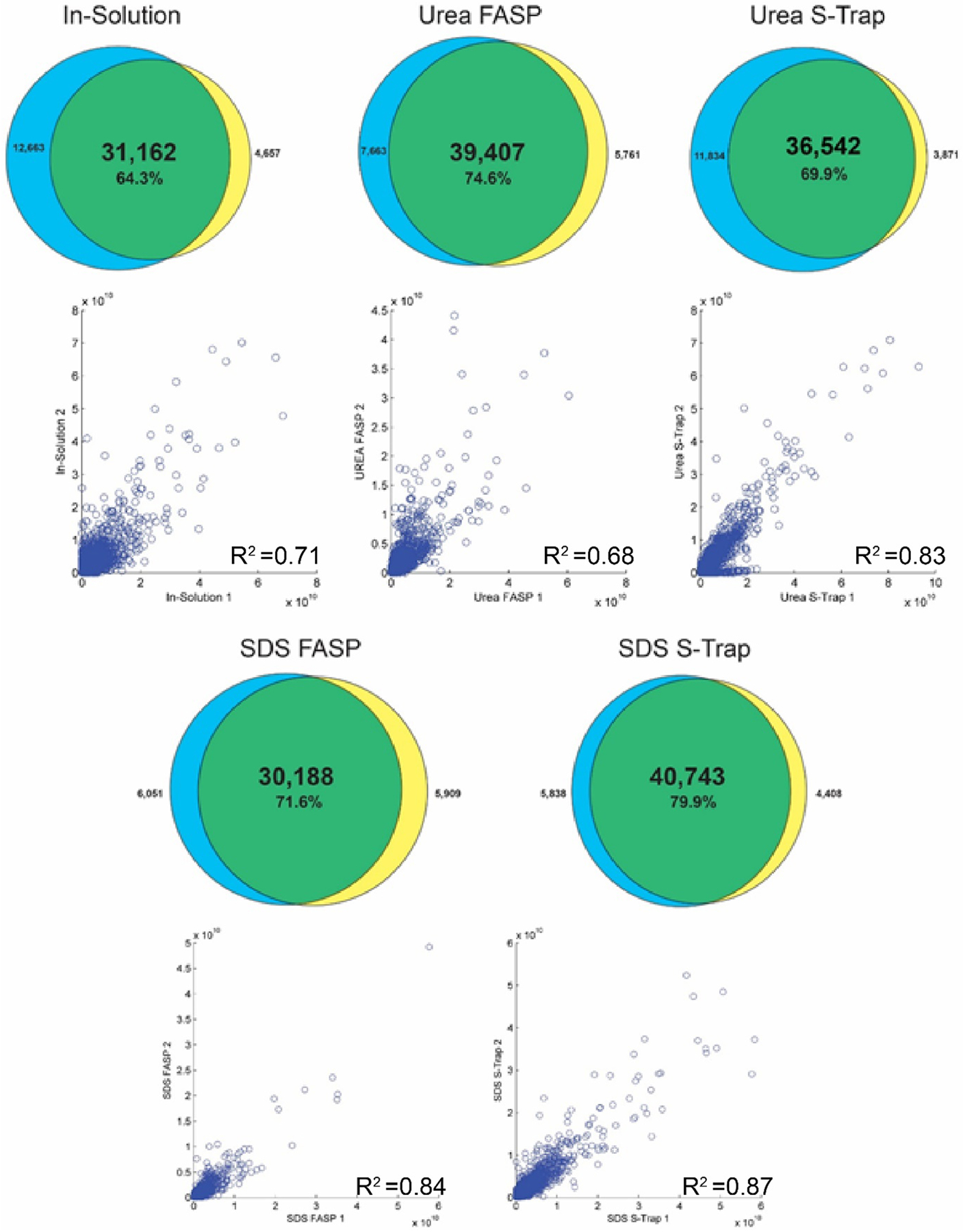
Experimental repeatability on the peptide level. Venn diagrams display the overlap in peptide identifications across two experimental replicates of each digestion condition. The scatter plots display the LFQ intensities of proteins detected in both replicates and their corresponding R2 values.
The in-solution digested samples had the least identification overlap (64.3%) and weakest correlation (R2 = 0.71). The urea FASP samples showed the weakest peptide quantification correlation overall, with an R2 of 0.68. Overall, the S-Trap digested samples showed the strongest correlations between replicates at both the protein and peptide levels, while in-solution digested samples were weakest, further illustrating their utility in bottom-up proteomics experiments.
Proteins from all FASP and S-Trap experiments were quantified relative to the in-solution digest and statistically analyzed using the online Proteosign interface.24 Volcano plots depicting this quantitative data are shown in Figure 6. Red dots indicate proteins that met a general threshold of differential regulation (log2 > 2 or log2 < −2 with a p-value <0.05). Clearly, each of the filter-based methods identifies a subset of proteins with a higher intensity than in-solution, as well as a subset with a lower intensity than in-solution. Given the vastly different mechanisms of digestion, it is expected that each method would preferentially enrich a distinct region of the proteome. Importantly, we compared quantification within each digestion type to examine the reproducibility of each digestion method (Figure 7). The volcano plots show a tight distribution of log2 fold change ratios around zero, with almost no proteins meeting the statistical threshold of significance when comparing experimental replicates. This tight correlation indicates that while there are large differences between the digestion methods, each digestion method has a high degree of reproducibility.
Figure 6.
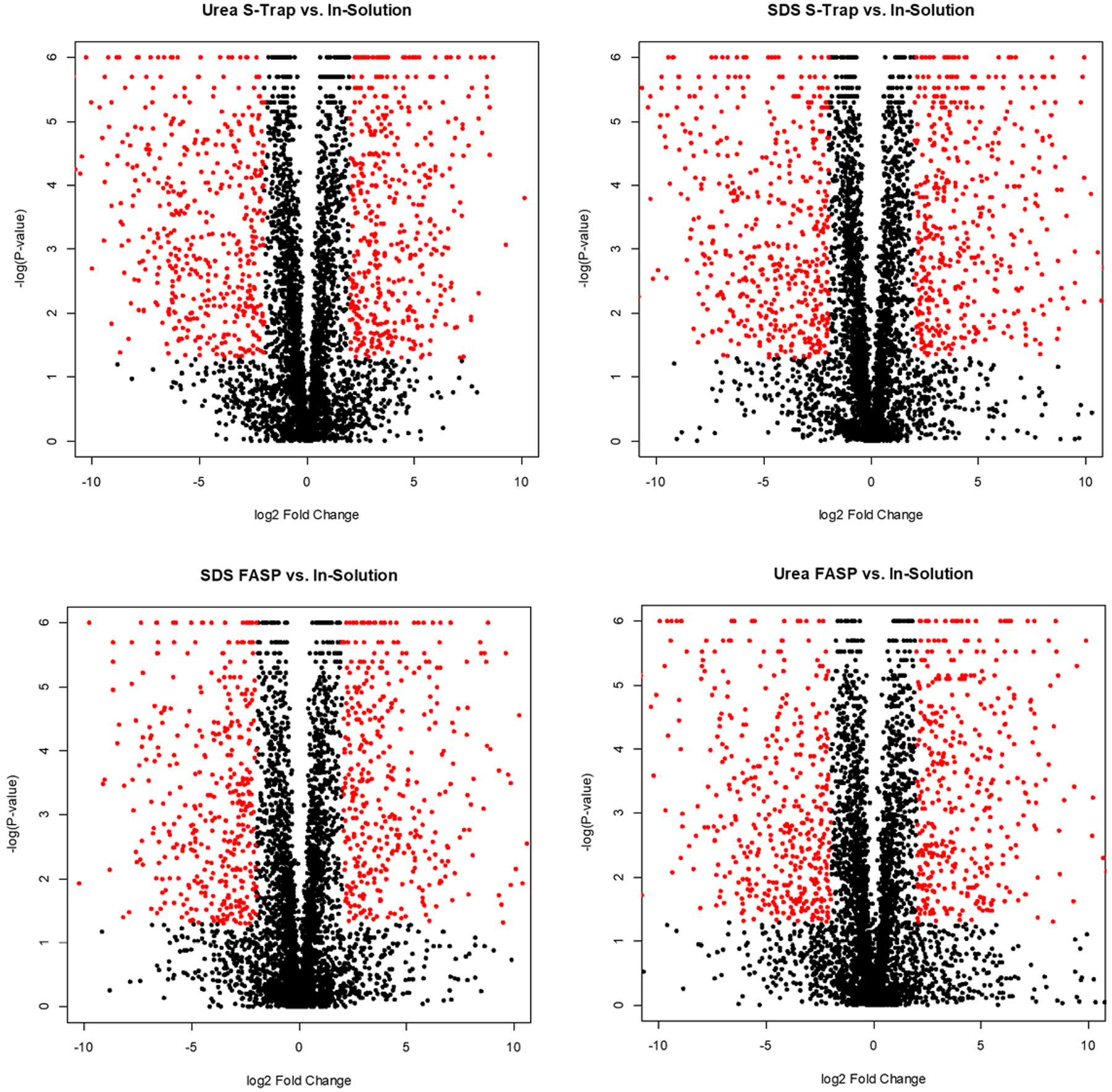
Volcano plots displaying the statistical p-value with the magnitude of abundance changes between each digestion mode compared to in-solution digestion. Red dots indicate proteins that meet the threshold for differential regulation (log2 > 2 or log2 < −2 with a p-value <0.05).
Figure 7.
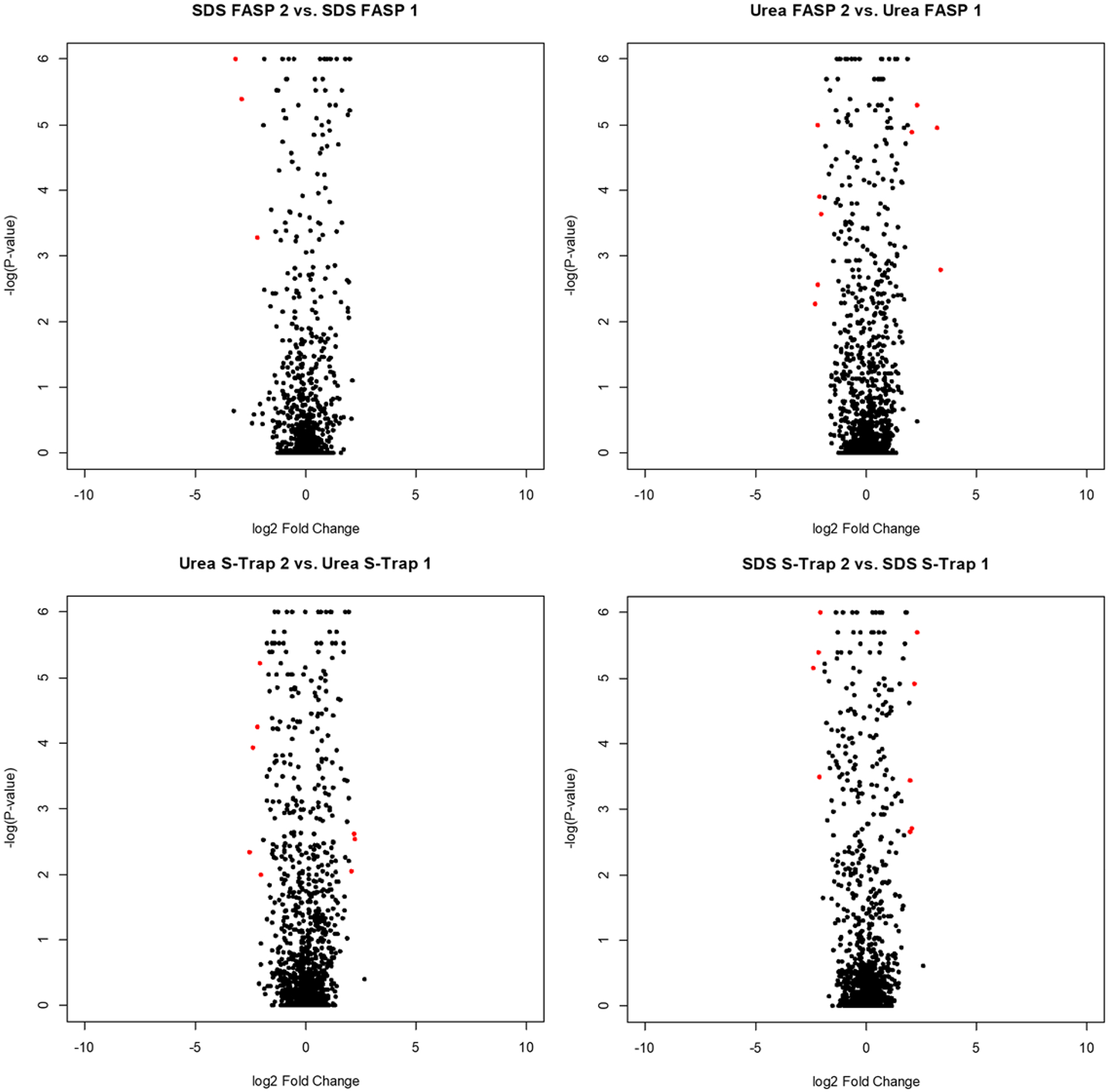
Volcano plots displaying the statistical p-value with the magnitude of abundance changes experimental replicates of each digestion method. Red dots indicate proteins that meet the threshold for differential regulation (log2 > 2 or log2 < −2 with a p-value <0.05).
Protein abundance values for each filter-based sample (urea FASP, SDS FASP, urea S-Trap, and SDS S-Trap digests) were compared to the values for the in-solution digestion and used to perform unsupervised hierarchical clustering (Figure 8A). Both the urea S-Trap and SDS S-Trap digests clustered together, while the FASP digests clustered separately from the S-Traps and each other, indicating a greater degree of reproducibility in the S-Trap digests despite the different modes of lysis. The proteins that exceeded the threshold for differential regulation compared to in-solution digestion (log2 < −2 or > 2, p < 0.05) for each method were analyzed to determine if all filter-based methods isolate a similar subset of the proteome. As seen in Figure 8B, each method had a relatively distinct set of proteins that met this threshold. Among the urea FASP, SDS FASP, urea S-Trap, and SDS S-Trap digests there were 182 proteins that commonly showed differential regulation (10.2%). Upon further examination, these 182 proteins showed a similar degree of up- or down-regulation compared to in-solution digestion in all four filter-based digests (Figure 8C), indicating that some consistent biases may be induced by all filter-based methods.
Figure 8.
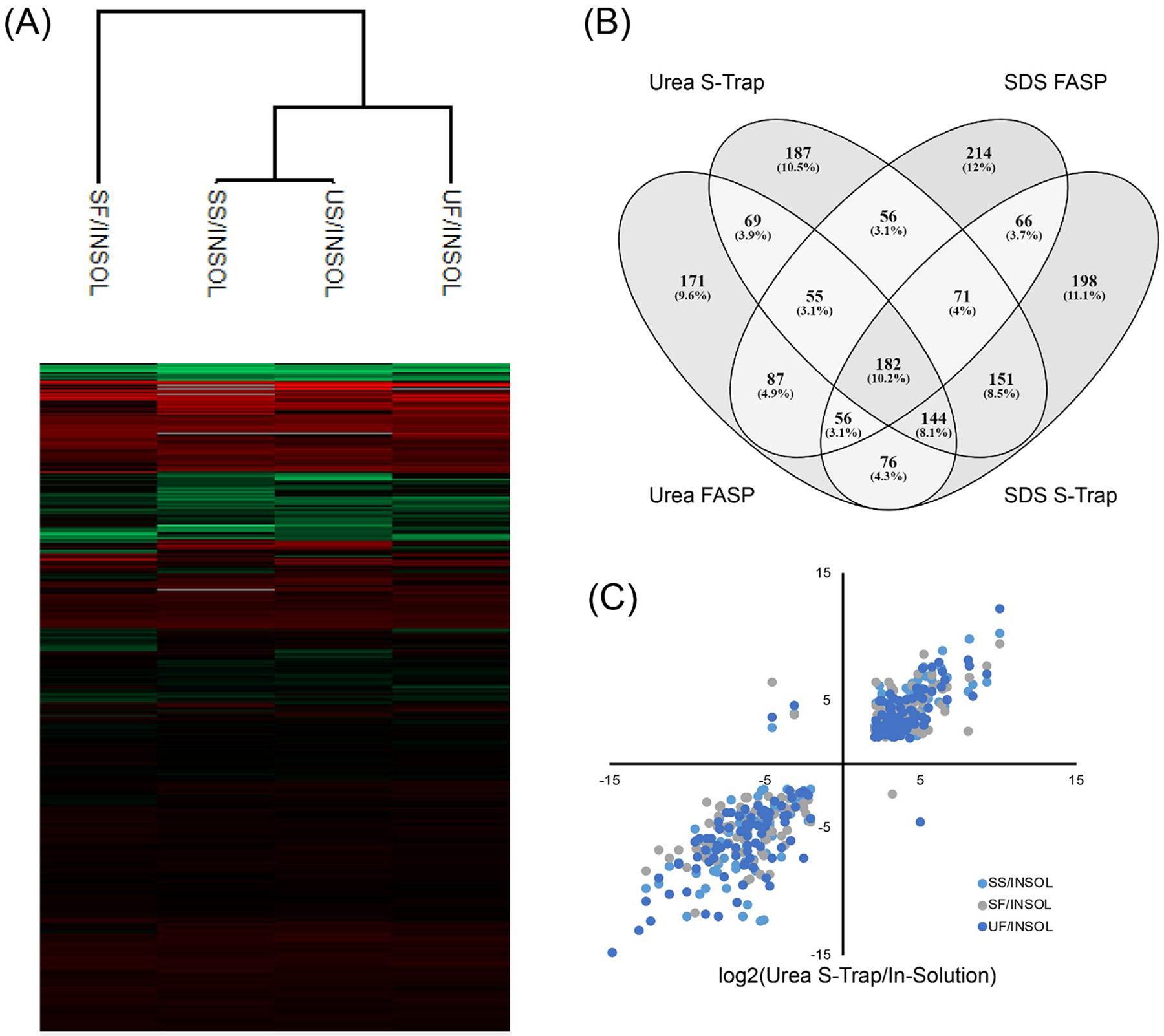
Examination of proteins that met the threshold of differential regulation relative to in-solution digestion (A). Unsupervised hierarchical clustering of protein fold changes relative to in-solution digestion (B). Correlation of proteins that met the threshold for differential regulation relative to in-solution digestion (C).
CONCLUSIONS
This report details the qualitative and quantitative differences in peptides and corresponding proteins identified by different digestion modes using bottom-up LC–MS/MS. Both urea and SDS-lysed and S-Trap digested methods outperformed FASP and in-solution based digestion methods in terms of total protein identifications and consistent quantification of proteins and peptides across experimental replicates. Urea-based S-Trap digestion identified 4662 proteins across two experimental replicates, while traditional in-solution based digests only identified 3981 proteins. Furthermore, the popular SDS FASP-based method only identified 3757 proteins across two experimental replicates. The urea-based FASP digestion identified 4278 proteins, indicating a lack of reproducibility with FASP-based digests. Trypsin performed with the greatest efficiency in S-Trap digested samples, based on the numbers of missed cleavages identified in the resulting peptides.
Each digestion method presents its own set of advantages and disadvantages. In-solution digestion is the most time and cost-effective, but our findings reveal it yields fewer protein identifications and has the lowest quantitative reproducibility. FASP-based methods, while effective, still require the most time, and have an additional cost from the filters. Furthermore, the urea-based FASP and SDS-based FASP produced very different numbers of protein identifications, indicating a lack of consistency in these filters. Finally, we found that the S-Traps demonstrated the best overall performance, with the largest numbers of protein identifications and quantitative reproducibility. The S-Trap protocol requires little more time than a typical in-solution digestion and provides the flexibility to use SDS in your proteomics sample. In addition, it outperformed all other methods regardless of lysis conditions. Therefore, S-Traps provide the best balance of time, cost, and performance of the bottom-up workflows examined in this study.
Successful bottom-up proteomic methods must be efficient and repeatable to provide an in-depth analysis of biological samples of interest. The successful performance of the S-Trap digestions under multiple lysis conditions suggests it would provide the most quantitative information when implemented in proteomic studies to answer biological questions.
Supplementary Material
ACKNOWLEDGMENTS
K.L. was supported by the National Institutes of Health (R01GM110406). M.S. was supported by National Institutes of Health Training Grant—Chemistry Biochemistry Biology Interface Program (T32GM075762). A.B.H. was supported by the National Institutes of Health (R01GM110406) and the National Science Foundation (CAREER Award, CHE-1351595). We gratefully acknowledge the assistance of Dr. Matthew Champion and the Notre Dame Mass Spectrometry and Proteomics Facility.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00235.
Figures S1–S3, Tables S4–S5 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Ong SE; Mann M Mass Spectrometry–Based Proteomics Turns Quantitative. Nat. Chem. Biol 2005, 1 (5), 252–262. [DOI] [PubMed] [Google Scholar]
- (2).Aebersold R; Mann M Mass spectrometry-based proteomics. Nature 2003, 422 (6928), 198–207. [DOI] [PubMed] [Google Scholar]
- (3).Ludwig KR; Dahl R; Hummon AB Evaluation of the mirn23a cluster through an iTRAQ-based quantitative proteomic approach. J. Proteome Res 2016, 15 (5), 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Washburn MP; Wolters D; Yates JR 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 2001, 19 (3), 242–247. [DOI] [PubMed] [Google Scholar]
- (5).Schroll MM; LaBonia GJ; Ludwig KR; Hummon AB lucose Restriction Combined with Autophagy Inhibition and Chemo-therapy in HCT 116 Spheroids Decreases Cell Clonogenicity and Viability Regulated by Tumor Suppressor Genes. J. Proteome Res 2017, 16 (8), 3009–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Labonia GJ; Lockwood SY; Heller AA; Spence DM Drug penetration and metabolism in 3D cell cultures treated in a 3D printed fluidic device : assessment of irinotecan via MALDI imaging mass spectrometry. Proteomics 2016, 16, 1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ludwig KR; Sun L; Zhu G; Dovichi NJ; Hummon AB Over 2300 Phosphorylated Peptide Identifications with Single-Shot Capillary Zone Electrophoresis-Tandem Mass Spectrometry in a 100 min Separation. Anal. Chem 2015, 87 (19), 9532–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sun L; Hebert AS; Yan X; Zhao Y; Westphall MS; Rush MJP; Zhu G; Champion MM; Coon JJ; Dovichi NJ Over 10000 peptide identifications from the hela proteome by using single-shot capillary zone electrophoresis combined with tandem mass spectrometry. Angew. Chem., Int. Ed 2014, 53 (50), 13931–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yan X; Sun L; Zhu G; Cox OF; Dovichi NJ Over 4100 protein identifications from a Xenopus laevis fertilized egg digest using reversed-phase chromatographic prefractionation followed by capillary zone electrophoresis–electrospray ionization–tandem mass spectrometry analysis. Proteomics 2016, 16 (23), 2945–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).León IR; Schwämmle V; Jensen ON; Sprenger RR Quantitative Assessment of In-solution Digestion Efficiency Identifies Optimal Protocols for Unbiased Protein Analysis. Mol. Cell. Proteomics 2013, 12 (10), 2992–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Weston LA; Bauer KM; Hummon AB Comparison of bottom-up proteomic approaches for LC–MS analysis of complex proteomes. Anal. Methods 2013, 5 (18), 4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Glatter T; Ahrné E; Schmidt A Comparison of different sample preparation protocols reveals lysis buffer-specific extraction biases in gram-negative bacteria and human cells. J. Proteome Res 2015, 14 (11), 4472–4485. [DOI] [PubMed] [Google Scholar]
- (13).Mostovenko E; Hassan C; Rattke J; Deelder AM; van Veelen PA; Palmblad M Comparison of peptide and protein fractionation methods in proteomics. EuPa Open Proteomics 2013, 1, 30–37. [Google Scholar]
- (14).Cao Z; Tang HY; Wang H; Liu Q; Speicher DW Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J. Proteome Res 2012, 11 (6), 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tanca A; Abbondio M; Pisanu S; Pagnozzi D; Uzzau S; Addis MF Critical comparison of sample preparation strategies for shotgun proteomic analysis of formalin-fixed, paraffin-embedded samples: Insights from liver tissue. Clin. Proteomics 2014, 11 (1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Burkhart JM; Schumbrutzki C; Wortelkamp S; Sickmann A; Zahedi RP Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteomics 2012, 75 (4), 1454–1462. [DOI] [PubMed] [Google Scholar]
- (17).Wisniewski JR; Zougman A; Nagaraj N; Mann M; Wi JR Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6 (5), 377–362. [DOI] [PubMed] [Google Scholar]
- (18).Manza LL; Stamer SL; Ham A-JL; Codreanu SG; Liebler DC Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5 (7), 1742–1745. [DOI] [PubMed] [Google Scholar]
- (19).Wiśniewski JR; Zougman A; Mann M Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res 2009, 8 (12), 5674–5678. [DOI] [PubMed] [Google Scholar]
- (20).Wiśniewski JR; Ostasiewicz P; Duś K; Zielińska DF; Gnad F; Mann M Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol 2012, DOI: 10.1038/msb.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wiśniewski JR; Nagaraj N; Zougman A; Gnad F; Mann M Brain phosphoproteome obtained by a fasp-based method reveals plasma membrane protein topology. J. Proteome Res 2010, 9 (6), 3280–3289. [DOI] [PubMed] [Google Scholar]
- (22).Zougman A; Selby PJ; Banks RE Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14 (9), 1006–1000. [DOI] [PubMed] [Google Scholar]
- (23).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- (24).Efstathiou G; Antonakis AN; Pavlopoulos GA; Theodosiou T; Divanach P; Trudgian DC; Thomas B; Papanikolaou N; Aivaliotis M; Acuto O; Iliopoulos I An end-user online differential proteomics statistical analysis platform. Nucleic Acids Res. 2017, 45 (W1), W300–W306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Artimo P; Jonnalagedda M; Arnold K; Baratin D; Csardi G; De Castro E; Duvaud S; Flegel V; Fortier A; Gasteiger E; Grosdidier A; Hernandez C; Ioannidis V; Kuznetsov D; Liechti R; Moretti S; Mostaguir K; Redaschi N; Rossier G; Xenarios I; Stockinger H ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40 (W1), W597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Huang DW; Sherman BT; Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 2009, 4 (1), 44–57. [DOI] [PubMed] [Google Scholar]
- (27).Cox J; Hein MY; Luber C.a.; Paron I Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.