

Abstract
Polyunsaturated fatty acyl chains (PUFAs) concentrate in the brain and give rise to numerous oxidative chemical degradation products. It is widely assumed that these products are the result of free radical chain reactions, and reactions of this type have been demonstrated in preparations where a single PUFA substrate species predominates. However, it is unclear whether such reactions can occur in the biologically complex milieu of lipid membranes where PUFA substrates are a minority species, and where diverse free radical scavengers or other quenching mechanisms are present. It is of particular interest to know whether they occur in brain, where PUFAs are concentrated and where PUFA oxidation products have been implicated in the pathogenesis of neurodegenerative disorders. To ascertain whether free radical chain reactions can occur in a complex brain lipid mixture, mouse brain lipids were extracted, formed into vesicles, and treated with a fixed number of hydroxyl radicals under conditions wherein the concentrations and types of PUFA-containing phospholipids were varied. Specific phospholipid species in the mixture were assayed by tandem mass spectrometry to quantify the oxidative losses of endogenous PUFA-containing phospholipids. Results reveal crosstalk between the oxidative degradation of ω3 and ω6 PUFAs that can only be explained by the occurrence of free radical chain reactions. These results demonstrate that PUFAs in a complex brain lipid mixture can participate in free radical chain reactions wherein the extent of oxidative degradation is not limited by the number of reactive oxygen species available to initiate such reactions. These reactions may help explain otherwise puzzling in vivo interactions between ω3 and ω6 PUFAs in mouse brain.
Introduction
It is well recognized that polyunsaturated fatty acyl chains (PUFAs) may undergo free radical chain reactions in vitro, and widely assumed that chain reactions of this sort are responsible for the oxidative degradation of PUFAs in vivo. However, PUFAs may undergo free radical-mediated oxidative degradation without the propagation of a chain reaction. Indeed, chain reactions have stringent kinetic requirements for propagation, and they are subject to quenching by a wide variety of naturally occurring free radical scavengers. Therefore, it is not immediately clear that free radical chain reactions can propagate in vivo, yet it is important to understand whether they can propagate in vivo because of their potential to amplify the chemical damage caused by free radicals and contribute to the manifestations of oxidative stress.
Oxidative stress is a recurring theme in human pathology, and of particular importance in the brain where it has been implicated in the pathogenesis of Alzheimer’s disease, amyotrophic lateral sclerosis, Friedreich’s ataxia, and other disorders of the central nervous system.1 Some forms of oxidative stress are enzymatically initiated and regulated, while other forms involve spontaneous and unregulated chemical reactions. The unregulated forms of oxidative stress are often implicated in the pathogenesis of neurodegenerative disease, and reviews of the literature in this field often focus on the role of metal ions and compounds that are susceptible to oxidative damage owing to their low reduction potentials.2−4
PUFAs comprise 0.5–0.7% of the total brain mass,5 and are particularly susceptible to the unregulated form of oxidative stress in which a bis-allylic hydrogen is removed by a hydroxyl radical (•OH), and molecular oxygen is added to form a lipid peroxyl radical.6−8 The peroxyl radicals formed in this way may be reduced to peroxide and undergo spontaneous decomposition. If PUFAs in the brain are oxidatively degraded solely by the direct action of a •OH radical, then one •OH radical should yield just one oxidatively degraded PUFA. Because most PUFAs in the brain are either arachidonate (ARA, ω3) or docosahexaenoate (DHA, ω6), it follows that a decrease in the concentration of one of these PUFA species should reduce the number of reactions between this PUFA species and the available •OH radicals, making more •OH radicals available to increase the oxidatively degradation of the other PUFA species. Paradoxically, however, a diet-induced deficiency of ω3 PUFAs reduces the in vivo oxidative degradation of ARA in the brains of mice.9
This paradox may be resolved if a significant amount of oxidative damage is caused by free radical chain reactions, also known as autoxidation (Figure 1A). In chain reactions, the extent of chemical damage will vary with the concentration of reactants; increasing the concentration of a reactant increases the efficiency of a chain reaction by shortening the diffusion distances between reacting molecules. In principle, a chain reaction may terminate before it propagates, yielding just one oxidatively degraded PUFA per initiating event. Conversely, there is no upper limit to the extent of oxidative damage via chain reactions following a single initiation event.10
Figure 1.

Chemical mechanisms. (A) PUFA peroxidation may be initiated by •OH radicals, and propagated indefinitely by a chain reaction in the presence of oxygen. The chain reaction is linear, that is, non-branching, so that oxidative damage does not necessarily accelerate with time, except insofar as additional chain reactions are initiated. It should be noted that PUFA-OOH species are unstable and may undergo spontaneous internal rearrangements and cleavages, as well as oxidations and reductions. (B) Bis-allylic PUFA hydrogens are readily abstracted by •OH radicals to yield a carbon-centered free radical; this process is markedly inhibited by deuterium substitution. (C) Under some conditions, a bis-allylic hydrogen may be abstracted by an α-tocopheryl radical, with regeneration of α-tocopherol in the course of peroxide formation.
However, chain reactions have not been demonstrated in the brain, and PUFA-based free radicals may be quenched by the many protective agents and free electron scavenging mechanisms operating in brain11 before they can participate in chain reactions. Chain reactions have been observed in low density lipoprotein particles, where α-tocopherol (αToc) appears to participate as a radical-transfer agent (Figure 1C).12−14 It has also been observed in erythrocytes15 and erythrocyte ghosts, where a kinetic chain reaction length of between 10 and 100 has been estimated, and the loss of PUFAs has been demonstrated.16,17 However, these systems do not have the high PUFA concentrations of brain, or the protective agents and free electron scavenging mechanisms of the brain.
It can be straightforward to demonstrate the operation of a free radical chain reaction in a chemically-defined system wherein reactant and product concentrations may be controlled and kinetics measured. In a biologically complex mixture, however, the demonstration is more challenging. The demonstration described herein relied on limiting the number of free radical initiators so that reactants could not be fully consumed by the direct action of free radicals, and altering the concentrations of reactants by the addition of synthetic analogs that varied in their susceptibility to free radical chain reactions. Isotope-substituted PUFAs that specifically hindered the type of chain reaction being considered, and that could be monitored in a complex mixture of brain lipid by mass spectrometry, were also employed.
Accordingly, unilamellar lipid vesicles were prepared from extracted brain lipids to create membranes that mimic the complex composition of lipid membranes in the brain. Synthetic PUFA-containing phospholipids (PUFA-PLs) were added to these extracts to increase the abundance of selected PUFA-PL species. The vesicles were then exposed to •OH radicals produced by the reaction of ascorbate and copper under conditions where the amounts of ascorbate and oxygen are limiting, and where the intrinsic reducing capacity of each sample is identical.
Two distinctly different outcomes could have emerged from these experiments, depending on whether chain reactions are occurring. If chain reactions are occurring, the addition of synthetic PUFA-PLs to the brain lipid mixture will accelerate the loss of endogenous PUFA-PLs by reducing the distances between PUFAs. Conversely, if chain reactions are not occurring, then •OH radicals would react preferentially with the superabundant synthetic PUFA-PLs and reduce the oxidative degradation of endogenous PUFAs.
We observed that the addition of synthetic PUFA-PLs markedly increased the degradation of endogenous PUFA-PLs, indicating that free radical chain reactions were indeed occurring. Oxidative PUFA degradation was not accelerated when the added synthetic PUFA-PLs had deuteriums in place of hydrogens at bis-allylic positions, which renders PUFA-PLs resistant to a chain reaction by the kinetic isotope effect. These results demonstrate that free radical chain reactions are possible in membranes composed of a complex brain lipid mixture. They highlight the potential for reactive oxygen species such as •OH radicals to cause chemical damage in the brain out of proportion to the rate at which they are created, and suggest that oxidative PUFA damage in the brain may be sensitive to PUFA concentration.
Materials & Methods
Reagents
Synthetic 1-stearoyl-2-arachidonyl-sn-glycero-3-phosphocholine (SAPC), 1-stearoyl-2-do-cosahexa-enoyl-sn-glycero-3-phosphocholine (SDPC), and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) in chloroform were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL) in sealed glass ampules and stored at −80 °C until day of use. Samples containing SAPC and SDPC were examined by mass spectrometry for the presence of preformed hydroperoxides, and none were detected. Synthetic 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was obtained as a powder from the same source and stored at −20 °C. Synthetic SAPC containing an esterified 7,7,10,10,13,13-d6-arachidonyl fatty acid, S(dA)PC, and synthetic SDPC containing an esterified 6,6,9,9,12,12,15,15,18,18-d10-docosahexa-enoyl fatty acid, S(dD)PC, were obtained as powders from Retrotope Inc. (Los Altos, CA). 2,6-Di-tert-butyl-p-cresol (BHT), diethylenetriamine pentaacetic acid (DTPA), and αToc were obtained from Fisher Scientific.
Animals
B6SJL/J mice were obtained from the Jackson Laboratory (Bar Harbor, ME), and were between 8 and 10 months of age when sacrificed by CO2 asphyxiation. Brains were removed within 3 min post mortem, and portions of the cortex were transferred to high-recovery clear borosilicate glass autosampler vials for weighing. All experimental procedures and animal care were in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
Lipid Extraction and Saponification
For each experiment, lipids were extracted from separate 5–10 mg tissue samples using a modified Bligh-Dyer extraction (BDx) procedure.18 Each tissue sample was homogenized in 760 μL of BD monophase (400 μL of methanol, 200 μL of dichloromethane, and 160 μL of 5 mM ammonium chloride) with a tip sonicator for 60 s in of BD. Then, 200 μL of dichloromethane, and 160 μL of water was added to break the monophase. After vortexing, the phases were separated by 1–2 min of low speed centrifugation. The lower phase was transferred to another glass tube, evaporated under argon, then redissolved in methanol and stored under argon at −80 °C.
Multiple-Reaction Monitoring–LC/MSMS
Chromatographic separations were performed with an Agilent XDB-C8 1.0 × 50 mm column (Agilent, Palo Alto, CA) and solutions flowing at 100 μL/min. Solution A was 50% methanol in water; solution B was 30:70 chloroform/methanol with 0.1% ammonium formate. Samples were injected into a column equilibrated with 67% B by volume. The B percentage was increased to 100% over 0.5 min, and then held at 100% solution B for 6 min. Multiple-reaction monitoring (MRM) liquid chromatography tandem mass spectrometry (LC/MSMS) analyses were performed on an ABI 4000 (Sciex, Toronto, Canada) with an electrospray source operating in positive mode with a declustering potential of +20 V, an ion source voltage of +5500 V, a 300 °C drying gas, and a 4 psi nitrogen collision gas. The characteristic collision-induced phosphocholine product ion was monitored at m/z 184 for each phospholipid species. Parent ion masses, elution times, and collision energies for each phospholipid species are listed in Table 1.
Table 1. Phospholipid Parametersa.
| species | parent ion (m/z) | collision energy (V) | elution time (min) |
|---|---|---|---|
| SAPC | 811.2 | 50 | 3.45 |
| SDPC | 835.2 | 40 | 3.55 |
| DMPC | 679.2 | 30 | 2.70 |
| DSPC | 790.6 | 30 | 3.70 |
| DOPC | 786.6 | 40 | 3.28 |
| S(dA)PC | 817.2 | 40 | 3.50 |
| S(dD)PC | 845.2 | 40 | 3.60 |
The product ion monitored in each case was the phosphocholine cation, m/z 184.
Phospholipid Assay
Phosphate concentrations were determined by the method of Bartlett.19 Since there was no MRM signal corresponding to DMPC detected in brain lipid extracts, the phosphate concentration in a solution of DMPC in methanol was determined and adjusted to 10 μM. This 10 μM solution was used to create calibration curves for MRM–LC/MSMS signals from SAPC, SDPC, and DOPC solutions in methanol, and to establish the linearity of these signals over a range of 1–50 μM. Known amounts of synthetic DMPC were added to brain lipid extracts and used as an internal standard to determine the concentrations of these phospholipid species.
Lipid Vesicle Preparation
As indicated in Table 2, a different mouse brain was used for each of the figures below. A total of 50 mg of cerebral cortex tissue was subjected to BDx, and the lower phase was removed, evaporated, and redissolved in 1 mL of methanol to yield the single brain extract (SBE). Three 10 μL aliquots of each SBE were assayed for phosphate as described above. The amount of endogenous DMPC in mouse brain is negligible. Therefore, a 10 μL aliquot of the SBE was mixed with 90 μL of synthetic DMPC in methanol to yield a diluted brain extract (DBE). Three 5 μL injections of the DBE were analyzed by MRM–LC/MSMS to determine the endogenous concentrations of SAPC and SDPC using DMPC as an internal standard. The results listed in Table 2 are the means of these 3 measurements on the DBE, divided by the means of the 3 phosphate determinations on each SBE.
Table 2. Lipid Vesicle Compositions in Each Figure.
| fraction
of endogenous phosphate |
added
synthetic phospholipid as a percentage
of endogenous phosphate |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| figure # | conditiona | SAPC | SDPC | SAPC | SDPC | DOPC | DMPC | S(dA)PC | S(dD)PC |
| +SAPC | 0.23 | 0.16 | 2.1 | 0 | 0 | 0 | 0 | 0 | |
| 2 | +SDPC | 0.21 | 0.16 | 0 | 1.9 | 0 | 0 | 0 | 0 |
| +DMPC | 0.22 | 0.16 | 0 | 0 | 0 | 2.0 | 0 | 0 | |
| +SAPC | 0.034 | 0.034 | 0.31 | 0 | 0 | 0 | 0 | 0 | |
| 3 | +SDPC | 0.051 | 0.052 | 0 | 0.47 | 0 | 0 | 0 | 0 |
| +DOPC | 0.041 | 0.041 | 0 | 0 | 0.39 | 0 | 0 | 0 | |
| 4 | n.d. | n.d. | 0 | 0 | 0 | 0 | 0 | 0 | |
| +BHT | n.d. | n.d. | 0 | 0 | 0 | 0 | 0 | 0 | |
| 5 | +SAPC ± BHT | 0.39 | 0.16 | 1.5 | 0 | 0 | 0 | 0 | 0 |
| +SDPC ± BHT | 0.39 | 0.16 | 0 | 3.4 | 0 | 0 | 0 | 0 | |
| +DMPC | 0.21 | 0.16 | 0 | 0 | 0 | 1.9 | 0 | 0 | |
| 6 | +S(dA)PC | 0.21 | 0.16 | 0 | 0 | 0 | 0 | 1.9 | 0 |
| +S(dD)PC | 0.21 | 0.16 | 0 | 0 | 0 | 0 | 0 | 1.9 | |
Symbols indicate the synthetic materials that were added prior to vesicle extrusion.
The remaining SBE was distributed into three plasma cleaned test tubes (∼480 μL each). In most experiments, an amount of synthetic SAPC, D-SAPC, SDPC, D-SDPC, or DOPC equal to 10 times the amount of endogenous SAPC or SDPC (whichever is higher) was added to each tube and evaporated. 1 mL HEPES buffer (pH 7.4) was added to each tube, bath-sonicated for 12 min, vigorously vortexed for 30 s, and extruded through 100 nm polycarbonate membranes to produce supplemented vesicle suspensions (SVS). Three 10 μL aliquots of each SVS were assayed for phosphate, and the remainder of each SVS was diluted such that the phosphate concentration was 100 μM, and 200 μL aliquots of this diluted SVS was placed into each of 4 autosampler vials. In experiments where BHT was added, it was added along with the synthetic SAPC and SDPC in an amount equal to 1% of the molar concentration of phosphate in the SBE.
To initiate oxidation, ascorbate and copper(II) phosphate were added to achieve final concentrations of 50 μM and 500 nM, respectively, which yields •OH radicals according to reaction (1)
| 1 |
Both initiation (reaction 1) and propagation (Figure 1A) depend on the availability of molecular oxygen, so that the rate and extent of lipid oxidation was sensitive to the surface area and volume of air above the samples. For the vials used, the liquid surface area was 0.95 cm2, and the volume of air in the sealed vial above the liquid surface was 1.3 mL. All oxidations were performed at room temperature (approximately 21 °C).
In experiments where αToc was used, an amount equal to 0.0062% of the molar concentration of phosphate in the SBE was added instead of synthetic PL. This amount of αToc was expected to yield 5 αToc molecules per vesicle. At the beginning of these experiments, ascorbate was added to a final concentration of 50 μM, and DTPA was added to a final concentration of 10 μM, but no copper was added.
The extent to which a PUFA-containing PC (PUFA-PC) lipid was lost under various conditions was determined by calculating the ratio of the signal from the lipid of interest to the signal from endogenous 1,2-distearoyl-sn-glycero-3-phospho-choline (DSPC), and normalizing these ratios to 1.0 at time = 0.
The error bars in the figures represent standard errors of the mean for 3 technical replicates at each time point; overlapping error bars indicate results that are not significantly different, while non-overlapping error bars indicate results for which P < 0.05 by Student’s t-test.
Results
The endogenous SAPC content of brain lipid extracts ranged from 0.5–1.6 nmol/mg brain tissue, while endogenous SDPC content ranged from 0.4 to 1.3 nmol/mg. These yields are comparable to the concentrations previously reported.18,20Figure 2 shows the effect of adding synthetic DMPC, SAPC and SDPC on the oxidative degradation of endogenous SAPC and SDPC. It should be noted that the loss of SAPC when synthetic SDPC was added represents the loss of endogenous SAPC. Similarly, the loss of SDPC when synthetic SAPC was added represents the loss of endogenous SDPC. With the addition of DMPC, the oxidative degradation of SAPC and of SDPC was minimal for 2 h, followed by losses to approximately 40–60% of original concentrations over the next 2 h. The addition of synthetic SAPC or SDPC markedly increased these losses so that only 10–20% of the SAPC or SDPC present originally remained after 4 h. Results indicate that PUFA-PC losses were similar in extent regardless of whether they represent the loss of an endogenous PUFA-PC, or the loss of endogenous plus synthetic PUFA-PC. We conclude that the susceptibility of PUFA-PLs to oxidation in these preparations is increased when overall PUFA-PL concentrations are increased.
Figure 2.

Oxidative PUFA-PL degradation in brain-derived-lipid vesicles to which synthetic SAPC, SDPC, or DMPC has been added. Measurements represent the integrated MRM–LC/MSMS signals of the designated PUFA-PL, divided by the signals from DSPC at each time point, normalized to 1.0 at time zero. Vesicle compositions are listed in Table 2. Lipid vesicle phosphate concentrations were 100 μM, and oxidizing agent concentrations were 500 nM Cu(II) and 50 μM ascorbate. All measurements were performed at room temperature, 21 °C. See methods section for statistical analysis. Left panel: SAPC. Right panel: SDPC.
Figure 3 compares the oxidative degradation of SAPC, SDPC, and DOPC in vesicles made from a brain sample that happened to have lower endogenous PUFA-PC concentrations as a fraction of total phosphate. In these experiments, SAPC and SDPC losses were less extensive; ∼40% of each PUFA-PL remained after 4 h, compared to between 10 and 20% remaining in Figure 2. Little or no DOPC was lost in all three conditions. The addition of DOPC dramatically reduced the oxidative degradation of SAPC and DOPC. These results suggest that the susceptibility of PUFA-PC to oxidative degradation is reduced when endogenous PUFA-PL concentrations are lower, and when PUFA-PL concentrations are further reduced by dilution with monounsaturated fatty acids.
Figure 3.
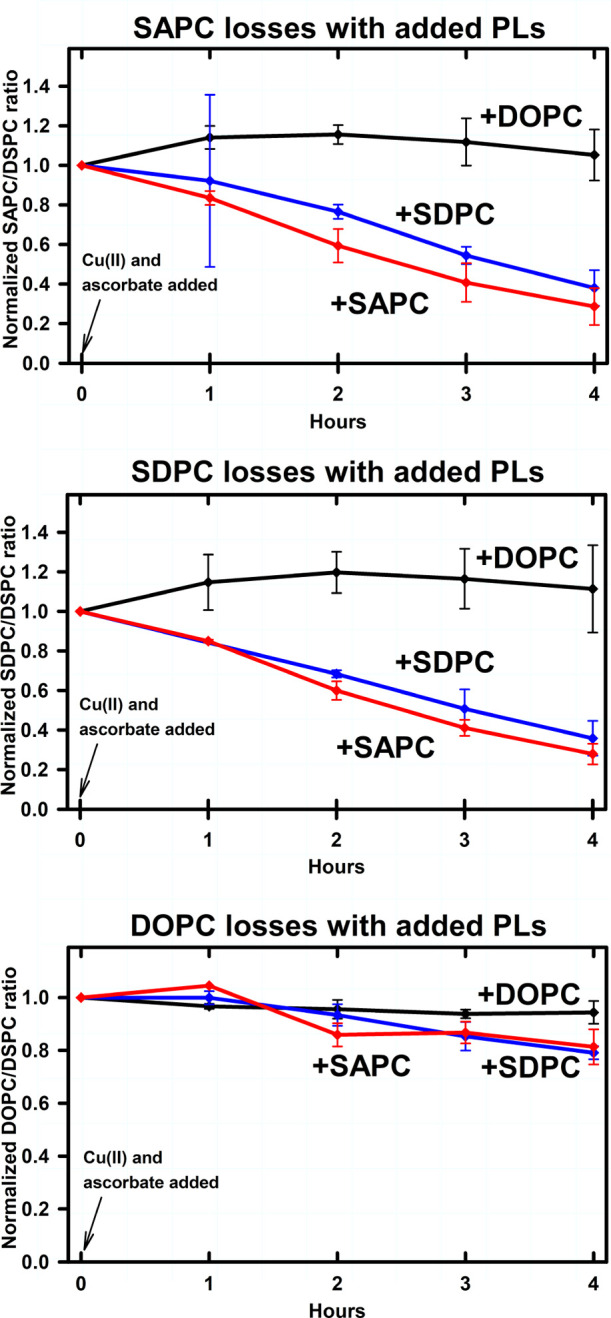
Oxidative PUFA-PL degradation in brain-derived-lipid vesicles to which synthetic SAPC, SDPC, or DOPC has been added. Vesicle compositions are listed in Table 2. Endogenous PUFA-PL concentrations were 15–33% of the concentrations in Figure 1; otherwise measurements, conditions, and statistical analysis were the same as for Figure 2. Top panel: SAPC. Middle panel: SDPC. Bottom panel: DOPC.
Figure 4 illustrates the effect of BHT on the oxidative degradation of endogenous SAPC and SDPC. The BHT concentration was 1 μM in a suspension of vesicles where the phosphate concentration was 100 μM, making the BHT/phospholipid ratio 1:100. BHT markedly reduced the oxidative degradation of endogenous SAPC and SDPC, indicating that BHT is effective at protecting PUFA-PC from oxidative degradation under these conditions.
Figure 4.

Effects of BHT on the oxidative degradation of endogenous PUFA-PLs. Vesicle compositions are listed in Table 2. Measurements, conditions, and statistical analysis were the same as for Figure 2. Left panel: SAPC. Right panel: SDPC.
The effects of BHT on the rates of oxidative degradation in the presence of added synthetic PUFA-containing phospholipids is illustrated in Figure 5. In each case, and regardless of whether synthetic SAPC or SDPC was added, or whether SAPC or SDPC concentrations were being measured, the inclusion of 1 μM BHT protected SAPC and SDPC against oxidative degradation. Likewise, substituting of DTPA (a copper chelator) for copper(II) phosphate, and the addition of αToc, protected both SAPC and SDPC from degradation (data not shown).
Figure 5.

Effects of BHT on oxidative PUFA-PL degradation in brain-derived-lipid vesicles to which synthetic SAPC or SDPC has been added. Vesicle compositions are listed in Table 2. Measurements, conditions, and statistical analysis were the same as for Figure 2. Top panels: added synthetic SAPC. Bottom panels: added synthetic SDPC. Left panels: SAPC degradation. Right panels: SDPC degradation.
The effect of deuterium-substituted PUFA-PLs on the oxidative degradation of endogenous PUFA-PLs is illustrated in Figure 6. Regardless of whether S(dA)PC or S(dD)PC was added, or whether SAPC or SDPC was measured, the addition of deuterium-substituted PUFA-PLs protected endogenous PUFA-PLs against oxidative degradation. It should be noted that the signals arising from endogenous PUFA-PLs are distinct from the signals arising from S(dA)PC and S(dD)PC, and the latter exhibited no oxidative losses over the course of the experiment (data not shown).
Figure 6.

Oxidative PUFA-PL degradation in brain-derived-lipid vesicles to which synthetic deuterium-substituted PUFA-PLs or synthetic DMPC have been added. Vesicle compositions are listed in Table 2. Measurements, conditions, and statistical analysis were the same as for Figure 2. Left panels: SAPC. Right panels: SDPC.
Discussion
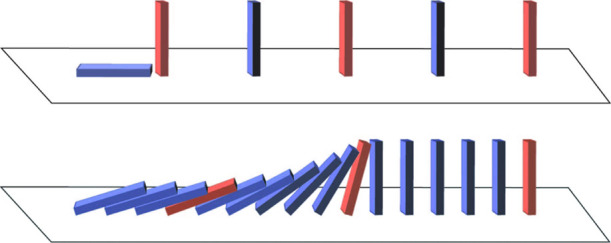
We considered two possible mechanisms for the oxidative degradation of endogenous PUFA-PLs in these experiments. If direct attack by •OH radicals was solely responsible for oxidative degradation, the synthetic PUFA-PLs added to the brain lipid extracts should have been preferentially degraded because of their greater concentration, thereby reducing the oxidative degradation of endogenous PUFA-PLs. Conversely, the addition of synthetic PUFA-PLs would increase the oxidative degradation of endogenous PUFA-PLs by shortening the time required for the diffusion of peroxyl radicals within the membrane to a susceptible PUFA-PL if free radical chain reactions significantly added to the degradation initiated by direct attack. This increase would be analogous to the “domino effect” wherein a larger number of dominoes topple per unit time as their separation distance is reduced (see table of contents graphic).21,22 In experiments where synthetic SAPC was added to a brain lipid extract, the oxidative degradation of endogenous SDPC could be distinguished from that of SAPC degradation. Conversely, when SDPC was added, the oxidative degradation of endogenous SAPC could be distinguished from that of SDPC degradation. We found that the addition of synthetic PUFA-PLs markedly increased the oxidative degradation of endogenous PUFA-PLs, indicating that free radical chain reactions were responsible for a substantial amount of oxidative PUFA-PL loss in these experiments (Figure 1A).
In contrast to the effects of added PUFA-PLs, the addition of PLs with saturated, monounsaturated, or deuterium-stabilized fatty acids (Figure 1B) did not increase the oxidative degradation of endogenous PUFA-PLs, demonstrating that the bis-allylic hydrogens in synthetic PUFA-PLs were essential for accelerating the oxidative degradation of endogenous PUFA-PLs. An alternative mechanism for propagating oxidative damage has been proposed in which an α-tocopheryl radical serves as mediator (Figure 1C),13,14,23 and αToc may have been present and effective in these experiments at concentrations too low to measure. However, the addition of synthetic αToc reduced (rather than increased) oxidative PUFA-PL degradation in these experiments.
It should be noted that the concentrations of ascorbate and Cu(II) used to generate •OH radicals in these experiments are comparable to in vivo concentrations. In brain, for example, ascorbate concentrations have been estimated at between 150 and 400 μM,24−26 while free copper ions are released into the synaptic cleft where small dimensions make its effective concentration quite high.27−29 It should also be noted that the ascorbate concentration in these experiments was 50 μM, and that three ascorbate molecules are required to reduce molecular oxygen to •OH radical. Therefore, it is unlikely that direct attack by •OH radicals could account for the near-complete oxidative degradation of PUFA-PLs in many of these experiments.
Clear demonstrations of free radical chain reactions in biological materials are uncommon, and reasons to doubt that they occur have been offered. For example, free radicals are often regarded as so reactive that they are likely to react with something else in the membrane before they encounter a PUFA. However, that kind of relatively indiscriminant reactivity is more likely for •OH radicals with a reduction potential of 2310 mV, than for alkylperoxy radicals (•OOCH) with reduction potentials of 770–1440 mV.30 Once formed, an alkylperoxy radical is unlikely to encounter many compounds with a lower reduction potential before it encounters a PUFA with a reduction potential of 600 mV.6,7
The oxidative degradation assay used in these experiments quantifies PL species by MRM–LC/MSMS, using saturated PLs as internal standards.31,32 SAPC and SDPC were chosen for study because they are common PUFA-PL species in the brain, easily detected by mass spectrometry, commercially available as synthetic forms, and available with deuterium-substituted PUFAs. Although •OH radicals have the potential to oxidize the saturated PLs that were used in these experiments as internal standards, this damage is likely to be quantitatively insignificant because of the many other species present that are equally or more susceptible. Conversely, the internal standards are not susceptible to free radical chain reactions because the reduction potentials of their C–H groups are substantially higher than those of alkylperoxyl radicals.6−8 Therefore, the MRM–LC/MSMS assay yields an unambiguous quantitative measure of the extent to which specific PL species are lost by oxidative mechanisms.
Deuterium-substituted PUFAs are inherently resistant to radical-mediated oxidative degradation due to kinetic isotope effects.33 They are not antioxidants in the traditional sense, but nevertheless effective at inhibiting overall lipid peroxidation even when they constitute only a small fraction of PUFAs in a system.34−36 It should be noted that this prior work showing that isotopic substitution could inhibit lipid peroxidation was attributed to the inhibition of autoxidation, although no attempt was made to show that peroxidation was occurring through autoxidation. In contrast, the results presented herein suggest that part of the ability of D-PUFAs to protect PUFA-PLs from free radical attack was most likely due to an overall attenuation of free radical chain reactions. The S(dA)PC and S(dD)PC used in these experiments were completely substituted at all of their bis-allylic positions, and such extensively substituted PUFAs have been previously shown to reduce the oxidative susceptibility of unsubstituted PUFAs to a degree that is out of proportion to their concentration in a membrane by unknown mechanisms.36
These experiments were prompted by previously reported in vivo results showing that a dietary omega-3-deficiency led to significantly lower rates of in vivo ARA degradation in mouse brain.9 This reduction might be explained in light of the results reported herein by reduced free radical chain reactions under conditions of omega-3 PUFA deficiency. The anti-oxidant effects of DHA in platelets at low concentrations, versus pro-oxidant effects of DHA at high concentrations, may have a similar explanation.37 However, dietary omega-3 supplementation has been associated with neuroprotection in cell and animal models, suggesting that measures of neuroprotection may not correlate with measures of oxidative stress.38−45 Moreover, DHA exhibits unexpected behaviors in some situations. For example, under some conditions it appears to have a relatively high oxidative stability, which has been attributed to a compact conformation that inhibits hydrogen abstraction.46,47 When it does undergo peroxidation, intramolecular propagation of the radical may be more likely than intermolecular,48 and peroxyl radicals may “float”49 or “snorkel”50 to the surface of a bilayer membrane where they are reduced rather than participate in free radical chain reactions.
Acknowledgments
This work was supported by grants from the NIH (AG057197), the Alzheimer’s Association, and the American Health Assistance Foundation (to P.H.A.). M.S.S. is an employee of Retrotope Inc.
Glossary
Abbreviations
- ARA
arachidonic acid
- DHA
docosahexaenoic acid
- PUFA
polyunsaturated fatty acid
- PUFA-PC
phosphatidylcholine with a PUFA esterified in the sn-2 position
- BDx
Bligh-Dyer extraction
- SAPC
stearoyl-arachidonoyl-phosphatidylcholine
- SDPC
stearoyl-docosahexaenoyl-phosphatidylcholine
- DMPC
dimyristoyl-phosphatidylcholine
- DSPC
distearoyl-phosphatidylcholine
- DOPC
dioleoyl-phosphatidylcholine
- S(dA)PC
SAPC in which deuterium has been substituted for the 6 bis-allylic hydrogens
- S(dD)PC
SDPC in which deuterium has been substituted for the 10 bis-allylic hydrogens.
The authors declare no competing financial interest.
References
- Reed T. T. Lipid peroxidation and neurodegenerative disease. Free Radical Biol. Med. 2011, 51, 1302–1319. 10.1016/j.freeradbiomed.2011.06.027. [DOI] [PubMed] [Google Scholar]
- Axelsen P. H.; Komatsu H.; Murray I. V. J. Oxidative Stress and Cell Membranes in the Pathogenesis of Alzheimer’s Disease. Physiology 2011, 26, 54–69. 10.1152/physiol.00024.2010. [DOI] [PubMed] [Google Scholar]
- Eskici G.; Axelsen P. H. Copper and Oxidative Stress in the Pathogenesis of Alzheimer’s Disease. Biochemistry 2012, 51, 6289–6311. 10.1021/bi3006169. [DOI] [PubMed] [Google Scholar]
- Cheignon C.; Tomas M.; Bonnefont-Rousselot D.; Faller P.; Hureau C.; Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe R. J. S.; Chouinard-Watkins R.; Bazinet R. P. Brain docosahexaenoic acid uptake and metabolism. Mol. Aspect. Med. 2018, 64, 109–134. 10.1016/j.mam.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H. Oxyradical Reactions - from Bond-Dissociation Energies to Reduction Potentials. FEBS Lett. 1990, 264, 165–167. 10.1016/0014-5793(90)80239-f. [DOI] [PubMed] [Google Scholar]
- Buettner G. R. The Pecking Order of Free-Radicals and Antioxidants - Lipid-Peroxidation, Alpha-Tocopherol, and Ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Yin H.; Xu L.; Porter N. A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Furman R.; Axelsen P. H. The effects of omega-3 fatty acid deficiency during development on oxidative fatty acid degradation during maturity in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2019, 79, 66–74. 10.1016/j.neurobiolaging.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimniak P. Relationship of electrophilic stress to aging. Free Radical Biol. Med. 2011, 51, 1087–1105. 10.1016/j.freeradbiomed.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B.; Gutteridge J. M. C.. Free Radicals in Biology and Medicine, 4th ed.; Oxford, 2007. [Google Scholar]
- Sato K.; Niki E.; Shimasaki H. Free Radical-Mediated Chain Oxidation of Low-Density-Lipoprotein and Its Synergistic Inhibition by Vitamin-e and Vitamin-C. Arch. Biochem. Biophys. 1990, 279, 402–405. 10.1016/0003-9861(90)90508-v. [DOI] [PubMed] [Google Scholar]
- Bowry V. W.; Stocker R. Tocopherol-Mediated Peroxidation - The Prooxidant Effect of Vitamin-E on the Radical-Initiated Oxidation of Human Low-Density-Lipoprotein. J. Am. Chem. Soc. 1993, 115, 6029–6044. 10.1021/ja00067a019. [DOI] [Google Scholar]
- Ingold K. U.; Bowry V. W.; Stocker R.; Walling C. Autoxidation of Lipids and Antioxidation by Alpha-Tocopherol and Ubiquinol in Homogeneous Solution and in Aqueous Dispersions of Lipids - Unrecognized Consequences of Lipid Particle-Size As Exemplified by Oxidation of Human Low-Density-Lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 45–49. 10.1073/pnas.90.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks J.; Dormandy T. L. The Autoxidation of Human Red Cell Lipids Induced by Hydrogen Peroxide. Br. J. Haematol. 1971, 20, 95–111. 10.1111/j.1365-2141.1971.tb00790.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y.; Niki E.; Eguchi J.; Kamiya Y.; Shimasaki H. Oxidation of Biological-Membranes and Its Inhibition - Free-Radical Chain Oxidation of Erythrocyte Ghost Membranes by Oxygen. Biochim. Biophys. Acta 1985, 819, 29–36. 10.1016/0005-2736(85)90192-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y.; Niki E.; Kamiya Y. Free-Radical Chain Oxidation of Erythrocyte Ghost Membranes by Oxygen. Chem. Lett. 1985, 14, 429–432. 10.1246/cl.1985.429. [DOI] [PubMed] [Google Scholar]
- Axelsen P. H.; Murphy R. C. Quantitative Analysis of Phospholipids Containing Arachidonate and Docosahexaenoate Chains in Microdissected Regions of Mouse Brain. J. Lipid Res. 2010, 51, 660–671. 10.1194/jlr.d001750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. R. Phosphorus Assay in Column Chromatography. J. Biol. Chem. 1959, 234, 466–468. 10.1016/s0021-9258(18)70226-3. [DOI] [PubMed] [Google Scholar]
- Axelsen P. H.; Murphy R. C.; Igarashi M.; Rapoport S. I. Increased Omega6-Containing Phospholipids and Primary Omega6 Oxidation Products in the Brain Tissue of Rats on an Omage3-Deficient Diet. PLoS One 2016, 11, e0164326 10.1371/journal.pone.0164326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. The Amateur Scientist. Sci. Am. 1984, 251, 122–129. 10.1038/scientificamerican0884-122. [DOI] [PubMed] [Google Scholar]
- van Leeuwen J. M. J.The Domino Effect. 2008, arXiv:physics/0401018. [Google Scholar]
- Bowry V. W.; Ingold K. U.; Stocker R. Vitamin-e in Human Low-Density-Lipoprotein - When and How This Antioxidant Becomes A Prooxidant. Biochem. J. 1992, 288, 341–344. 10.1042/bj2880341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski W. K.; Hyde J. S. Concentration Of Oxygen In Lipid Bilayers Using A Spin-Label Method. Biophys. J. 1983, 41, 283–286. 10.1016/s0006-3495(83)84439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiber H.; Ruff M.; Uhr M. Ascorbate Concentration in Human Cerebrospinal-Fluid (CSF) and Serum - Intrathecal Accumulation and Csf Flow-Rate. Clin. Chim. Acta 1993, 217, 163–173. 10.1016/0009-8981(93)90162-w. [DOI] [PubMed] [Google Scholar]
- Miele M.; Fillenz M. In vivo determination of extracellular brain ascorbate. J. Neurosci. Methods 1996, 70, 15–19. 10.1016/s0165-0270(96)00094-5. [DOI] [PubMed] [Google Scholar]
- Harter C.; Baechi T.; Semenza G.; Brunner J. Hydrophobic photolabeling identifies BHA2 as the subunit mediating the interaction of bromelain-solubilized influenza virus hemagglutinin with liposomes at low pH. Biochem 1988, 27, 1856–1864. 10.1021/bi00406a010. [DOI] [PubMed] [Google Scholar]
- Kardos J.; Kovács I.; Hajós F.; Kálmán M.; Simonyi M. Nerve-Endings from Rat-Brain Tissue Release Copper Upon Depolarization - A Possible Role in Regulating Neuronal Excitability. Neurosci. Lett. 1989, 103, 139–144. 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- Frederickson C. J.; Giblin L. J.; Rengarajan B.; Masalha R.; Frederickson C. J.; Zeng Y.; Lopez E. V.; Koh J.-Y.; Chorin U.; Besser L.; Hershfinkel M.; Li Y.; Thompson R. B.; Krężel A. Synaptic release of zinc from brain slices: Factors governing release, imaging, and accurate calculation of concentration. J. Neurosci. Methods 2006, 154, 19–29. 10.1016/j.jneumeth.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.; Butler J. Energetics of interconversion reactions of oxyradicals. Adv. Free Radical Biol. Med. 1985, 1, 91–131. 10.1016/8755-9668(85)90005-5. [DOI] [Google Scholar]
- Murray I. V. J.; Sindoni M. E.; Axelsen P. H. Promotion of oxidative lipid membrane damage by amyloid beta proteins. Biochem 2005, 44, 12606–12613. 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray I. V. J.; Liu L.; Komatsu H.; Uryu K.; Xiao G.; Lawson J. A.; Axelsen P. H. Membrane mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J. Biol. Chem. 2007, 282, 9335–9345. 10.1074/jbc.m608589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberson C. R.; Xu L.; Muchalski H.; Montenegro-Burke J. R.; Shmanai V. V.; Bekish A. V.; Mclean J. A.; Clarke C. F.; Shchepinov M. S.; Porter N. A. Unusual Kinetic Isotope Effects of Deuterium Reinforced Polyunsaturated Fatty Acids in Tocopherol-Mediated Free Radical Chain Oxidations. J. Am. Chem. Soc. 2014, 136, 838–841. 10.1021/ja410569g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S.; Lamberson C. R.; Xu L.; To R.; Tsui H. S.; Shmanai V. V.; Bekish A. V.; Awad A. M.; Marbois B. N.; Cantor C. R.; Porter N. A.; Clarke C. F.; Shchepinov M. S. Small amounts of isotope-reinforced polyunsaturated fatty acids suppress lipid autoxidation. Free Radical Biol. Med. 2012, 53, 893–906. 10.1016/j.freeradbiomed.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev A. Y.; Tsui H. S.; Milne G. L.; Shmanai V. V.; Bekish A. V.; Fomich M. A.; Pham M. N.; Nong Y.; Murphy A. N.; Clarke C. F.; Shchepinov M. S. Isotope-reinforced polyunsaturated fatty acids protect mitochondria from oxidative stress. Free Radical Biol. Med. 2015, 82, 63–72. 10.1016/j.freeradbiomed.2014.12.023. [DOI] [PubMed] [Google Scholar]
- Firsov A. M.; Fomich M. A.; Bekish A. V.; Sharko O. L.; Kotova E. A.; Saal H. J.; Vidovic D.; Shmanai V. V.; Pratt D. A.; Antonenko Y. N.; Shchepinov M. S. Threshold protective effect of deuterated polyunsaturated fatty acids on peroxidation of lipid bilayers. FEBS J. 2019, 286, 2099–2117. 10.1111/febs.14807. [DOI] [PubMed] [Google Scholar]
- Véricel E.; Polette A.; Bacot S.; Calzada C.; Lagarde M. Pro- and antioxidant activities of docosahexaenoic acid on human blood platelets. J. Thromb. Haemostasis 2003, 1, 566–572. 10.1046/j.1538-7836.2003.00076.x. [DOI] [PubMed] [Google Scholar]
- Green P.; Glozman S.; Weiner L.; Yavin E. Enhanced free radical scavenging and decreased lipid peroxidation in the rat fetal brain after treatment with ethyl docosahexaenoate. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2001, 1532, 203–212. 10.1016/s1388-1981(01)00132-9. [DOI] [PubMed] [Google Scholar]
- Green P.; Glozman S.; Yavin E. Ethyl docosahexaenoate-associated decrease in fetal brain lipid peroxide production is mediated by activation of prostanoid and nitric oxide pathways. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2001, 1531, 156–164. 10.1016/s1388-1981(01)00101-9. [DOI] [PubMed] [Google Scholar]
- Yavin E.; Glozman S.; Green P. Docosahexaenoic acid accumulation in the prenatal brain - Prooxidant and antioxidant features. J. Mol. Neurosci. 2001, 16, 229–236. 10.1385/jmn:16:2-3:229. [DOI] [PubMed] [Google Scholar]
- Lim G. P.; Calon F.; Morihara T.; Yang F. S.; Teter B.; Ubeda O.; Salem N.; Frautschy S. A.; Cole G. M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. 2005, 25, 3032–3040. 10.1523/jneurosci.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F.; Lim G. P.; Yang F.; Morihara T.; Teter B.; Ubeda O.; Rostaing P.; Triller A.; Salem N.; Ashe K. H.; Frautschy S. A.; Cole G. M. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron 2004, 43, 633–645. 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F.; Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. Prostaglandins, Leukotrienes Essent. Fatty Acids 2007, 77, 287–293. 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Lukiw W. J.; Cui J. G.; Marcheselli V. L.; Bodker M.; Botkjaer A.; Gotlinger K.; Serhan C. N.; Bazan N. G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Invest. 2005, 115, 2774–2783. 10.1172/jci25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P. K.; Marcheselli V. L.; Serhan C. N.; Bazan N. G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 8491–8496. 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita K. Paradox of omega-3 PUFA oxidation. Eur. J. Lipid Sci. Technol. 2014, 116, 1268–1279. 10.1002/ejlt.201400114. [DOI] [Google Scholar]
- Kazuo M. Prevention of Fish Oil Oxidation. J. Oleo Sci. 2019, 68, 1–11. 10.5650/jos.ess18144. [DOI] [PubMed] [Google Scholar]
- Else P. L.; Kraffe E. Docosahexaenoic and arachidonic acid peroxidation: It’s a within molecule cascade. Biochim. Biophys. Acta Biomembr. 2015, 1848, 417–421. 10.1016/j.bbamem.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Barclay L. R. C.; Ingold K. U. Autoxidation of Biological Molecules .2. the Autoxidation of A Model Membrane - A Comparison of the Autoxidation of Egg Lecithin Phosphatidylcholine in Water and in Chlorobenzene. J. Am. Chem. Soc. 1981, 103, 6478–6485. 10.1021/ja00411a036. [DOI] [Google Scholar]
- Marquardt D.; Williams J. A.; Kučerka N.; Atkinson J.; Wassall S. R.; Katsaras J.; Harroun T. A. Tocopherol activity correlates with its location in a membrane: A new perspective on the anti-oxidant Vitamin E. J. Am. Chem. Soc. 2013, 135, 7523–7533. 10.1021/ja312665r. [DOI] [PubMed] [Google Scholar]