

Abstract
Background:
A decrease in the lumacaftor-mediated increase in F508del-CFTR function and expression upon prolonged exposure to ivacaftor has previously been described. However, the efficacy observed with ivacaftor-containing CFTR modulator therapies in vivo is in conflict with these reports. We hypothesized that a portion of the apparent decrease in CFTR function observed after prolonged ivacaftor (VX-770) exposure in vitro was due to an increase in constitutive CFTR-mediated ion transport.
Methods:
Human nasal epithelial (HNE) cells were obtained by brushings from three CF individuals homozygous for the F508del CFTR mutation. Differentiated epithelia were pre-treated with prolonged (24 h) exposure to either lumacaftor (VX-809; 3 μM), tezacaftor (VX-661; 3 μM), elexacaftor (VX-445; 3 μM), and/or ivacaftor (0.1–6.4 μM) or DMSO (vehicle control), and CFTR function was assayed by Ussing chamber electrophysiology.
Results:
In cells treated with lumacaftor, constitutive CFTR activity was not increased at any concentration of co-treatment with ivacaftor. Constitutive CFTR activity was also unchanged in cells treated with the combination of tezacaftor and elexacaftor. An increase in constitutive CFTR activity above the DMSO controls was only observed in cells treated with the combination of tezacaftor and elexacaftor and co-treated with at least 0.1 μM ivacaftor.
Conclusions:
These results demonstrate that ivacaftor is a critical component in the triple combination therapy along with tezacaftor and elexacaftor to increase constitutive CFTR function. This work further elucidates the mechanism of action of the effective triple combination therapeutic that is now the primary clinical tool in treating CF.
Keywords: Cystic fibrosis, CFTR modulators, ivacaftor, tezacaftor, elexacaftor, Trikafta, orkambi, symdeko
1. Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by loss-of-function mutations to the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) [1]. Dysfunctional CFTR can cause improper airway surface hydration leading to an over-accumulation of viscous secretions resulting in chronic inflammation and progressive lung disease [1]. Over 2,000 mutations of CFTR have been identified and over 400 of these identified mutations are known to cause CF when present with another CF-causing allele [2]. Of these, the most common CF-causing mutation is F508del, which has an allelic frequency of approximately 70% and is present in 90% of patients with CF [1,2] (these numbers vary across races and ethnicities [3]). F508del-CFTR dysfunction is characterized by improper protein folding, making it a target for cellular degradation before it can pass through the Golgi complex and be trafficked to the cell surface. Pharmacologically rescued F508del-CFTR that does reach the plasma membrane still exhibits a gating defect rendering it non-functional.
The recent and growing clinical application of combination CFTR modulator therapies to treat CF has brought about remarkable progress in treating CF [4]. To date, four drugs have come to market to precisely rectify functional defects in mutant CFTR. Lumacaftor (VX-809) and tezacaftor (VX-661) are CFTR correctors that abate the folding defects exhibited by F508del-CFTR. Ivacaftor (VX-770) is a CFTR potentiator that amends the gating and conductance defects such as those caused by the CFTR mutations G551D and R117H, respectively. Elexacaftor (VX-445) is a more recently introduced CFTR modulator that has also been shown to act as both a CFTR corrector and a CFTR potentiator [5–7]. Recently, the clinical introduction of the triple combination therapeutic comprised of tezacaftor/ivacaftor/elexacaftor has shown remarkable success in treating CF caused by F508del-CFTR, and has largely replaced double modulator combination therapies, such as lumacaftor/ivacaftor and tezacaftor/ivacaftor, in the clinic.
The modest clinical effectiveness of double modulator combination therapies in early clinical studies led investigators to examine the molecular interaction between CFTR correctors and ivacaftor. In these works, prolonged (24 h) exposure to ivacaftor led to decreases in mature F508del-CFTR expression and plasma membrane stability as measured by Western blotting and decreases in F508del-CFTR-mediated ion transport as measured by electrophysiological methods [8–11]. It was concluded by the authors of these studies that prolonged (but not acute) exposure to ivacaftor decreases the correction of F508del-CFTR by tezacaftor and/or lumacaftor. These findings inspired a search for alternative CFTR potentiators that do not inhibit this correction [12–15]. However, the abrogating effect of prolonged ivacaftor exposure is concentration dependent and it is debated whether the concentrations of ivacaftor reported to interfere with F508del-CFTR correction are physiologically relevant [11,16,17]. Furthermore, the hypothesis that prolonged ivacaftor exposure nullifies correction of CFTR is not supported by clinical trial data. Clinical trials that have compared the combination therapy of lumacaftor/ivacaftor or tezacaftor/ivacaftor to monotherapy of lumacaftor or tezacaftor alone, respectively, showed an improvement in lung function with the combination therapy [18–20].
Recent research from our laboratory and elsewhere has demonstrated that changes in constitutive CFTR activity (i.e. spontaneously active CFTR-mediated ion transport prior to cAMP stimulation by exogenous forskolin) is a quantifiable measure of CFTR modulator efficacy [21–23]. Considering these recent works, we sought to re-examine the apparent abrogating effect of ivacaftor on CFTR function. We hypothesized that a portion of the apparent decrease in CFTR function observed after prolonged ivacaftor exposure was due to an increase in constitutive CFTR-mediated ion transport in the presence of ivacaftor. We predicted that prolonged ivacaftor exposure would result in decreased CFTR-mediated currents as measured by stimulation of CFTR with forskolin, as has been previously observed, but that these results would be explained by increases in constitutive CFTR activity. We predicted that prolonged exposure to ivacaftor during modulator treatments results in a measurable net benefit to CFTR function in cultured airway epithelia.
2. Methods
2.1. Cell expansion and maintenance
Under informed consent as approved by the Institutional Review Board (HS-2832) of National Jewish Health, human nasal epithelial (HNE) cells were obtained by nasal brushing from three CF individuals homozygous for the F508del CFTR mutation. HNE cells were cultured as previously described [21,24]. Briefly, HNE cells captured by nasal brushings were expanded on an irradiated NIH 3T3 feeder layer in a complete F-media in the presence of the RhoA kinase inhibitor Y-27632 (ApexBIO). Once expanded, cells were then plated on Type I bovine collagen-coated Transwell inserts (Corning) for differentiation in the absence of Y-27632. After initial seeding on inserts (2.5 × 104 cells cm−2), cells remained submerged in PneumaCult™-Ex Plus (STEMCELL Technologies) media for 2 d, then the apical media was removed and the basal media was replaced with PneumaCult™-ALI media (STEMCELL Technologies). Cells remained at ALI for 21–28 d prior to electrophysiological analysis, with basal media changes every 2–3 d. Well-differentiated cultures were defined by the presence of beating cilia and secreted mucus.
2.2. Drug treatment and electrophysiological analyses
Prior to electrophysiological analyses, differentiated epithelia were pre-treated with prolonged (24 h) exposure to either VX-661 (3 μM), VX-809 (3 μM), VX-445 (3 μM), and/or VX-770 (0.1–6.4 μM) or DMSO (vehicle control). After prolonged drug treatment, cells were mounted in an Ussing chamber apparatus (Physiological Instruments) pre-warmed to 37°C and allowed to stabilize between apical and basolateral baths filled with Ringer’s solution (120 mM NaCl, 10 mM D-glucose, 3.3 mM KH2PO4, 0.83 mM K2HPO4, 1.2 mM MgCl2, 1.2 mM CaCl2, saturated with 95% O2/5% CO2, pH 7.4) under short-circuit conditions with continuous measurement of transepithelial short-circuit current (ISC; μA cm−2) and intermittent measurement of transepithelial electrical resistance (TEER; Ω cm−2). Once stable, the Ringer’s solution was removed from the apical bath and replaced with a Cl− free, gluconate-substituted Ringer’s solution (115 mM Na+ gluconate, 5 mM Ca2+ gluconate, 10 mM D-glucose, 3.3 mM KH2PO4, 0.83 mM K2HPO4, 1.2 mM MgSO4, saturated with 95% O2/5% CO2, pH 7.4) and again allowed to stabilize before commencing acute drug additions. Acute test compound additions to the epithelia consisted of the following: apical 100 μM amiloride; apical/basolateral 20 μM forskolin (Fsk)/100 μM 3-isobutyl-1-methylxanthine (IBMX) (Fsk/IBMX); apical 1 μM VX-770; apical 10 μM CFTRinh-172, and/or apical 100 μM ATP. Although the potency of CFTR activation by Fsk and IBMX, and CFTR inhibition by CFTRinh-172 can vary by primary tissues, cell-lines, and experimental conditions, these compounds and concentrations were chosen as they are the most widely-used in electrophysiological experiments investigating CFTR [25]. While it is not known if complete activation and inhibition of CFTR were achieved in these experiments, the differences observed amongst the treatment groups demonstrate the impacts on these CFTR modulators on CFTR expression and function.
2.3. Calculations and statistical analysis
Statistical analyses were performed by either one- or two-way ANOVA with Tukey’s multiple comparisons test. To control for donor-to-donor variation, statistical analyses were carried out on donor-normalized data, wherein the mean ΔISC value for the replicates of the 0.1 μM VX-770 treatment within each donor was set to a value of 1. All data are presented as mean ± standard error. For transparency, all technical replicates from all donors are shown (3 technical replicates for each condition from 3–4 unique donors). All statistics were carried out on average values for each donor (n = 3–4 unique donors). Correlation analysis was carried out by a simple linear regression and values for slope (m), coefficient of variation (R2), and significance (P) were presented. Figure assembly and all statistical analyses were completed in Prism 6.0 (GraphPad Software, Inc.).
3. Results
3.1. Bioelectrical properties of monolayers
All cell cultures had visibly beating cilia and mucus secretions and formed electrically tight and polarized epithelia. Baseline values (prior to establishment of Cl− gradient) for TEER, transepithelial membrane potential difference (TEPD), and ISC were (approximate range): TEER = 200 to 400 Ω cm−2; TEPD = −5 to −10 mV; ISC = 20 to 50 μA cm−2. Acute additions of test compounds resulted in rapid and quick-to-stabilize responses in ISC. As expected, additions of amiloride (prior to presented data) or ATP (after presented data) resulted in a rapid decrease or increase in ISC, respectively. A summary of non-normalized baseline, gradient-responsive, and amiloride-responsive ISC values for each donor are presented in Supp. Fig. 1.
3.2. Prolonged ivacaftor exposure reduces maximal CFTR-mediated ion currents in F508del HNE cells treated with lumacaftor
To confirm previous reports that prolonged VX-770 reduces VX-809-rescued CFTR function [8,9], cells were pre-treated with either DMSO, VX-809 alone, or VX-809 and 6.4 μM VX-770 for 24 h, and the cells treated with VX-809 alone were then acutely treated with VX-770 during the electrophysiological assay (Fig. 1A). CFTR function was greatest in cells that had been treated with the CFTR modulators compared to the DMSO control (Fig. 1B–C). As expected based on the prior reports, compared to cells exposed to VX-770 acutely, the cells pre-treated with VX-770 exhibited reduced CFTR function (by approximately 20–50%) as measured by the ΔISC response to additions of Fsk/IBMX and CFTRinh-172.
Figure 1: Comparison of the effects of chronic and acute VX-770 treatment in the presence of VX-809 on F508del-CFTR function.
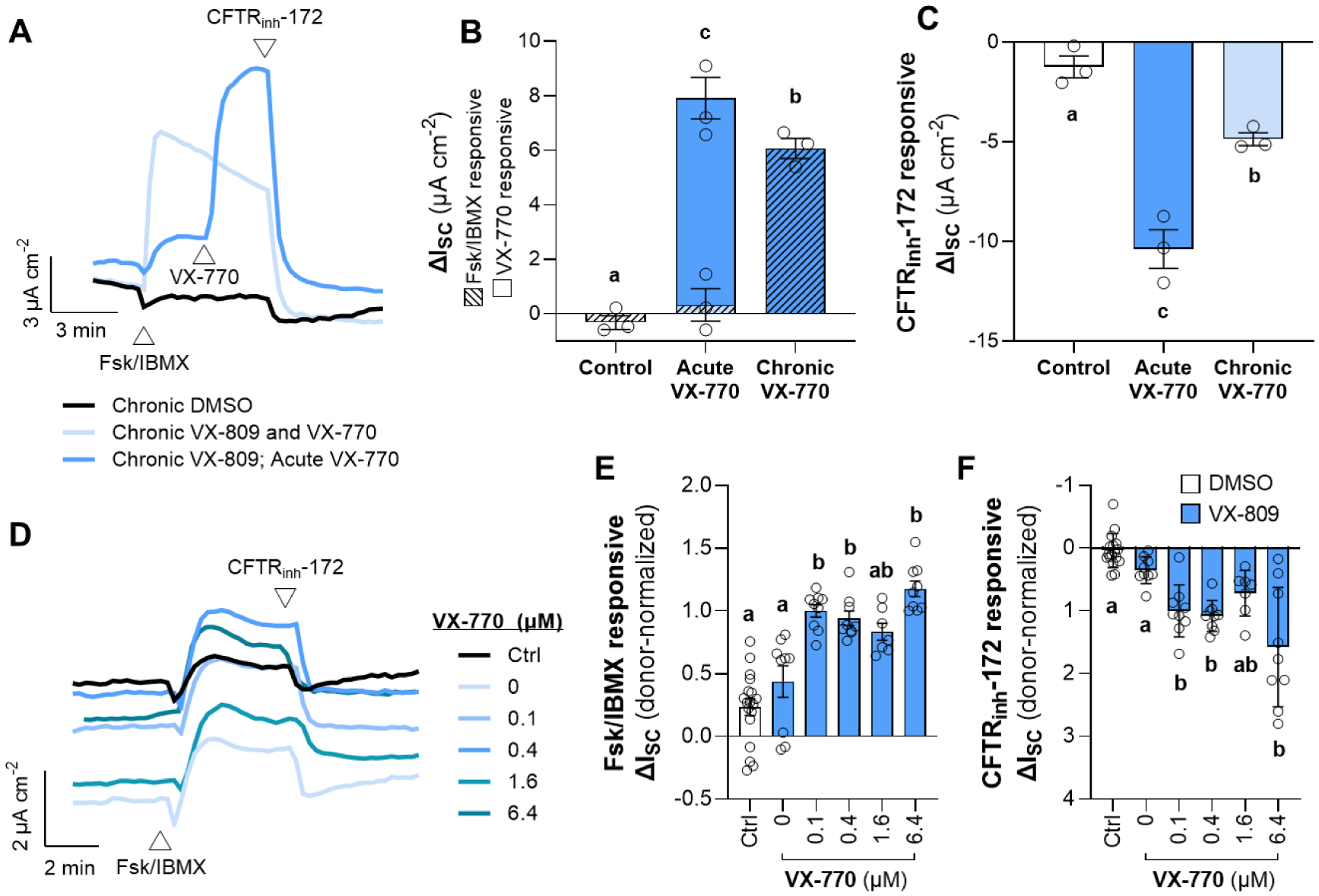
(A–C) F508del/F508del HNE cells were chronically (24 h) treated with either DMSO (control), VX-809 alone, or VX-809 and 6.4 μM VX-770. All cells were acutely exposed to Fsk/IBMX, and the cells treated with VX-809 alone were then acutely exposed to VX-770 (A). Representative traces of activation, potentiation, and inhibition of CFTR by Fsk/IBMX, VX-770, and CFTRinh-172, respectively. (B–C) Changes in ISC in response to Fsk/IBMX or CFTRinh-172. In Panel B, hatched bars represent the Fsk/IBMX-responsive ΔISC and solid bars represent the VX-770-responsive ΔISC. (D–F) F508del/F508del HNE cells were chronically (24 h) treated with either DMSO (Ctrl) or VX-809 (3 μM) (blue), and chronically co-treated with either DMSO (0 μM) or increasing concentrations of VX-770 (0.1–6.4 μM). (D) Representative Ussing trace. (E–F) Changes in ISC in response to Fsk/IBMX or CFTRinh-172. Data are presented as mean ± standard error. Groups with different letters are significantly different (P < 0.05) from each other (n = 3 unique donors; one-way ANOVA; Tukey’s post hoc).
In a second experiment, cells were pre-treated for 24 h with either DMSO (Ctrl), or VX-809, and co-treated with either DMSO (0 μM VX-770) or increasing concentrations of VX-770 (0.1–6.4 μM) (Fig. 1D). The additions of Fsk/IBMX or CFTRinh-172 resulted in slightly but significantly greater ΔISC (approximately 2-fold) across epithelia treated with VX-809 and at least 0.1 μM VX-770 (Fig. 1E–F). Both Fsk/IBMX and CFTRinh-172 responsive ΔISC generally increased with increasing concentrations of VX-770, with the 6.4 μM VX-770 conditions exhibiting the greatest ΔISC values. In this experiment, our results did not replicate those in previous studies reporting a decrease in forskolin-responsiveness after prolonged ivacaftor treatment [8,9]. This discrepancy may be due to several experimental differences between these studies, including cell type (bronchial epithelial cells were utilized in the prior studies), length of exposure to CFTR modulators, composition of the media employed (previously reported to impact constitutive CFTR activity [23]), and method of data analysis.
3.3. Prolonged ivacaftor abrogates maximal CFTR-mediated ion currents in F508del HNE cells treated with tezacaftor and elexacaftor
In this experiment, cells were pre-treated for 24 h with either DMSO, or the combination of VX-661 and VX-445, and co-treated with either DMSO (0 μM VX-770) or increasing concentrations of VX-770 (0.1–6.4 μM) (Fig. 2A). Cells treated with VX-661, VX-445, and at least 0.1 μM VX-770 exhibited 20–30 μA cm−2 greater ISC after the addition of amiloride (before the addition of Fsk/IBMX) (Fig. 2A), demonstrating that basal ion transport was increased. The additions of Fsk/IBMX or CFTRinh-172 resulted in much greater ΔISC (approximately 20-fold) across cells treated with VX-661 and VX-445 (Fig. 2B–C). The greatest Fsk/IMBX responsive ΔISC was observed in the 0 μM VX-770 condition, and Fsk/IBMX-responsive ΔISC decreased with increasing doses of VX-770 (Fig. 2B). Cells pre-treated with 6.4 μM VX-770 had approximately 60% the Fsk/IBMX-responsive ΔISC as cells treated with 0.1 μM VX-770. However, CFTRinh-172 responsive ΔISC increased at 0.1–0.4 μM VX-770 and slightly but significantly decreased from this maximum at 6.4 μM VX-770 (Fig. 2C).
Figure 2: Effect of chronic VX-770 treatment in the presence of VX-661 and VX-445 on F508del-CFTR function.
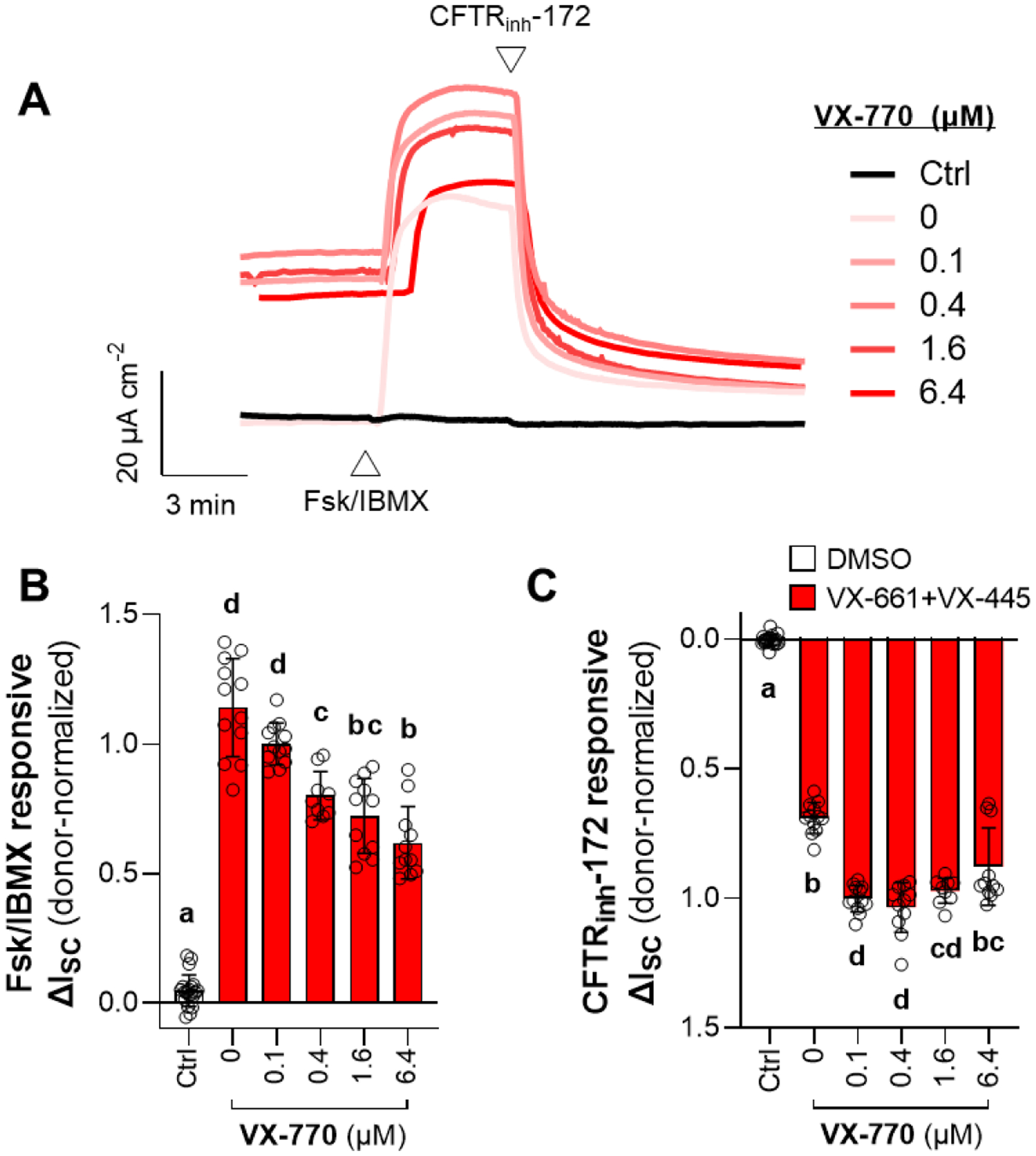
F508del/F508del HNE cells were chronically (24 h) treated with either DMSO (Ctrl) or the combination of VX-661 (3 μM) and VX-445 (3 μM) (red), and chronically co-treated with either DMSO (0 μM) or increasing concentrations of VX-770 (0.1–6.4 μM) (A) Representative traces of activation and inhibition of CFTR by Fsk/IBMX and CFTRinh-172, respectively. (B–C) Changes in ISC in response to Fsk/IBMX (B) or CFTRinh-172 (C). Data are presented as mean ± standard error. Groups with different letters are significantly different (P < 0.05) from each other (n = 3–4 unique donors; one-way ANOVA; Tukey’s post hoc).
3.4. Prolonged ivacaftor exposure in the presence of tezacaftor and elexacaftor increases constitutive CFTR activity
The constitutively active CFTR activity was estimated as the difference in ISC from before CFTR activation to after CFTR inhibition in the presence of amiloride [22,23], and presented as donor-normalized data (Fig. 3). In cells treated with VX-809, constitutive CFTR activity was not increased at any concentration of co-treatment with VX-770 (Fig. 3B). Constitutive CFTR activity was also unchanged in cells treated with the combination of VX-661 and VX-445. An increase in constitutive CFTR activity above the DMSO controls was only observed in cells treated with the combination of VX-661 and VX-445 and co-treated with at least 0.1 μM VX-770. To test the hypothesis that the apparent reduction in Fsk/IBMX responsive ΔISC was due to an increase in constitutive CFTR activity, we regressed the two metrics. There was a significant and negative correlation between constitutive CFTR activity and Fsk/IBMX responsive ΔISC (m = −0.181; R2 = 0.82; P < 0.001) (Fig. 3C).
Figure 3: Increased constitutive F508del-CFTR activity in the presence of chronic VX-661, VX-445, and VX-770 explains apparent decrease in Fsk/IBMX responsiveness.
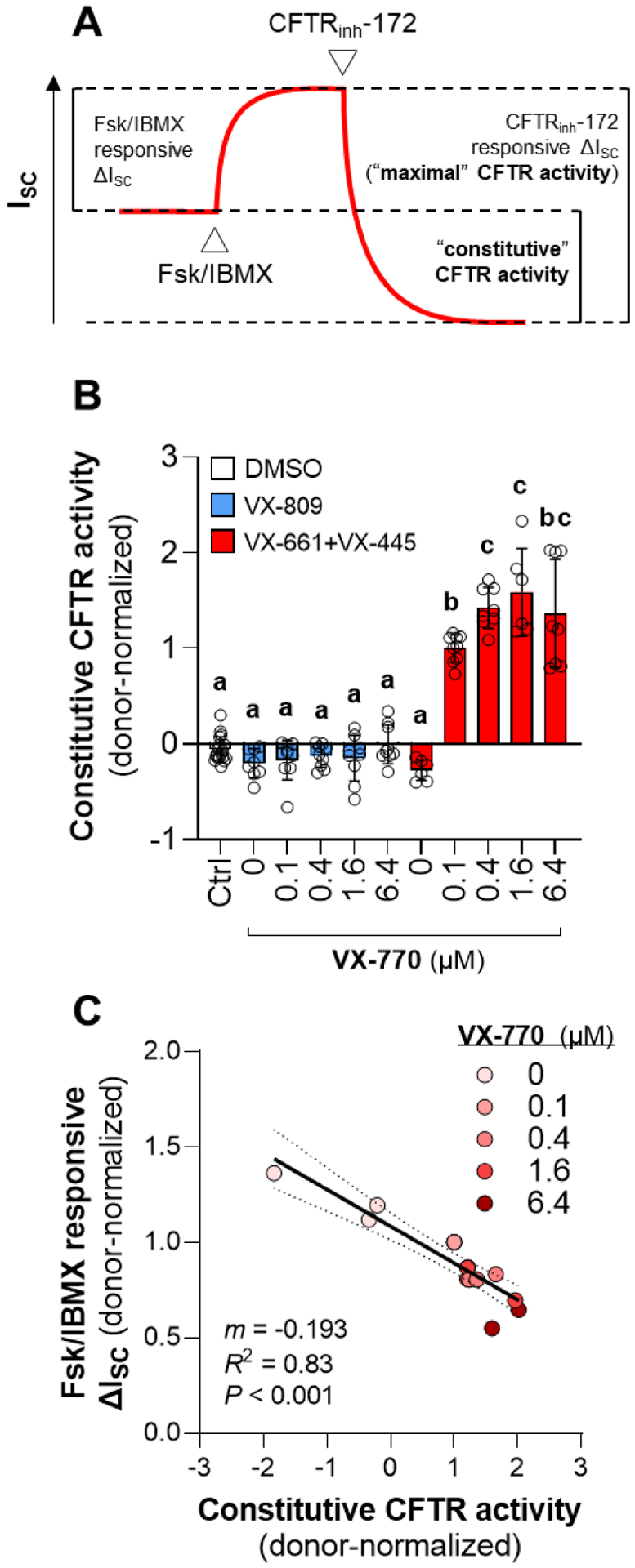
(A) Values for constitutive CFTR activity were calculated as the difference between ISC before Fsk/IBMX addition and after CFTRnh-172 addition in the presence of amiloride. (B) Constitutive CFTR activity calculated from data presented in Figs. 1 and 2. Data are presented as mean ± standard error. Groups with different letters are significantly different (P < 0.05) from each other (n = 3–4 unique donors; two-way ANOVA; Tukey’s post hoc). (C) Relationship between Fsk/IBMX responsive ISC and constitutive CFTR activity data from experiment presented in Fig. 2.
4. Discussion
The recent discovery of elexacaftor and prodigious clinical success of the triple combination therapeutic in the treatment of CF caused by the F508del CFTR mutation [26] has inspired the rapid execution of several recent studies investigating the interactions and combined mechanisms of action of elexacaftor, tezacaftor, and ivacaftor [5–7]. Here, we add to this growing body of literature by identifying a potentially useful functional explanation for the effectiveness of the triple combination in rescuing F508del-CFTR in vivo. That is, the combination of elexacaftor, tezacaftor, and ivacaftor increases the constitutive activity of CFTR. We provide a possible explanation for the apparent counter-productive effect of prolonged ivacaftor exposure that has been reported elsewhere [8–11]. When using constitutive CFTR activity as the functional metric, prolonged exposure to ivacaftor does not appear to abrogate CFTR correction by the triple combination of CFTR modulators, but instead results in a net increase in CFTR function.
It has been suggested that ivacaftor may not be an optimal potentiator for use in combination modulator therapies aimed at increasing F508del-CFTR function in individuals with CF [8–15]. In support of this view, it was recently reported that F508del-CFTR in cells treated chronically with the triple combination therapeutic of tezacaftor/ivacaftor/elexacaftor cannot be acutely potentiated by ivacaftor [22]. However, potentiators such as ivacaftor are known to tightly bind to CFTR [27], so it makes intuitive sense that CFTR already saturated with ivacaftor during chronic exposure could not be further potentiated by acute exposure to ivacaftor. A similar behavior, that chronic treatment precludes acute responsiveness, has recently been shown of the potentiating action of elexacaftor [7]. Perhaps the most concerning observation forming the basis of this view is that prolonged ivacaftor exposure decreases modulator-rescued F508del-CFTR protein expression and function in vitro due to increased protein instability [8–11]. However, it is not yet known whether alternative CFTR potentiators that do not nullify modulator correction F508del-CFTR [8,12,13] will exhibit any greater in vivo efficacy in treating CF than ivacaftor. In the present study, our results reproduce the well-described observation that prolonged ivacaftor exposure reduces the effectiveness of lumacaftor; this is demonstrated in Fig. 1 by the decrease in maximal CFTR function (observed in the responses to acute stimulation of F508del CFTR by Fsk/IBMX and ivacaftor and the subsequent inhibition of CFTR by CFTRinh-172) upon co-treatment with ivacaftor in lumacaftor-treated cells. Additionally, we surmise that even greater Fsk/IBMX and CFTRinh-172 responsiveness would have been observed in the 0 μM ivacaftor cells exposed to prolonged tezacaftor/elexacaftor exposure (depicted in Fig. 2) if these cells had been acutely exposed to ivacaftor, similar to the results shown in Fig. 1. Despite these potential drawbacks of ivacaftor, we have demonstrated that prolonged exposure to ivacaftor increases the constitutive activity of CFTR, which may be an underlying explanation for its known clinical efficacy.
The U.S. Food and Drug Administration is now recognizing in vitro studies as sufficient evidence to expand drug labels for CF modulators to rarer CF-causing CFTR mutations [28–30]. Thus, it is imperative that in vitro analyses use the most appropriate metrics to assess the impact of modulators on CFTR function and epithelial osmoregulatory function in general. On the airway epithelium in vivo, some portion of the endogenous CFTR that is present on the luminal surface is actively transporting chloride. This is the constitutively active CFTR, which is regulated in part by endogenous pathways controlling cellular cAMP/PKA signaling. When exogenous forskolin is added to the airway epithelial cultures in vitro, it bypasses the endogenous cAMP/PKA regulation of CFTR activity and abstracts the true nature of CFTR function on the epithelium. Studies aimed at understanding the endogenous regulation of CFTR activity are scarce, which may contribute to the lack of use of constitutive CFTR activity as a common metric for evaluating CFTR function. Only recently has the potential value of the measurement of constitutive CFTR activity been explicitly proposed [22,23]. Importantly, the in vitro studies utilized to support the expanded use of CFTR modulators were performed in Fischer Rat Thyroid cells exogenously expressing mutant CFTRs; while this model has proven very valuable to drug development, the physiologically relevant endogenous expression and activity of human airway epithelial CFTR is absent from this model.
Our results demonstrating that prolonged ivacaftor exposure increases constitutive CFTR function could explain some of the perplexing results of previous works investigating effects of prolonged ivacaftor treatment. In one study, 3 out of the 4 rare CFTR mutations tested that showed a decrease in Fsk/IBMX responsiveness after prolonged ivacaftor exposure appeared, on closer inspection of the electrophysiological tracings presented, to have an increase in constitutive CFTR activity (though, it was not measured in the study) [15]. In another study, it was reported that prolonged ivacaftor exposure abrogated the effect of the CFTR corrector C18 as measured by Fsk/IBMX activation of CFTR-mediated ISC [31]. However, the same study reported that prolonged ivacaftor exposure during co-treatment with C18 improved more functionally relevant metrics such as ciliary beat frequency, mucociliary transport, and air surface liquid depth, and it appeared that constitutive CFTR activity (though it was not measured in this study either) was greatest in the epithelia pre-treated with prolonged exposure to ivacaftor, reflecting the functional osmoregulatory improvements that were reported.
The interaction between ivacaftor and elexacaftor appears to be critical in increasing CFTR function. An important observation made in the present study is that prolonged ivacaftor treatment resulted in an increase in Fsk/IBMX responsiveness in cells co-treated with lumacaftor (Fig. 1E), but a decrease in Fsk/IBMX responsiveness in cells co-treated with tezacaftor/elexacaftor (Fig. 2B). We further showed that the decreased Fsk/IBMX responsiveness in the cells co-treated with tezacaftor/elexacaftor/ivacaftor for 24 h is correlated with increased constitutive CFTR activity (Fig. 3B). Lumacaftor and tezacaftor are structural analogs with highly similar mechanisms of action in correcting F508del-CFTR [32], therefore this difference is likely due to the incorporation of elexacaftor. Recent studies in our lab and elsewhere have demonstrated that ivacaftor and elexacaftor synergistically interact as co-potentiators of CFTR [6,7]. We posit that this unique interaction between ivacaftor and elexacaftor is at least partially responsible for the observed increases in constitutive CFTR function.
In our examination of the effects of prolonged ivacaftor treatment on CFTR function, we demonstrated the potential for contrasting interpretations between metrics of CFTR function. We have shown that, during co-treatment with tezacaftor and elexacaftor, prolonged ivacaftor treatment strongly reduces CFTR function when measured by Fsk/IBMX responsiveness (Fig. 2B), moderately improves CFTR function when measured by subsequent CFTRinh-172 responsiveness (Fig. 2C), and strongly improves CFTR function when measured by constitutive CFTR activity (Fig. 3B). Given that ivacaftor and the triple combination therapeutic are inherently chronic treatments in vivo and are known to have tremendous clinical success in the treatment of CF [18,26,33], the metrics that are used to measure the impact of these modulators on CFTR function in vitro should reflect their known clinical impact. We propose here, in agreement with findings of recent related studies [22,23], that the measurement of constitutive CFTR activity will be a useful functional metric for assessing CFTR modulator efficacy in vitro.
5. Prospective
With the growing use of high throughput in vitro electrophysiological experimentation in the development and approval of CFTR modulating compounds, a more complete understanding of the electrophysiological metrics that best reflect endogenous CFTR function is needed. Our work here illustrates the possible discrepancy between electrophysiological measurements of CFTR function, and we suggest that a measurement of constitutive CFTR activity may be among the most functionally relevant metrics to consider. Using this metric to evaluate CFTR function, we have shown that prolonged ivacaftor treatment does not abrogate the beneficial effects of CFTR correctors, but rather may be critical in conferring the dramatic gain of CFTR function observed from treatment with the triple combination therapeutic. There is a need to better understand the endogenous mechanisms regulating constitutive CFTR activity and to better understand which electrophysiological metrics in vitro best reflect clinical efficacy of CFTR modulators in vivo.
Supplementary Material
Highlights.
Changes in in vitro constitutive CFTR activity (i.e. spontaneously active CFTR-mediated ion transport prior to cAMP stimulation by exogenous forskolin) are a quantifiable measure of CFTR modulator efficacy.
The combination of elexacaftor, tezacaftor, and ivacaftor increases the constitutive activity of CFTR in human nasal airway epithelial cells.
In agreement with previous studies, cells that were pre-treated with prolonged (24 h) exposure to ivacaftor exhibited reduced responsiveness to additions of Fsk/IBMX.
When using constitutive CFTR activity as the functional metric, prolonged ivacaftor treatment does not appear to abrogate CFTR function, but instead results in a net benefit to CFTR function.
Acknowledgments
This research was supported by the Cystic Fibrosis Foundation (BRATCH16I0 and BRATCH21I0 to P.E.B. and ZEITLI2010 to P.L.Z.), the Eugene F. and Easton M. Crawford Charitable Lead Unitrust (C.A.S. and P.E.B.), the Gilead Sciences Research Scholars Program in Cystic Fibrosis (P.E.B), the National Institutes of Health (F32HL158174 to C.A.S. and R01HL155325 to P.E.B), and the Department of Pediatrics at National Jewish Health. The authors would also like to thank the Cystic Fibrosis Foundation for the contribution of compounds to this work through the CFTR Chemical Compound Distribution Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Ciaran A. Shaughnessy: Methodology, Formal analysis, Investigation, Writing – Original Draft, Visualization
Pamela L. Zeitlin: Writing – Review & Editing, Funding acquisition
Preston E Bratcher: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Resources, Writing – Original Draft, Supervision, Project administration, Funding acquisition
Declaration of Competing Interest
Authors declare no competing interests.
References
- [1].Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005;352:1992–2001. [DOI] [PubMed] [Google Scholar]
- [2].The Clinical and Functional TRanslation of CFTR (CFTR2) n.d. http://www.cftr2.org.
- [3].Rohlfs EM, Zhou Z, Heim RA, Nagan N, Rosenblum LS, Flynn K, et al. Cystic fibrosis carrier testing in an ethnically diverse US population. Clin Chem 2011;57:841–8. 10.1373/clinchem.2010.159285. [DOI] [PubMed] [Google Scholar]
- [4].Lopes-Pacheco M CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front Pharmacol 2020;10:1–29. 10.3389/fphar.2019.01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Laselva O, Bartlett C, Gunawardena TNA, Ouyang H, Eckford PDW, Moraes TJ, et al. Rescue of multiple class II CFTR mutations by elexacaftor+ tezacaftor+ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur Respir J 2021:2002774. 10.1183/13993003.02774-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Veit G, Vaccarin C, Lukacs GL. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J Cyst Fibros 2021. 10.1016/j.jcf.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shaughnessy CA, Zeitlin PL, Bratcher PE. Elexacaftor is a CFTR potentiator and acts synergistically with ivacaftor during acute and chronic treatment. Sci Rep 2021:1–11. 10.1038/s41598-021-99184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, et al. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med 2014;6:1–14. 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cholon DM, Quinney NL, Fulcher ML, Esther CR, Das J, Dokholyan NV., et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med 2014;6:1–12. 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Veit G, Roldan A, Hancock MA, da Fonte DF, Xu H, Hussein M, et al. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020;5:1–14. 10.1172/JCI.INSIGHT.139983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matthes E, Goepp J, Carlile GW, Luo Y, Dejgaard K, Billet A, et al. Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). Br J Pharmacol 2016;173:459–70. 10.1111/bph.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Phuan PW, Veit G, Tan JA, Finkbeiner WE, Lukacs GL, Verkman AS. Potentiators of defective DF508-CFTR gating that do not interfere with corrector action. Mol Pharmacol 2015;88:791–9. 10.1124/mol.115.099689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gees M, Musch S, Van Der Plas S, Wesse AS, Vandevelde A, Verdonck K, et al. Identification and characterization of novel CFTR potentiators. Front Pharmacol 2018;9:1–12. 10.3389/fphar.2018.01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bali V, Lazrak A, Guroji P, Matalon S, Bebok Z. Mechanistic Approaches to Improve Correction of the Most Common Disease-Causing Mutation in Cystic Fibrosis. PLoS One 2016;11:1–13. 10.1371/journal.pone.0155882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Avramescu RG, Kai Y, Xu H, Bidaud-Meynard A, Schnúr A, Frenkiel S, et al. Mutation-specific downregulation of CFTR2 variants by gating potentiators. Hum Mol Genet 2017;26:4873–85. 10.1093/hmg/ddx367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guimbellot JS, Ryan KJ, Anderson JD, Liu Z, Kersh L, Esther CR, et al. Variable cellular ivacaftor concentrations in people with cystic fibrosis on modulator therapy. J Cyst Fibros 2020;19:742–5. 10.1016/j.jcf.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guhr Lee TN, Cholon DM, Quinney NL, Gentzsch M, Esther CR. Accumulation and persistence of ivacaftor in airway epithelia with prolonged treatment. J Cyst Fibros 2020;19:746–51. 10.1016/j.jcf.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med 2018;197:214–24. 10.1164/rccm.201704-0717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial. Lancet Respir Med 2014;2:527–38. 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- [20].Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012;67:12–8. 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bratcher PE, Yadav S, Shaughnessy CA, Thornell IM, Zeitlin PL. Effect of apical chloride concentration on the measurement of responses to CFTR modulation in airway epithelia cultured from nasal brushings. Physiol Rep 2020;8:e14603. 10.14814/phy2.14603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Becq F, Mirval S, Carrez T, Lévêque M, Billet A, Coraux C, et al. The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. Eur Respir J 2021:2100671. 10.1183/13993003.00671-2021. [DOI] [PubMed] [Google Scholar]
- [23].Nick HJ, Zeitlin PL, Yadav S, Bratcher PE. Measurements of spontaneous CFTR-mediated ion transport without acute channel activation in airway epithelial cultures after modulator exposure. Sci Rep 2021;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yadav S, Shaughnessy CA, Zeitlin PL, Bratcher PE. Downregulation of epithelial sodium channel (ENaC) activity in human airway epithelia after low temperature incubation. BMJ Open Respir Res 2021;8:e000861. 10.1136/bmjresp-2020-000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zegarra-Moran O, Galietta LJV. CFTR pharmacology. Cell Mol Life Sci 2017;74:117–28. 10.1007/s00018-016-2392-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018;379:1612–20. 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu F, Zhang Z, Levit A, Levring J, Touhara KK, Shoichet BK, et al. Structural identification of a hotspot on CFTR for potentiation. Science (80- ) 2019;364:1184–8. 10.1126/science.aaw7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kingwell K FDA OKs first in vitro route to expanded approval. Nat Rev Drug Discov 2017;16:591–2. 10.1038/nrd.2017.140. [DOI] [PubMed] [Google Scholar]
- [29].Durmowicz T, Pacanowski M. Novel Approach Allows Expansion of Indication for Cystic Fibrosis Drug. FDA News Events 2017. https://www.fda.gov/Drugs/NewsEvents/ucm559051.htm. [Google Scholar]
- [30].Durmowicz AG, Lim R, Rogers H, Rosebraugh CJ, Chowdhury BA. The U.S. food and drug administration’s experience with ivacaftor in cystic fibrosis: Establishing efficacy using in vitro data in lieu of a clinical trial. Ann Am Thorac Soc 2018;15:1–2. 10.1513/AnnalsATS.201708-668PS. [DOI] [PubMed] [Google Scholar]
- [31].Birket SE, Chu KK, Houser GH, Liu L, Fernandez CM, Solomon GM, et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am J Physiol - Lung Cell Mol Physiol 2016;310:L928–39. 10.1152/ajplung.00395.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fiedorczuk K, Chen J. Mechanism of CFTR correction by type I folding correctors. BioRxiv 2021:2021.06.18.449063. [DOI] [PubMed] [Google Scholar]
- [33].Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in Persons with Cystic Fibrosis and the G551D-CFTR Mutation. N Engl J Med 2010;363:1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.