
Figure 1.
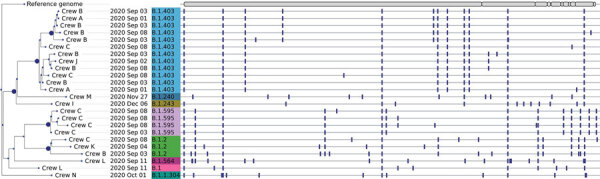
Phylogenetic tree of SARS-CoV-2 consensus whole-genome sequences from 24 of 42 positive specimens from Cameron Peak firefighters available at the Colorado State Public Health Lab with >89% genome coverage. Nodes with at least 95% ultrafast bootstrap support are labeled. Firefighter crew, sample collection date, and lineage are displayed at the tips. A visualization of the reference genome is depicted at the top of the phylogeny. Vertical bars shown across each consensus sequence indicate positions of nucleotide changes relative to the reference genome. High-quality consensus sequences were defined as sequences with >89% genome coverage (10× sequence coverage depth for Illumina [https://www.illumina.com] and 20× for Oxford Nanopore [https://nanoporetech.com]) and minimum base quality of 20. Prior to phylogenetic inference, consensus sequences were aligned to the reference genome (Genbank accession no. NC_045512.2), and insertions were removed so that all sequences were 29,903 nt in length. Phylogenetic inference of the consensus sequences was performed using IQTree version 2.0.3 (http://www.iqtree.org) with 1,000 ultrafast bootstrap replicates and phylogenetic tree visualization was performed using the python module ete3 version 3.1.2 (https://pypi.org/project/ete3). Pangolin v.2.4.25 (9) and Nextstrain’s Nextclade tools (10) were used to assign lineage and clade designations to each assembled genome.