

Summary
Poly(ADP-ribose) (PAR) is an RNA-like polymer that regulates an increasing number of biological processes. Dysregulation of PAR is implicated in neurodegenerative diseases characterized by abnormal protein aggregation, including Amyotrophic Lateral Sclerosis (ALS). PAR forms condensates with FUS, an RNA-binding protein linked with ALS, through an unknown mechanism. Here, we demonstrate that a strikingly low concentration of PAR (1 nM) is sufficient to trigger condensation of FUS near its physiological concentration (1 μM), which is three orders of magnitude lower than the concentration at which RNA induces condensation (1 μM). Unlike RNA, which associates with FUS stably, PAR interacts with FUS transiently, triggering FUS to oligomerize into condensates. Moreover, inhibition of a major PAR-synthesizing enzyme, PARP5a, diminishes FUS condensation in cells. Despite their structural similarity, PAR and RNA co-condense with FUS, driven by disparate modes of interaction with FUS. Thus, we uncover a mechanism by which PAR potently seeds FUS condensation.
eTOC Blurb
The Myong and Leung labs demonstrate that PAR is a potent inducer of the condensation of FUS – a disordered protein linked to neurodegenerative diseases such as ALS and FTLD.
Graphical Abstract
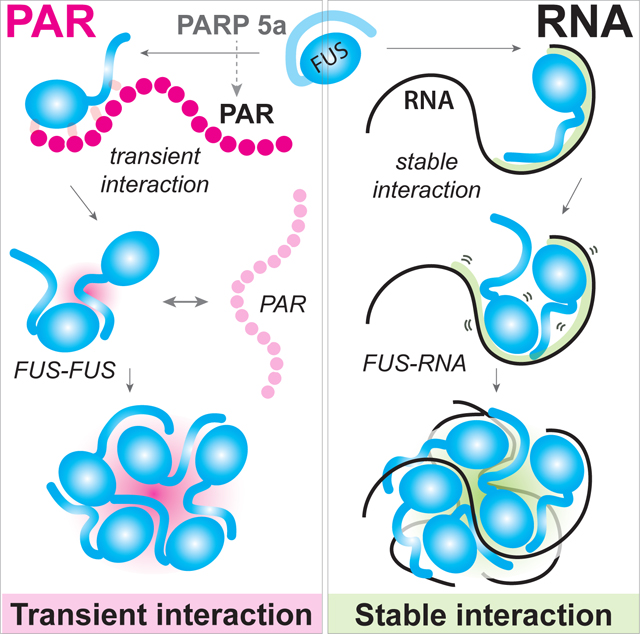
Introduction
Biomolecular condensates form through multivalent interactions that promote the assembly of proteins, RNAs, and other molecules into a dense phase (Alberti et al., 2019). Multivalent scaffolds such as RNA promote condensation of RNA-binding proteins (Harmon et al., 2017; Li et al., 2012; Mitrea et al., 2016; Rhine et al., 2020b). Significant effort has identified pathogenic mutations in disordered RNA-binding proteins, as well as disruptions to pathways such as DNA repair and autophagy, as key drivers of neurodegenerative phenotypes (Elbaum-Garfinkle, 2019; Fujioka et al., 2020; Harrison and Shorter, 2017).
Recently, a family of druggable enzymes known as ADP-ribosyltransferases, commonly termed PARPs, were also identified as contributors to neurodegenerative pathologies (Kam et al., 2018; McGurk et al., 2019; Rulten et al., 2014). Some PARPs use NAD+ to synthesize an RNA-like polymer called poly(ADP-ribose) (PAR). PAR is attached as a post-translational modification of proteins (Gibson and Kraus, 2012), which is known primarily as a transient signal for facilitating DNA strand break repair. PARylation of proteins is now recognized to have additional biological roles and to be enriched among the protein components of biomolecular condensates such as stress granules and nucleoli (Leung, 2014; Leung et al., 2011; Leung, 2020). Elevated levels of PARylation are detected in the motor neurons of ALS patients as well as the cerebrospinal fluid of Parkinson’s patients (Kam et al., 2018; McGurk et al., 2018b). Knockdown of PARP5a prevents disease in a fly model of Amyotrophic Lateral Sclerosis (ALS), and small-molecule inhibitors of PARP1 or PARP5a/b protect primary rodent neurons from TDP-43-associated neurotoxicity (McGurk et al., 2018a; McGurk et al., 2020). In vitro, free PAR chains seed the formation of droplets or aggregates of key disease proteins, such as TDP-43, hnRNPA1, FUS, and α-synuclein (Duan et al., 2019; Kam et al., 2018; McGurk et al., 2018a; Patel et al., 2015). These in vivo and in vitro data demonstrate the relevance of PAR in neurodegeneration but a mechanism by which PAR–protein interactions lead to condensation remains unexplored.
Solid-like aggregates of the disordered RNA-binding protein FUS are found in the cytoplasm of degenerating neurons in a subset of ALS and frontotemporal lobar dementia (FTLD) patients (Kwiatkowski et al., 2009; Vance et al., 2009). FUS is present in both the cytoplasm and the nucleus (Dormann et al., 2010) with roles in microRNA processing, RNA export, DNA damage response, and stress granule formation (Daigle et al., 2013; Deng et al., 2014; Dini Modigliani et al., 2014; Hyman et al., 2014; Murray et al., 2017; Shelkovnikova et al., 2014; Svetoni et al., 2016; Zhang et al., 2018). FUS forms liquid-like droplets with RNA and other proteins (Niaki et al., 2020; Patel et al., 2015; Wang et al., 2018). Although PAR is similar to RNA in some respects, there are important structural differences: each ADP-ribose unit consists of a ribose connected to an adenosine via two phosphate groups, as opposed to the single phosphate group found in RNA that joins the ribose group of adjacent bases. Thus, there are twice as many ribose and phosphate groups per base than in RNA (Leung, 2014). PAR binds to FUS in vitro and recruits FUS to DNA damage foci in cells (Altmeyer et al., 2015; Rulten et al., 2014; Singatulina et al., 2019). PAR binding requires the positively-charged RGG domains of FUS (Altmeyer et al., 2015; Dasovich et al., 2021; Singatulina et al., 2019), which likely interact with the negatively-charged PAR chains. Here, we investigate the role of PAR in FUS liquid-liquid phase separation (LLPS) to better understand the molecular FUS–PAR interaction and how it might lead to ALS-associated protein aggregation.
We find that PAR induces FUS condensation with a potency one thousand times higher than RNA. Strikingly, short-lived FUS–PAR interactions are sufficient to trigger FUS–FUS oligomerization and condensation, and PAR-seeded FUS condensates remain stable even after the PAR is degraded or removed from the reaction. By contrast, RNA-mediated FUS condensates completely dissolve after the RNA is digested. Mutations in FUS linked to ALS or FTLD alter the properties of FUS–PAR condensates. Furthermore, we show that inhibition or knockdown of PARP5a, but not PARP1 decreases FUS granule formation in neuronal cells. Together, our data support a mechanism by which PAR acts by transiently interacting with FUS to trigger formation of FUS condensates that do not require PAR to maintain their structural integrity. Despite the low abundance and transient nature of PARylation in cells, our data suggest that PAR could significantly impact FUS LLPS. Given the elevated levels of PARylation in patients with neurodegeneration, our data support further investigations into causal roles for PAR in neurodegenerative diseases.
Results
Low concentrations of PAR are sufficient to promote FUS condensation
We first carried out in vitro phase separation assays to test if PAR promotes FUS condensation. Linear PAR polymers of varying lengths were synthesized by PARP5a and labeled with Cy5 to enable fluorescent visualization of condensates (STAR Methods, Figure S1A–B). We combined purified FUS protein (1 μM, which is close to the range of physiological FUS concentration) with different PAR lengths and concentrations (Figure 1A) (Patel et al., 2015). As a comparison, we applied Cy3 labeled poly-uracil RNAs of comparable lengths that we used in our previous work (Niaki et al., 2020; Rhine et al., 2020a). Condensates were visualized with 10 nM fluorescently labeled RNA or PAR (STAR Methods). After incubating FUS with PAR or RNA for 4 hours, condensates were imaged using brightfield and fluorescence microscopy.
Figure 1: Substoichiometric concentrations of PAR are sufficient to promote condensation of FUS.

(A) Schematic of purified FUS added to Cy5-labeled PARs or Cy3-labeled poly-uracil RNAs of varying lengths. (B) Representative wide-field fluorescence microscopy images of 1 μM wild-type FUS incubated with decreasing concentrations of PARs or poly-uracil RNAs for 4 h. Asterisks denote images with increased contrast to better visualize weakly fluorescent droplets. Scale bar, 5 μm. (C) Quantification and heatmap of the area density (the fraction of area covered by condensates) of the data from (B). The density of the heatmap was normalized from 0 to 1 based on the condition with the highest coverage (1 μM U40). (D) Intradroplet concentration of varying lengths of PAR from the FUS + 100 nM PAR conditions. Concentrations were interpolated from a free dye standard curve (see Figure S1G). Error bars are standard error of the mean (n > 30). (E) Top, brightfield still images from C-Trap fusions of FUS-U40 or FUS–PAR16 condensates. The scale bar is 3 μm. Bottom left, aspect ratio over time plot of one FUS–PAR16 fusion event. Bottom right, average fusion time (τ) of FUS-U40 and FUS–PAR16 condensates. Error bars represent standard error of the mean (n > 5). Statistics were calculated using Welch’s t-test (d.f. = 4) where ns = not significant. (F) Average fluorescence recovery after photobleaching of 1 μM PAR16 with 10 nM Cy5-PAR16 (magenta) or 1 μM U40 with 10 nM Cy3-U40 (green) incubated with 1 μM FUS. Error bars are standard deviation (n>8).
Strikingly, we found that low concentrations of PAR (1–100 nM) led to condensation of FUS (Figure 1B–C, and even as low as 10 pM for PAR16; Figure S1C). By contrast, equivalent concentrations of RNA did not induce FUS condensation (Figure 1B–C). The PAR effect is protein-specific, as neither bovine serum albumin nor maltose binding protein formed condensates with PAR16. FUS–PAR droplets are also length-dependent, as monomeric and dimeric ADP-ribose were insufficient for FUS LLPS (Figure S1D–E), and very few observable droplets formed in PAR4 reactions (Figure 1B–C, Figure S1F). These data suggest that a minimal length of greater than four units of PAR is required to stimulate FUS condensation, and that PAR more potently stimulates FUS LLPS than RNA does. Overall, longer PAR chains and higher PAR concentrations promoted more FUS condensation, although high concentrations of FUS and PAR32 (1 μM each) led to aggregation (Figure 1B). Shorter chains of PAR were more highly enriched in the condensates than the longer PAR chains (Figure 1D, Figure S1G), indicating that a change in multivalent FUS–PAR interactions may impact the nature of condensation.
To test if FUS–PAR condensates are liquid-like, we performed controlled fusions of FUS–PAR droplets with a dual-laser optical trap (Figure 1E) (Rhine et al., 2021). We observed that FUS–PAR16 condensates and FUS–U40 condensates fuse with similar kinetics, demonstrating that FUS–PAR droplets are indeed liquid-like (Rhine et al., 2020a). Fluorescence recovery after photobleaching (FRAP) experiments likewise showed that PAR16 rapidly exchanges into and out of FUS condensates as is expected for liquid-like droplets (Figure 1F). Overall, this evidence supports that FUS–PAR16 droplets have liquid-like properties.
PAR transiently interacts with FUS
RNA-mediated FUS condensation requires prolonged and stable FUS–RNA interactions (Rhine et al., 2020a; Schwartz et al., 2013). Therefore, we tested whether the formation of FUS–PAR condensates was also driven by a stable association. Unlabeled FUS was incubated with Cy5-labeled PARs of varying lengths, and the resulting FUS–PAR complexes were resolved on electrophoretic mobility shift assay (EMSA) gels. All lengths of PAR induced the formation of higher-order FUS complexes that collected near the top of the gel (Figure 2A–B, Figure S2A). In addition, PAR16 and PAR32 formed discrete complexes reminiscent of FUS–RNA complexes found in our previous study (Figure 2A) (Niaki et al., 2020). Because unbound PAR8 and PAR16 were too small to be visualized on these EMSA gels for proper quantification, fluorescence anisotropy was instead used to estimate the apparent Kd (Kd_app) value of the FUS–PAR interaction. Compared to FUS and RNA (Kd_app ~5 nM), FUS and PAR exhibited a weaker affinity with a Kd_app up to forty-fold higher value (Figure 2C–D). Notably, the PAR8 concentration that led to FUS condensation (e.g., 10 nM) was twenty-fold lower than the Kd_app (>200 nM), suggesting the potent condensation facilitated by PAR does not require high-affinity binding.
Figure 2: PAR interacts with FUS primarily through transient interactions.

(A) Electrophoretic mobility shift assays (EMSAs) of increasing concentrations of FUS incubated with different lengths of 1 nM PAR. The annotations denote the following complexes: M = multimers, C2 = complex 2 (FUS dimer), C1 = complex 1 (FUS monomer). (B) Quantification of normalized aggregation observed on an EMSA gel of all PARs and U50 RNA incubated with FUS (see Figure S2A). (C) Fluorescence anisotropy binding isotherms of increasing concentrations of FUS incubated with 10 nM Cy5-PAR or Cy5-U40. Error bars represent standard error of the mean (n = 3), and best fit lines were plotted by fitting with a four-parameter total binding equation in Prism 8. (D) Apparent FUS–PAR/RNA Kd values derived from the fits in (C). Error is standard error of the mean (n = 3), and each PAR Kd was pairwise compared with the Kd of U40 using Welch’s t-test where * = p < 0.05. (E) Schematic of the single-molecule nucleation assay with pdPAR constructs. (F) Representative single-molecule traces of increasing concentrations of Cy3-FUS added to pdRNA or pdPAR constructs. Examples of long-lived and transient binding interactions are denoted in the first trace. (G) Schematic of analysis of single-molecule traces. (H) Quantification of the average number of transient binding events per trace (i.e. per 300 s) for FUS with the pdPAR and pdRNA constructs. Error bars are standard error of the mean (n > 100), and significance of pairwise pdU50-pdPAR averages was calculated using Welch’s t-test. (I) Same as H, but with long-lived binding events.
To better understand the nature of the FUS–PAR interaction, we performed a single-molecule assay in which a Cy5-labeled partially duplexed form of PAR or RNA (pdPAR or pdRNA, respectively) was immobilized to a quartz slide via biotin-neutrAvidin linkage (Figure 2E). Cy3-labeled FUS was applied to the single-molecule surface prepared with tethered pdPAR9, pdPAR22, or pdU50 RNA, and total-internal reflection microscopy was used to visualize individual FUS, PAR, and/or RNA molecules (Rhine et al., 2021) (STAR Methods). As observed previously, FUS stably associated with pdU50 RNA, indicated by the long-lasting Cy3 signal on a single RNA (Rhine et al., 2020a) (Figure 2F). Conversely, FUS interacted with both pdPAR9 and pdPAR22 in predominantly transient bursts (<1 s), evidenced by a series of spikes in the Cy3 signal (Figure 2F). Quantification of the single-molecule traces revealed that FUS–PAR binding was dominated by transient interactions whereas FUS–RNA binding was primarily long-lived (Figure 2H–I). The frequency and intensity of the FUS–PAR interactions increased as a function of FUS concentration (Figure 2F–I, Figure S2B–D). Together, these data suggest that FUS interacts with PAR in a fundamentally disparate manner compared to RNA.
Transient FUS–PAR interactions drive FUS condensation
The sub-stoichiometric and transient nature of the FUS–PAR interaction led us to ask if PAR stimulates the FUS–FUS contacts that give rise to FUS condensation. If so, PAR may not be needed after seeding condensate formation to sustain the FUS droplets. To test this possibility, we formed FUS–PAR droplets and applied a PAR-degrading enzyme, PAR glycohydrolase (PARG) (Hatakeyama et al., 1986). While the Cy5-PAR signal was efficiently removed upon addition of PARG, the FUS condensates remained intact when visualized by the brightfield images or using Cy3-FUS (Figure 3A, Figure S2E), clearly indicating the sustained FUS droplets after the degradation of PAR. In stark contrast, FUS–RNA (U40) droplets treated with RNase resulted in a complete disappearance of both the Cy3-RNA and FUS droplets (Figure 3A, upper panel). Thus, PAR must be promoting FUS–FUS contacts that are distinct from the contacts induced in the presence of RNA and can support sustained LLPS. Dynamic light scattering of FUS–PAR condensates showed rapid nucleation into large particles, which was a distinct kinetic behavior compared to the formation of FUS–RNA condensates. (Figure S2F).
Figure 3: Transient FUS–PAR interactions are responsible for FUS condensation.

(A) Representative wide-field images of wild-type FUS droplets that were treated with either RNase (for U40 sample) or PARG (for PAR samples) at the 3 h time point. Scale bar, 5 μm. (B) Schematic of the bead droplet experiment. (C) Wide-field images of wild-type FUS incubated with DNA, RNA, or PAR-treated beads for 4 h. Scale bar, 10 μm. (D) Quantification of the area coverage for bead reactions and for free polymer (see Figure S2H). (E) Schematic of how droplet reactions are fractionated midway through the reaction. (F) Multicolor images of the pellet and supernatant fractions of the bead droplet experiments. For the pellet fraction, the Cy5 signal is the DNA conjugated to the beads. For the supernatant fraction, the Cy5 signal is Cy5-labeled FUS added after separating the beads. Scale bar, 10 μm. (G) Schematic of wild-type, Y>S, and R>G FUS constructs. Black lines denote the locations of substitutions. (H) Representative fluorescence wide-field images of FUS with either RNA (1:1 FUS:RNA) or PAR (10:1 FUS:PAR) after 4 h. Scale bar, 5 μm. (I) Quantification of the droplet area coverage of the FUS constructs. Error bars denote standard error of the mean (n = 5), and pairwise comparisons between wild-type and the other constructs were performed with Welch’s t-test. Orange points and text denoting significance level are Y>S FUS and gray are R>G FUS. (J) Quantification of the number of transient and long-lived binding events of 1 nM FUS constructs with pdPAR22. Error bars are standard error of the mean (n > 100), and significance was determined with Welch’s t-test. (K) Representative single-molecule nucleation traces of 1 nM Cy3-Y>S or Cy3-R>G FUS with pdPAR22. (L) Images of florescence wide field images of 1 μM Y>S or R>G FUS with DNA-, RNA-, or PAR-treated beads after 4 h. (M) Quantification of bead reactions with wild-type and mutant FUS. Error bars denote standard error of the mean (n = 5), and significance was calculated using Welch’s t-test.
These findings led us to hypothesize that transient FUS–PAR contacts are sufficient to induce FUS–FUS interactions, which form FUS condensates that no longer require PAR. To test this hypothesis, we confined PAR by preparing PAR-coated beads, which were spun down to remove free PAR in solution (STAR Methods). We then combined the PAR-beads with Cy3-FUS and visualized the beads with an inert Cy5-DNA (Figure 3B–D, Figure S2G–H). Strikingly, when Cy3-FUS was applied to the solution containing PAR-coated beads, we observed the robust formation of FUS droplets that were physically separated from the PAR-beads (Figure 3C–D), consistent with our hypothesis that transient interactions between PAR and FUS lead to FUS condensation. In contrast, DNA- and RNA-coated beads were insufficient to form FUS condensates (Figure 3C–D).
To further test if the PAR-treated FUS can stimulate FUS condensation, we removed the PAR-beads by centrifugation after 1 hour of incubation with Cy3-FUS; unreacted Cy5-FUS was applied to the supernatant (PAR-bead-free) fraction (Figure 3E). The PAR-treated Cy3-FUS recruited Cy5-FUS into FUS condensates, indicating that the PAR-treated FUS is active in inducing condensates on its own (Figure 3F). This finding strongly supports the role of PAR in altering FUS into an LLPS-prone form that can recruit other FUS molecules into a condensate (Figure 3F).
Role of arginine and tyrosine in PAR-FUS phase separation
The RGG domains of FUS, which are enriched with arginine and glycine residues, are necessary for PAR interactions (Dasovich et al., 2021; Singatulina et al., 2019). In addition, previous studies showed the role of arginine and tyrosine as sticker residues that enable condensation of FUS and other phase separating proteins (Choi et al., 2020; Martin et al., 2020; Qamar et al., 2018; Wang et al., 2018). Therefore, we investigated the FUS–RNA and FUS–PAR interactions with two FUS mutants: one in which all arginine residues in the C-terminal RGG domains were mutated to glycine (R>G), and one in which all tyrosine residues in the N-terminal QGSY-rich domain were mutated to serine (Y>S) (Bogaert et al., 2018; Wang et al., 2018) (Figure 3G).
As expected, wild-type FUS condensed on its own at ≥4 μM and with RNA or PAR at all tested concentrations ≥1 μM (Figure 3H–I, Figure S3A). R>G FUS showed no condensation alone and very minimal condensation with PAR at ≥4 μM concentrations Figure 3H–I, Figure S3A); EMSA gels and single-molecule assays showed that R>G FUS did not bind to PAR (Figure 3J–K, Figure S3B). Y>S FUS alone also had minimal condensation (Figure S3A), which is consistent with previous data (Wang et al., 2018). However, addition of RNA and PAR both led to the formation of wild-type-like, albeit smaller, condensates (Figure 3H–I), likely because Y>S FUS still retains normal RNA and PAR binding (Figure S3B). We interestingly observed that Y>S FUS shifted to predominantly sustained interactions with PAR (Figure 3J–K, S3C).
Interestingly, when we performed the PAR-coated bead experiment with Y>S FUS, it preferentially interacted with the beads and formed very few droplets beyond those associated with the beads (Figure 3L–M). The increased interaction of Y>S FUS with the PAR-coated beads may arise from the long-lived Y>S FUS–PAR interactions we observed above (Figure 3H–K). Y>S FUS also had decreased condensation with lower concentrations of PAR (Figure S3D). As expected, R>G FUS was unable to form any droplets with PAR-coated beads (Figure 3L–M). Together, these data support our model that PAR promotes FUS condensation through transient interactions and suggest that tyrosine FUS–FUS contacts may play a role in oligomerization mediated by transient PAR binding.
PARP5a knockdown reduces FUS condensation in cells
To determine whether the FUS–PAR interaction leads to FUS condensation in cells, we created a FUS-Halo stable line of SH-SY5Y human neuroblastoma cells (Figure 4A). FUS-Halo was visualized with JF549 (Figure S4A). Sodium arsenite (0.5 mM) treatment induced the formation of primarily cytoplasmic FUS granules (Figure 4A, Figure S4B–C). The FUS-Halo was only mildly overexpressed (1.3–1.4 fold) in relation to endogenous FUS (Figure S4D). The stress granule (SG) markers eIF3B and G3BP1 overlapped with FUS granules (Figure S4C), indicating that FUS granules may share some or all components with SGs. The FUS granules also stained positive for ADP-ribosylation (Figure S4C).
Figure 4: PARP5a activity promotes FUS condensation in cells.

(A) Representative confocal images of FUS-Halo SH-SY5Y cells treated with shRNAs for the indicated genes (see STAR Methods and timeline at the bottom of the panel). FUS was visualized with JF549 (red), and nuclei were visualized with Hoechst (blue). Cells were stressed with sodium arsenite for 1 h, and cells were live-imaged at 37 °C. The scale bar is 8 μm. (B) Quantification of the FUS granules per cell in (A). The error bars denote standard error of the mean (n > 30), and Welch’s t-test was used to calculate the significance of the indicated pairwise comparisons. NC-1, noncoding shRNA control. (C) Same as (A) but instead of transfected shRNAs, cells were treated with PARP inhibitors for the indicated time period (see timeline at the bottom of the panel). All images show live cells that were stressed with sodium arsenite for 1 h. (D) Same as (B) but for the data in (C). Significance was calculated for each condition compared to untreated FUS-Halo cells. (E) Airyscan confocal images of FUS-Halo cells transfected with GFP-PARP5a (green). The inset shows the dashed-line box. The scale bar is 5 μm. (F) Normalized intensity plots show the enrichment of the FUS-Halo (red) and GFP-PARP5a channels for the yellow dashed lines in the inset in (E).
To identify the possible source of PARylation, we treated the FUS-Halo cells with shRNAs specific for the major nuclear and cytoplasmic PAR synthesis enzymes, PARP1 and PARP5a, respectively (Figure S4E). Knockdown of PARP5a, but not PARP1, led to a significant reduction in FUS granule size and number (Figure 4A–B, Figure S4F–G), indicating that PARP5a is required for cellular FUS condensation. Moreover, overexpression of the GFP-tagged version of the cytoplasmic isoform of PARG led to a reduction in FUS granule formation (Figure S3H–I). To determine whether the reduction in FUS granule formation was due to the PARP activity, FUS-Halo cells were treated with the inhibitors of PARP5a/b, G007-LK or IWR-1 (Huang et al., 2009; Voronkov et al., 2013), or the PARP1 inhibitor Olaparib. While Olaparib did not reduce FUS granule formation, PARP5a/b inhibitor treatment resulted in a significant reduction in FUS condensation upon stress (Figure 4C–D). Altogether, these data indicate that PARP5a enzymatic activity is required for FUS granules and underscores the physiological relevance of PAR in forming stress-responsive condensates.
Next, we tested the localization of FUS and PARP5a in FUS-Halo cells, which were transfected with GFP-PARP5a. Super-resolution microscopy revealed that GFP-PARP5a puncta, like endogenous PARP5a, did not completely colocalize with FUS puncta (Figure 4E–F; Figure S4C). Live-cell imaging during the stress process revealed transient colocalization between FUS and PARP5a, but most FUS and PARP5a granules remained discrete (Figure S4J). The weak PAR enrichment in FUS granules suggests that a combination of PARP5a-mediated PARylation of other proteins and noncovalent interactions with these PAR chains may together contribute to FUS LLPS in cells (Figure S4C).
PARP5a activity promotes FUS LLPS
Based on our cellular data, we tested if PARP5a activity is sufficient to induce FUS condensation in vitro. Cy3-labeled FUS and AlexaFluor488-labeled PARP5a catalytic domain (1 μM each) were combined in vitro with increasing concentrations of its substrate NAD+ (0–1 mM). We observed FUS and PARP5a droplets only when NAD+ was added, indicating that the synthesis of PAR in situ can also promote FUS condensation (Figure 5A–D). Interestingly, we found that high, non-physiological concentrations of NAD+ formed very few condensates, likely due to the high concentrations of PAR buffering FUS condensation as had been described for RNA (Maharana et al., 2018) (Figure S4K). PARP5a alone also led to robust droplet formation, but this buffering effect was not observed at high NAD+ concentrations (Figure 5E–G), indicating the distinct nature of these condensates.
Figure 5: PARP5a activity promotes the condensation of FUS in vitro.

(A) Representative wide-field fluorescence microscopy images of 1 μM wild-type FUS (10 nM Cy3-FUS) and 1 μM PARP5a (10 nM A488-PARP5a) incubated with increasing concentrations of NAD+ for 4 h. Scale bar, 10 μm.
(B) Western blot analyses of the PARylation reactions from (A).
(C) Quantification of droplet area (the fraction of area covered by condensates) of the data from (A). Error bars are standard error of the mean (n = 3).
(D) Number of co-localized, FUS-only, and PARP5-only droplets based on Cy3-FUS and A488-PARP5a signals in (A). Error bars are standard error of the mean (n = 3).
(E) Representative wide-field fluorescence microscopy images of 1 μM PARP5a (10 nM A488-PARP5a) incubated with increasing concentrations of NAD+ for 4 h. Scale bar, 10 μm.
(F) Western blot analyses of the PARylation reactions from (E).
(G) Quantification of droplet area (the fraction of area covered by condensates) of the data from (E). Error bars are standard error of the mean (n = 3).
We next assessed the extent of PARylation under these conditions and found that only the highest NAD+ concentration (1 mM) resulted in extensive PARylation of FUS (Figure 5B). Similarly, we observed a negligible amount of FUS PARylation in cellular conditions when FUS granules were formed (Figure S4L). These results suggest that PAR binding, but not PARylation, of FUS is the main mechanism of condensation.
PAR does not hinder FUS–RNA condensation
FUS has an RNA recognition motif, a zinc finger, and several RGG domains that contribute to RNA binding (Deng et al., 2014; Loughlin et al., 2019; Schwartz et al., 2013). Given that PAR interactions with FUS are largely transient and appear to be necessary for proper FUS granule formation in cells, we next asked what effect PAR had on FUS–RNA condensation. We reasoned that either (1) PAR and RNA would compete for FUS binding and decrease droplet formation by independently sequestering FUS, or that (2) the two molecules would condense synergistically. Thus, we performed phase separation assays using equimolar (1 μM each) FUS and U40 RNA with or without PAR (100 nM). Using turbidity as a readout, PAR did not appear to significantly impact the condensation kinetics of FUS with RNA (Figure 6A–B). Interestingly, we found that adding free PAR in single-molecule nucleation assays increased the number of transient FUS binding events with pdU50 RNA, indicating that PAR may stimulate short-lived FUS–RNA interactions (Figure S5A–B). There was no corresponding shift in the long-lived FUS-RNA interactions (Figure S5B).
Figure 6: PAR co-condenses with FUS and RNA.

(A) Normalized turbidity measurements of 1 μM FUS and 1 μM U40 supplemented with 500 nM additional PAR (magenta) or U40 RNA (green). Error bars indicate standard error of the mean (n = 3). (B) The t1/2 value of the turbidity measurements in (A). Error bars denote standard error of the mean of the fitted t1/2 values (n = 3), and Welch’s t-test was used to calculate the significance for the indicated comparisons. (C) The number of 1 nM Cy3-FUS transient binding events on pdU50 RNA supplemented with 100 pM free PAR. (D) Schematic of possible outcomes of FUS–RNA–PAR co-condensate formation. (E) Representative wide-field images of 1 μM FUS incubated with 1 μM U40 RNA (10 nM Cy3-U40) and 100 nM PAR (10 nM Cy5-PAR). The scale bar is 5 μm. (F) Colocalization scores of the Cy3 and Cy5 channels in (E). Error bars are standard error of the mean (n = 5), and significance was calculated using Welch’s t-test. (G) The area density of the Cy5 channel in (E) (white bars) compared to RNA alone (green) or PAR alone (magenta) at comparable RNA or PAR concentrations. Error bars denote standard error of the mean (n = 5), and significance was calculated using Welch’s t-test for the indicated pairwise comparisons.
To test whether FUS, RNA, and PAR co-condense into droplets, we combined 1 μM FUS with Cy3-U40 RNA (1 μM) and Cy5-PAR of various lengths (100 nM) and monitored droplet formation with fluorescence microscopy (Figure 6D). We observed robust co-condensation of FUS, RNA and PAR for all PAR lengths (Figure 6E–F). FUS–RNA condensation area was not impacted by adding long PARs. However, short PARs significantly enhanced FUS LLPS in the presence of RNA, when compared to FUS with PAR only (Figure 6G). Together, these data suggest that PAR does not inhibit FUS–RNA condensation.
RNA and PAR formed distinct complexes with FUS on EMSA gels, indicating that they may compete for binding at low protein concentration (Figure S5E–F). To further test this apparent binding competition, we performed fluorescence anisotropy with bound FUS-Cy5-RNA or FUS-Cy5-PAR complexes titrated with the other unlabeled polymer (i.e., PAR and RNA, respectively). We found that RNA was able to effectively outcompete PAR for FUS binding, but not vice versa (Figure S5G). This result suggests that RNA has stronger affinity to FUS than PAR, consistent with their apparent Kd (cf. Figure 2C–D). But more importantly, these data indicate that the RNA and PAR binding sites at least partially overlap (Figure S5G), as had been suggested by previous reports (Portz et al., 2021; Singatulina et al., 2019).
How do FUS, RNA, and PAR form stable co-condensates if RNA and PAR are competing for FUS binding? We hypothesized that the co-condensed FUS-RNA-PAR may form a stable tripartite condensate by linking RNA-bound FUS and PAR-bound FUS with FUS-FUS contacts. FRAP measurements supported this hypothesis; the apparent U40 exchange rate in FUS-PAR16-U40 co-condensates decreased compared to FUS-U40 droplets (Figure S5H–I). Notably, EMSA analyses revealed that FUS also forms high molecular-weight complexes with both RNA and PAR (Figure S5E–F) Therefore, the multivalency of the FUS–RNA–PAR interaction may limit diffusion of RNA within condensates, and a network of FUS–RNA and FUS–PAR interactions may underly the architecture and component dynamics of these FUS–RNA–PAR droplets.
PAR is weakly incorporated into FUS–RNA condensates
To decipher the interaction network within the tripartite FUS–RNA–PAR co-condensate, we performed a series of dissolution assays on droplets consisting of FUS, U40 RNA and different lengths of PAR. Four different treatments were used: (1) 1,6-hexanediol, which disrupts hydrophobic interactions in phase-separated condensates (Kroschwald et al., 2017); (2) Karyopherin-β2 (Kapβ2), which extricates FUS from condensates (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018); (3) RNase; and (4) PARG (Figure 7A). Real-time videos of the Cy3-RNA and Cy5-PAR signals were recorded for each treatment and the intensity of droplets over time was plotted for both fluorescent signals (Figure 7A–B, Figure S6A–D).
Figure 7: The architectural network of FUS–RNA–PAR co-condensates.

(A) Schematic showing how FUS–RNA–PAR co-condensates were treated with various proteins/chemicals. (B) Representative fluorescence wide-field images of FUS–RNA–PAR co-condensates before and after treatment. The Cy5 (magenta) signal is PAR and the Cy3 signal (green) is RNA. The scale bar is 5 μm. (C) Stills from representative dissolution videos of FUS–RNA–PAR16 co-condensates. The Cy5 (magenta) signal is PAR and the Cy3 signal (green) is RNA. The treatments were added just after the 0 s time point. For Kapβ2, time points are shown for 0, 5, 15, and 30 s. The scale bar is 5 μm. (D) Intensity plots of the Cy3-RNA and Cy5-PAR signals for each of the indicated co-condensate reactions treated with Kapβ2. Error bars denote standard deviation (n=10). The plot on the right shows the fitted t1/2 value for the Cy3 and Cy5 dissolution intensity curves. (E) Representative intensity plots of individual FUS–RNA–PAR condensates treated with RNase. The lighter lines indicate later time points during the video acquisition, and bolded lines are highlighted times as indicated. (F) Wide-field fluorescence images of FUS-Cy3-RNA droplets immediately after adding Cy5-PAR. (G) Same as (F) but for FUS-Cy5-PAR droplets after adding Cy3-RNA. The inset below shows more timepoints of the Cy3-RNA displacement of Cy5-PAR. The scale bar is 5 μm, which also applies to panel (F). (H) Quantification of the Cy3 and Cy5 fluorescence signals in (F) and (G).
FUS–RNA–PAR droplets treated with 1,6-hexanediol uniformly disintegrated with no biases toward RNA or PAR, though PAR32-containing droplets resisted dissolution (Figure 7B–C, Figure S6E). By contrast, Kapβ2 treatment led to the loss of the Cy5-PAR signal first for all conditions except PAR32,, (Figure 7B–D, Figure S6F, Video S1). These data suggest that Kapβ2 and PAR may bind to FUS in a similar manner, consistent with previous work demonstrating that Kapβ2 interacts with the RGG motifs of FUS to promote disassembly (Qamar et al., 2018). Interestingly, Kapβ2-mediated PAR and RNA loss was inversely correlated as a function of PAR length: longer PARs led to faster RNA ejection but slower PAR loss (Figure 7C–D). Furthermore, this direct relationship between PAR length and RNA release may indicate that as PAR length increases, its contribution to the stability of FUS condensates becomes more pronounced, and thus its removal disrupts the integrity of the three-component system more.
As expected, RNase immediately degraded RNA, removing it from the droplets (Figure 7B–C, 7E top panels, Figure S6G). Intriguingly, RNase also temporarily removed PAR from FUS droplets, but the FUS–PAR droplets gradually recovered to form condensates that mimicked PAR-only condensates (Figure 7E, Figure S1F, S6G, Video S2). PARG treatment degraded PAR signal without producing any change in the RNA signal (Figure 7B–C, Figure S6H), indicating the importance of the FUS–RNA network in maintaining the architecture of FUS–RNA–PAR condensates.
We also found that RNA can enter pre-formed FUS–PAR droplets, displacing most of the existing PAR (Figure 7F–H, Video S3). By contrast, PAR cannot enter pre-formed RNA condensates (Figure 7F–H). Together, these results support a model in which PAR potently promotes LLPS but is not stably engaged with FUS in the condensate, allowing for facile penetration of RNA.
ALS/FTLD-linked FUS mutants have altered PAR LLPS
Over 70 mutations in FUS are linked with the neurodegenerative diseases ALS/FTLD (Deng et al., 2014), and these mutations can impact the material properties of FUS condensates. Mutations in arginine residues increase LLPS propensity whereas mutations in glycine produce gel-like condensates with RNA (Niaki et al., 2020; Rhine et al., 2020a). We incubated R244C FUS and G156E FUS, both of which are linked to ALS, with PAR16 and U40 RNA (Figure S7A–B). Both R244C and G156E formed co-condensates with RNA and PAR (Figure S7A–B), but we found that PAR was more highly enriched in G156E FUS compared to wild-type and R244C FUS (Figure S7B). G156E FUS also displayed low fluorescence recovery of both RNA and PAR, suggesting that increased PAR enrichment may drive solidification of the FUS condensate (Figure S7C). As with wild-type FUS, RNA addition to mutant FUS-PAR condensates led to PAR displacement (Figure S7D). Therefore, PAR may contribute to a gel-like transition in G156E FUS, but RNA can still displace this PAR.
Discussion
Here, we demonstrate that PAR can potently drive condensation of the ALS/FTLD-linked disordered protein FUS. Unlike the equimolar concentration requirement for RNA-mediated FUS condensation, one-thousand-fold lower concentrations of PAR are sufficient to induce formation of liquid-like droplets with FUS in vitro. Our data demonstrate that PAR initiates condensation through a unique molecular mechanism in a length-dependent manner.
The catalyst-like mechanism of PAR-mediated condensation
We propose a model in which primarily transient interactions between PAR and the RGG-domains of FUS drive condensation. For longer PARs (>10 units), FUS can use PAR as a scaffold for multivalent interactions that drive condensation as has been described for FUS and RNA (Harmon et al., 2017; Mitrea et al., 2016; Niaki et al., 2020). However, PARs of all lengths can also prime FUS for condensation through transient interactions, and the primed FUS is sufficient for formation and maintenance of FUS condensate even in the absence of PAR. The exact mechanism by which this occurs is unclear, but our data suggest that PAR may interact with the RGG domains of FUS, liberating the tyrosine residues of the disordered QGSY-rich domain to promote condensation (Murray et al., 2017; Portz et al., 2021). Notably, PAR binders are enriched with RGG motifs in the human proteome, where FUS and its related proteins EWSR1 and TAF15 are three of the most enriched PAR binders (Dasovich et al., 2021). We propose that PAR, like RNA, can act like a nucleator for FUS condensation, but also distinctly as a transient catalyst. This distinct PAR property bears resemblance to the cyclical turnover of an enzyme which can perform multiple rounds of reactions without being consumed or sequestered by the substrate.
Such a trans-acting catalyst-like mechanism may explain why PARP5a activity is required for proper FUS condensation, yet without maintaining their physical association in cells. Three possible scenarios may underlie such a mechanism. First, PARP5a synthesizes PAR, which may then be released as a soluble polymer that prompts FUS condensation. In vitro data indicate that PAR can be released by PARG through its endo-glycosidic activity or by the ADP-ribosylhydrolase TARG1, which removes entire PAR chains from PARylated proteins (Sharifi et al., 2013). However, definitive evidence of long-lived soluble PAR in cells is currently lacking (Mashimo et al., 2013; Pourfarjam et al., 2020). Second, PARP5a may PARylate FUS, which then becomes proficient at condensation. However, we did not observe an appreciable PARylation of FUS by PARP5a in vitro at physiological NAD+ concentrations or when FUS granules are formed in cells, but we cannot exclude the possibility that a low yet undetectable level of FUS PARylation is sufficient for condensation. Third, FUS may assemble on PARylated PARP5a, as it does on auto-PARylated PARP1 at DNA damage foci in the nucleus (Altmeyer et al., 2015; Patel et al., 2015; Singatulina et al., 2019). Alternatively, FUS can non-covalently bind PAR conjugated to other PARP5a-modified substrates. In all these contexts, the transient interactions between PAR and FUS may prime FUS to drive homotypic FUS–FUS multimerization.
Notably, this mechanism is inherently different from the FUS–RNA interaction, which exclusively relies upon the nucleation of FUS on RNA molecules (Rhine et al., 2020a). Considering the highly dynamic synthesis and degradation of cellular PAR and its functional role in recruiting proteins, this unique switch-like mechanism that “turns on” the protein to condense via a short-lived contact reveals a highly efficient pathway built for a rapid and robust cellular response. These condensates made from primed FUS can subsequently be resolved by cellular chaperones, such as Kapβ2. Taken together, RNA and PAR enhance the otherwise weak FUS–FUS contacts but do so through distinct mechanisms despite their biochemical similarities. Our data further indicates that modulating the ratio of RNA and PAR changes the physical size and dynamics of these condensates; therefore, it is important to investigate how cellular condensates are regulated by proteins that can bind to both RNA and PAR. This class of proteins may help condensate formation by cross-linking protein networks mediated by these two functionally distinct nucleic acid polymers.
The effect of PAR length in binding, condensation, and neurodegeneration
One unique aspect of this study is that we analyzed for the first time how protein condensation can be regulated by the length of PAR. Although previous studies have demonstrated that PAR can induce the phase separation of proteins (Duan et al., 2019; Kam et al., 2018; McGurk et al., 2018a; Patel et al., 2015), these reports used a mixture of PAR chains ranging from 2 to 300 units in length, making it impossible to evaluate the relationship between PAR length and its potency in protein condensation. Increasing evidence has revealed that cellular pathways are only activated when PAR length exceeds a certain threshold. For example, initiation of the programmed cell death pathway parthanatos, DNA repair factor recruitment, and activation of the cell cycle checkpoint kinase Chk1 are all more strongly promoted by longer PAR chains (Andrabi et al., 2006; Fahrer et al., 2007; Fahrer et al., 2010; Min et al., 2013).
Here, we demonstrate that PAR length determines the threshold and physical properties of protein condensation. Monomers, dimers and tetramers of ADP-ribose are insufficient for FUS condensation. For chains longer than eight units, the degree of phase separation increases as a function of PAR chain length. As PAR length is tightly controlled in cells by opposing classes of enzymes and co-factors, the polymer length may constitute part of the “PAR code” which directs the biological outcome of condensates (Leung, 2020).
Notably, PAR levels are elevated in ALS patient motor neurons, Parkinson’s patients and animal models of Alzheimer’s diseases (Kam et al., 2018). Furthermore, human genetic analyses revealed that mutations in PAR-degrading enzymes (which result in abnormally high PAR levels) are linked to several yet-to-be-named, rare hereditary diseases with neurodegenerative phenotypes (Ghosh et al., 2018; Mashimo et al., 2019; Sharifi et al., 2013). Some of these phenotypes can be recapitulated in animal models (Ghosh et al., 2018; Mashimo et al., 2019), which show an increased number of protein aggregates in cells (Hanai et al., 2004). Together with these biochemical, cellular and genetic data, our study demonstrating PAR as a potent inducer of protein condensation indicate a strong possibility that PAR contributes to pathologic protein aggregation in neurodegeneration.
We surprisingly found that PAR enrichment is also elevated in G156E condensates in vitro. These condensates display gel-like properties, and G156E FUS has previously been implicated in aggressive cases of ALS coupled with FTLD (dementia) symptoms (Patel et al., 2015; Ticozzi et al., 2009). Our results indicate that the PARP5a inhibitors we tested, G007-LK and IWR-1, may be candidate therapeutics for reducing the neurotoxicity of ALS/FTLD-linked FUS. Previous reports have likewise identified PARP inhibitors as possible treatments for TDP-43-mediated neurodegeneration (McGurk et al., 2018a), highlighting the deleterious role that PAR may play in promoting the oligomerization of aggregate-prone proteins.
Limitations of Study
Many of our experiments on protein condensation rely on purified PAR, and the protein-conjugated PAR may behave differently. Indeed, we observed that FUS and PARP5a are co-condensed in vitro, only partially co-localized in cells. In addition, although FUS has been identified as an ADP-ribosylated substrate (Ayyappan et al., 2021), it is unclear as to which PAR-conjugated substrates are critical for the formation of these FUS granules. An additional limitation of the current study is that we only used linear PAR, which is made from PARP5a, whose activity is critical for the formation of FUS granules. Given that other biomolecular condensates such as DNA repair foci are dependent on PARP1, which makes linear and branched PAR, it is unclear how branching affects protein condensation. We note that there can be minute amounts of residual free PAR present in PAR-bead solution even after the centrifugation. Nevertheless, the robust formation of FUS droplets only seen in PAR-bead but not in RNA-bead supports our hypothesis that transient PAR-FUS interaction is the main driving force for FUS-FUS interaction and FUS condensation.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sua Myong (smyong@jhu.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Microscopy, single-molecule, and other raw data materials are available from the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication at the following DOI: https://doi.org/10.5281/zenodo.5866686.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. Coli Culturing
BL21 or NEB Turbo E. coli were transformed with antibiotic resistant plasmids and cultured in Lysogeny broth. Cultures (5 mL) from individual colonies were grown overnight for use in protein purification (see below) or for generating frozen stocks by combining 1:1 with 50% (v/v) glycerol and storing at −80 °C.
SH-SY5Y Cell Culturing
Female human SH-SY5Y neuroblastoma cells (ATCC, Manassas, VA) were stably transfected with a wild-type FUS-Halo plasmid which conferred resistance to the antibiotic geneticin. The piggybac retrotransposon system was used to insert the FUS coding sequence into a random location in the genome. Stably-transfected cells were grown at 37 °C with 5% CO2 in a DMEM solution supplemented with 10% FBS, 2 mM glutamate, 0.15% sodium bicarbonate, 1 mM sodium pyruvate, and 200 ng/μL G418. Cells were periodically tested with a mycoplasma-detecting test, and the absence of mycoplasma was also confirmed through Hoechst staining and fluorescence microscopy (see below). Cells were passaged and prepared for long-term storage as described previously (Rhine et al., 2020a).
METHODS DETAILS
FUS Purification
The bacterial expression plasmids for WT FUS, R244C FUS, and G156E FUS were designed for previous research (Niaki et al., 2020). The Y>S and R>G FUS genes were synthesized by Genscript (Piscataway, NJ) and inserted into the WT FUS plasmid backbone. All FUS plasmids consisted of an N-terminal maltose binding protein (MBP) tag and 6xHis tag, which were separated from the FUS ORF by a tobacco etch virus (TEV) protease recognition site. The plasmids were transformed into BL21 (DE3) bacteria, and FUS was purified as described previously (Rhine et al., 2020a). All FUS used for the following experiments was <2 weeks old.
In brief, the 6xHIS-MBP-FUS plasmids were transformed into competent BL21(DE3) E. coli cells (New England Biolabs, Ipswich, MA). A starter culture (5 mL) was incubated overnight in lysogeny broth (LB) and 50 mg/L kanamycin sulfate at 37 °C with 200 rpm shaking. The next day, the starter culture was added to LB (0.5–2 L) and allowed to incubate further at 37 °C with shaking. Once the OD600 of the culture reached 0.4 (after approximately 2–3 h), the cells were induced with 0.25 mM IPTG and proteins were expressed at 30 °C for 2 h. The cells were centrifuged at 5000 x g at 4 °C for 10 min, and the pellets were collected for immediate purification or stored at −80 °C.
The pellets were resuspended in FUS Lysis Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 10 mM imidazole, 1.5 mM β-mercaptoethanol, 5% (v/v) glycerol, 1% (v/v) NP-40, 5 mg/L culture RNAse A, half-tablet EDTA-free protease inhibitor) and lysed by sonication. The lysate was centrifuged at 23644 x g at 4 °C for 30 min, and the supernatant was filtered through a 0.22 μm syringe filter.
The supernatant was loaded into an ÄKTA pure 25 M FPLC system (GE Healthcare/Cytiva) and applied to a 5 mL HisTrap HP column (GE Healthcare/Cyvita). The column was washed with 10 column volumes of FUS Binding Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 10 mM imidazole, 1.5 mM β-mercaptoethanol, 5% (v/v) glycerol). FUS was eluted by linearly increasing the imidazole concentration with FUS Elution Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 500 mM imidazole, 1.5 mM β-mercaptoethanol). The fractions containing substantial FUS were stored in FUS Elution Buffer with 25% glycerol at 4 °C.
The HisTrap HP column was stripped every 5–10 purifications by incubating in Column Stripping Buffer (500 mM NaCl, 50 mM EDTA, pH 8.0, 20 mM Na3PO4, pH 7.0) for 20 min, then in 1 M NaOH for 2 h. Nickel (100 mM NiSO4) was flowed through the column at 0.5 mL/min for 50 min to regenerate the HisTrap HP column.
PARG Purification
Bacterial expression vectors for PARG were codon-optimized for bacterial expression and included a His-SUMO tag at the N-terminus. The vectors were transformed into competent BL21(DE3) E. coli cells (New England Biolabs, Ipswich, MA). A 30 mL starter culture was grown overnight at 37 °C. Following overnight incubation, the 30 mL starter culture was added to a 1 L culture and grown to an OD600 of ~0.4. The culture was cooled to 4 °C for 1 h, and IPTG was added to a final concentration of 0.5 mM. The cells were incubated overnight at 16 °C with shaking. Next, the cells were pelleted at 5000 x g at 4 °C for 30 min, and the pellet was resuspended in PARG Lysis Buffer (250 mM NaCl, 50 mM HEPES pH 7.5, 30 mM imidazole, fresh 1 mM DTT, half tablet EDTA-free protease inhibitor) for storage at −80 °C.
The resuspended cells were thawed, DTT was added to 1 M, and NP-40 was added to 0.1% (v/v). The cells were lysed by sonication and centrifuged at 17000 x g at 4 °C for 30 min. The supernatant was filtered through a 0.22 μm syringe filter and loaded onto an ÄKTA pure 25 M FPLC system (GE Healthcare/Cytiva, Marlborough, MA). The supernatant was applied to a 5 mL HisTrap HP column (GE Healthcare/Cytiva, Marlborough, MA), and the column was washed with 10 column volumes of PARG Lysis Buffer. PARG was eluted by linearly increasing imidazole concentration using PARG High Imidazole Buffer (250 mM NaCl, 50 mM HEPES pH 7.5, 300 mM imidazole, fresh 1 mM DTT). The fractions containing PARG were pooled, concentrated using an Amicon filter, and centrifuged at 16000 x g for 5 min at 4 °C to remove aggregates.
The concentrated PARG was then run on a Superdex 200 10/300 column (GE Healthcare/Cytiva, Marlborough, MA), which was equilibrated and eluted with PARG SEC Buffer (50 mM HEPES, pH 7.5, 400 mM NaCl, 5 mM DTT). The PARG-containing fractions were pooled, concentrated, and centrifuged at 16000 x g again as described above. PARG protein was aliquoted, flash-frozen with liquid nitrogen, and stored at −80 °C.
Purification of the PARP5a Catalytic Domain
The catalytic domain of PARP5a (residues 1093–1327) was expressed and purified as described previously, with modification (Tan et al., 2012). Bacterial expression vectors for PARP5a include a His tag at the N-terminus. The vectors were transformed into competent BL21(DE3) E. coli cells (New England Biolabs, Ipswich, MA). A 50 mL starter culture was grown overnight at 37 °C. Following overnight incubation, the 10 mL starter culture was added per 1 L culture and grown to an OD600 of ~0.8. The culture was cooled to 4 °C for 1 h, and IPTG was added to a final concentration of 0.5 mM. The cells were incubated overnight at 16 °C with shaking. Next, the cells were pelleted at 5000 x g at 4 °C for 30 min, and the pellet was resuspended in PARP5a Lysis Buffer (300 mM NaCl, 50 mM HEPES pH 8.0, 20 mM imidazole, 10 mM benzamide, 0.5 mM TCEP, 10% glycerol) for storage at −80 °C.
The resuspended cells were thawed, 1X EDTA-free protease inhibitor cocktail was added and NP-40 was added to 1% (v/v). The cells were lysed by sonication and centrifuged at 17000 x g at 4 °C for 30 min. The supernatant was filtered through a 0.22 μm syringe filter and HisPur Ni-NTA resin (Thermo Scientific, Waltham, MA) equilibrated in lysis buffer was added (1.25 mL slurry per L culture, 1 CV). The resin and lysate were gently stirred for 2 h on ice, then the mixture was transferred to a disposable polypropylene gravity column. The resin was washed with PARP5a Low-Salt Wash Buffer (500 mM NaCl, 50 mM HEPES pH 8.0, 20 mM imidazole, 0.5 mM TCEP, 10% glycerol, 5 CV), PARP5a High-Salt Wash Buffer (1 M NaCl, 50 mM HEPES pH 8.0, 20 mM imidazole, 0.5 mM TCEP, 10% glycerol, 5 CV), then PARP5a Low-Salt Wash Buffer (5 CV). The protein was then eluted with PARP5a Elution Buffer (500 mM NaCl, 50 mM HEPES pH 8.0, 300 mM imidazole, 0.5 mM TCEP, 10% glycerol, 5 CV), concentrated to ≤2 mL with an Amicon Spin Filter (10,000 MWCO, Millipore, Burlington, MA). The protein was desalted into Storage Buffer (200 mM NaCl, 50 mM HEPES pH 8.0, 0.1 mM TCEP, 10% glycerol) using a HiTrap Desalting Column (2 x 5 mL, GE Healthcare/Cytiva, Marlborough, MA) hooked up to an NGC chromatography system (Bio-Rad, Hercules, CA). PARP5a was further purified with a Superdex 200 column equilibrated with PARP5 activity buffer (50 mM NaCl, 20 mM HEPES pH 7.5, 0.1 mM TCEP, 5 mM MgCl2, 5% v/v glycerol). Fractions containing pure PARP5 were aliquoted, flash-frozen with liquid nitrogen, and stored at −80 °C.
Kapβ2 Purification
Karyopherin-β2 was expressed and purified as described previously (Guo et al., 2018). To purify GST-TEV-Kapβ2, BL21 DE3 RIL E. coli cells transformed with the appropriate plasmid were grown at 37 ºC in LB supplemented with ampicillin until cells reached an OD600 of ~0.6. Expression was induced overnight at 25 ºC with 1 mM IPTG. Cells were pelleted by spinning at 4000 rpm for 20 min at 4 ºC and resuspended with resuspension buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1mM EDTA, 2mM DTT, 20% glycerol, supplemented with protease inhibitors) before lysing by sonication. Cell lysate was separated by spinning at 16000 rpm at 4 ºC for 45 min. Cell lysate was then loaded onto Glutathione Sepharose® 4 Fast Flow (Cytiva) and incubated in batch at 4 ºC for 90–120 min. Beads were then spun at 4000 rpm for 3 min and washed with 20 CV resuspension buffer. Beads were then incubated with ATP buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 1 mM EGTA, 0.5 mM MgCl2, 5 mM ATP, 2 mM DTT, 20% glycerol, supplemented with protease inhibitors) for 10 minutes at room temperature, then washed with 10 CV ATP buffer. Beads were then washed with 5 CV Buffer A (20 mM imidazole, pH 6.5, 75 mM NaCl, 1 mM EDTA, 2 mM DTT, 20% glycerol). Protein was eluted in batch by incubating 2 CV Buffer A with 20 mM glutathione (pH 6.5) for 30 minutes at 4 ºC before eluting by spinning at 4000 rpm for 5 min. Eluant was then incubated with TEV protease (1:100 molar ratio) overnight at 30 ºC with shaking at 300 rpm. Protein was further purified using a salt gradient (Buffer A and Buffer B (20 mM imidazole, pH 6.5, 1 M NaCl, 1 mM EDTA, 2 mM DTT, 20% glycerol)) over a HiTrap Q HP column (Cytivia). Protein was then collected and frozen in liquid nitrogen and stored at −80 ºC before use.
PAR Preparation and Labeling
PAR was synthesized, purified, and fractionated as described previously (Tan et al., 2012). PAR was labeled with Cy5 and purified as described before (Dasovich et al., 2021). pdPAR was prepared via four steps: alkyne conjugation to PAR, DNA attachment to PAR, Cy3 labeling of DNA-PAR chimera, and annealing (Abraham et al., 2020). Briefly, 1 nmol of PAR in 100mM imidazole-HCl pH 7, 750 mM alkyne linker pH ~7, 100 mM EDC were prepared (total volume 50 μL) and incubated at 20 °C overnight. The reaction mixture was cleaned up using Monarch PCR & DNA Cleanup Kit following the manufacturer’s instructions for oligonucleotide clean-up. Alkyne-PAR was attached to azide-18nt DNA under Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAc). For 10 μL CuAAc reaction, 200 μM DNA, 100 μM PAR, 100 mM phosphate buffer pH 7.4, 1 mM CuSO4, 5 mM THPTA, and 10 mM sodium ascorbate were mixed in a PCR tube and incubated at room temperature for 2 hours. DNA-PAR was purified by ion exchange chromatography on Agilent 1260 infinity II HPLC equipped with Agilent Bio SAX column (NP5, Non-porous, 5 μm, 4.6 x 250 mm), mobile phase A (25 mM Tris-HCl, pH 9), and mobile phase B (25 mM Tris-HCl, 1 M NaCl pH 9). DNA-PAR was eluted at 1 mL/min, 25 °C by a linear gradient of mobile phase B (30% to 80% over 20 min). The DNA-PAR fraction was ethanol precipitated and purity assessed on 15% urea-PAGE. The HPLC purified DNA-PAR chimera was fluorescently labeled at 2’-end of PAR with dATP-Cy3 using ELTA). The details of ELTA method were described previously (Ando et al., 2019). Briefly 20 μL labeling reaction, 20–50 pmol of PAR, dATP-Cy3 (3–5 x of [PAR]), 20 mM Tris-HCl pH 7.5, 20 mM Mg(Oac)2, 2.5 mM DTT, 50 ng/μL of LMW polyIC, 50 ng/μL OAS1 were mixed and incubated at 37 °C, 2 h, 750 RPM. Poly IC was digested over 1 h, 37 °C by RNase R treatment and unreacted dATP-Cy3, OAS1, and salt was removed by Monarch PCR & DNA Cleanup Kit to give DNA-PAR-Cy3 (strand1). Partially complementary biotin-DNA-Cy5 (strand2) and DNA-PAR-Cy3 (strand1) were annealed by mixing them at 1.1:1 molar ratio in 10 mM Tris-HCl, pH 7.5, 100 mM KCl, heating to 95 °C for 2 min, followed by slow cooling (2 °C per min) to room temperature. Annealing was verified by gel electrophoresis on 6% DNA retardation gel.
RNA Preparation
RNA was synthesized, labeled, and annealed as previously described (Niaki et al., 2020). Briefly, RNA was synthesized by Integrated DNA Technologies with 5’ or 3’ amine modifications for labeling with Cy3 or Cy5 NHS esters. Unlabeled RNA was stored as a stock at 1 mM at −20 °C, and 100 μM aliquots were used for preparing droplet and other experiments.
To label the RNA with the NHS esters, 100 μM unlabeled RNA was combined with 0.1 mg Cy3- or Cy5-NHS and 10 mM sodium bicarbonate. Labeling reactions were incubated overnight in the dark at room temperature with rotation. The labeled RNA was purified by performing two successive ethanol precipitations: ice-cold ethanol was added to 70% (v/v) and incubated at −20 °C for 30 min; the RNA was centrifuged at 21000 x g for 30 min at 4 °C; the supernatant fraction was discarded and the pellet was washed twice with 70% (v/v) ethanol; and the pellet was resuspended in 50 μL T50 (10 mM Tris-HCl pH 8.0, 50 mM NaCl) buffer. The purified labeled RNA stock was stored at −20 °C, and 1 μM aliquots were used for most experiments.
RNA annealing between complementary strands was performed by mixing the FRET acceptor (Cy5) and donor (Cy3) strands at a 1.2:1 molar ratio and heating to 95 °C. The RNA was then cooled to 4 °C at a rate of 2 °C/min. RNA stocks were stored at −20 °C, and 10 nM single-use aliquots were prepared for single-molecule experiments. RNA prepared for single-molecule nucleation experiments followed this general protocol but did not use a Cy3-labeled strand because it would interfere with the Cy3-FUS signal.
Protein Labeling
Purified FUS was labeled as described previously (Rhine et al., 2020a). Briefly, four 0.5 mL Zeba Spin Desalting Columns (7K MWCO) columns were washed with 300 μL FUS Labeling Buffer (1 M Urea, 1 M KCl, 1X PBS, 5% (v/v) glycerol) three times at 1500 x g, following the manufacturer’s protocol. FUS was exchanged into this buffer by flowing through two of the Zeba columns. The buffer-exchanged protein was reacted with ~20 μM Cy3-NHS for 45 min in the dark at room temperature with rotation in 0.1 M sodium bicarbonate. Excess dye was removed using the remaining two Zeba columns, and the labeled protein was stored in the dark at 4 °C for up to 2 weeks.
PARP5a catalytic domain was diluted to 2 mg/mL with 0.1 M sodium bicarbonate pH 8.3 in a volume totaling 250 μL. Alexa Fluor 488 TFP ester (ThermoFisher A37563, 0.1 mg) was added and the reaction was stirred at room temperature for 1 hr. PARP5a catalytic domain was separated from excess dye with a 5 mL HiTrap desalting column on an NGC chromatography system (Bio-Rad) equilibrated in PARP5a activity buffer (50 mM NaCl, 20 mM HEPES pH 7.5, 0.1 mM TCEP, 5 mM MgCl2, 5% (v/v) glycerol). The Alexa Flour 488 concentration of the labeled PARP5a was determined with a NanoDrop OneC, then the protein was aliquoted, flash-frozen with liquid nitrogen, and stored at −80 °C.
In Vitro Phase Separation Reactions
All phase separation assays were performed by cleaving the 6x-His and MBP tags from purified FUS proteins with TEV protease as in previous studies (Rhine et al., 2020a; Rhine et al., 2021). Successful cleavage of all proteins was verified with SDS-PAGE and Coomassie staining (data not shown). FUS protein was buffer-exchanged from the elution buffer into 20 mM Na3PO4, pH 8.0, using successive spins in Amicon filters. In general, PAR phase separation reactions used FUS (1 μM), unlabeled PAR (90 nM), and Cy5-labeled PAR (10 nM) in 1X Cleavage Buffer (100 mM NaCl, 50 mM Tris pH 7.4, 1 mM DTT, and 1 mM EDTA, pH 8), unless otherwise indicated. RNA droplet reactions generally used 1 μM RNA and 10 nM Cy3-RNA. For conditions with 10 nM or less PAR, all PAR was Cy5-labeled and there was no unlabeled PAR. Reactions (200 μL) were incubated at room temperature for 4 h in Nunc Lab-Tek 8-well chambers. Images were acquired with a Nikon Ti Eclipse wide-field microscope in the brightfield, Cy3, and/or Cy5 channels.
Fluorescence Recovery After Photobleaching (FRAP)
FRAP experiments were performed by photobleaching entire droplets or granules with a 50 mW bleaching laser (405 nm) and Bruker Galvano mirror scanner as described previously (Niaki et al., 2020). Movies were acquired in either the Cy3 (RNA) or Cy5 (PAR) channels, depending on the experiment. Eight droplets were bleached for each experiment, and the fluorescence recovery was measured over the course of 10 min by acquiring Cy3 or Cy5 images ever 3 s for 2 min then every 10 s for 8 min.
Bead Phase Separation Experiments
Phase separation reactions containing PAR-conjugated beads were performed essentially as described above (see In Vitro Phase Separation Reactions). StreptAvidin-coated 4.0 μm beads (0.1%; Spherotech, Lake Forest, IL) were combined with biotinylated PAR or RNA (100 nM) and/or Cy5-labeled DNA duplex (10 nM). After reacting for 5 min at room temperature, the beads were pelleted by centrifuging at 21000 x g for an additional 5 min. The pelleted beads were washed three times with T50 buffer, resuspended in T50 buffer, and added to phase separation reactions at a final concentration of 0.001% (w/v). We estimated that the local concentration of polymer on the beads was ~10–100 nM, and the total concentration of RNA or PAR in the reaction could not exceed 1 nM based on the initial quantity of polymer reacted with the beads. For experiments in which the droplet reaction was pelleted, droplet reactions were incubated in 1.5 mL Eppendorf tubes for 1 h and pelleted at 21000 x g. The supernatant and pellet were fractionated; after an additional 3 h incubation, droplets were visualized as described above.
PARP5a Catalysis Phase Separation Reactions
All phase separation assays were performed by cleaving the and 6x-His and MBP tags from purified FUS proteins as explained in previous studies (Rhine et al., 2020a; Rhine et al., 2021). In general, PARP5a phase separation reactions used FUS (1 μM), PARP5a (1 μM), and Cy3-labeled FUS (10 nM) with the indicated NAD+ concentration in 1X PARylation Buffer (50 mM NaCl, 20 mM HEPES pH 7.5, 0.1 mM TCEP, and 5 mM MgCl2). Reactions were incubated at room temperature for 4 h. Images were acquired with a DeltaVision CoolSnap HQ microscope in the TRITC and FITC channels. For western blotting, samples used for microscopic visualization were mixed with lithium dodecyl sulfate sample buffer to a final concentration of 1X, separated with 4–8% Bis-Tris gels in MOPS-Tris running buffer, then transferred to PVDF membranes. Membranes were dried, then blocked with 5% non-fat dry milk in TBS-T overnight. Membranes were probed with poly/mono-ADP ribose (Cell Signaling Technology #83732) and anti-FUS (Santa Cruz sc-47711) diluted 1:1000 in 5% non-fat dry milk in TBS-T for 1 h at ambient temperature, then anti-Rabbit IRDye680 RD (Li-Cor Biosciences) and anti-Mouse IRdye800 CW (Li-Cor Biosciences) diluted 1:10,000 in 5% non-fat dry milk in TBS-T for 1 h at ambient temperature. Data were acquired with an Odyssey (Li-Cor Biosciences).
Dissolution of In Vitro Droplets
Phase separation reactions were carried out as described above (see In Vitro Phase Separation Reactions) and incubated at room temperature for 3 h. While acquiring fluorescence microscopy videos with a framerate of 1 fps, dissolution agents were added by pipetting directly into the reaction approximately 5 s after the start of the video. The videos continued for 3 min with frame acquisition slowing to 0.2 fps after the first minute. Each agent was added to the following final concentrations: 5% (v/v) 1,6-hexanediol; 1 μM Kapβ2; 125 μg/mL RNAse A; and 2 μM PARG. Dissolution of FUS–PAR droplets were imaged in the Cy5 channel only, whereas dissolution of FUS–RNA–PAR droplets were imaged in the Cy3 and Cy5 channels.
RNA/PAR Spike Droplets
Phase separation reactions were prepared as described in In Vitro Phase Separation Reactions and incubated for 3 h with RNA (1 μM unlabeled, 10 nM labeled) or PAR (90 nM unlabeled, 10 nM labeled). The other polymer was added to the final concentrations listed above by pipetting directly into the reaction just after the first frame of a 30 min fluorescence microscopy video (1 frame every 10 s).
Optical Trapping and Fusion of Droplets
Droplets were formed as described above (see In Vitro Phase Separation Reactions) and flowed into a C-Trap quad-trap laser setup. Two of the optical traps were used to weakly trap a pair of droplets, which were then moved into a separate flow channel consisting of 1X Fusion Buffer (1X Cleavage Buffer with 10 mM Trolox) supplemented with RNA (1 μM) or PAR (100 nM), depending on the reaction. One of the trapped droplets was slowly moved into the other droplet’s trap until the two droplets either successfully or unsuccessfully fused, as described previously (Rhine et al., 2020a). Fluorescence confocal images were acquired before and after each fusion event.
Electrophoretic Mobility Shift Assays (EMSAs)
Stoichiometric FUS–PAR and FUS–RNA interactions were resolved by EMSAs on 6% polyacrylamide retardation gels as described previously (Sarkar and Myong, 2018). Concentrations of FUS, PAR, and RNA were varied for each well. Most often, increasing concentrations of FUS were added to 1 nM Cy5-labeled PAR in 1X EMSA Binding Buffer (100 mM β-mercaptoethanol, 100 mM KCl, 50 mM Tris, pH 7.4, 2 mM MgCl2, 100 μg/mL BSA). In reactions with RNA, FUS was kept at 500 nM while PAR and Cy3-labeled U40 concentrations were varied. The reactions were covered from light and incubated at room temperature for 1 h. Loading dye was added to the reactions before undergoing electrophoresis at 100 V for ~1.5 h. The gels were imaged on a Typhoon 5 fluorescent scanner (GE Healthcare/Cytiva).
Fluorescence Anisotropy
FUS–RNA or FUS–PAR isothermal binding plots were tested and constructed as described previously (Rhine et al., 2020a). Briefly, anisotropy reactions were performed in 1X EMSA Binding Buffer for 1 h in Thermo Scientific Nunc MicroWell 96-well plates. Increasing concentrations of FUS were added to Cy3-U40 or Cy5-PAR, which was fixed at 10 nM. Fluorescence polarization was measured by a Tecan Spark 10M plate reader.
Competition assays were performed with the same general workflow, but 100 nM FUS was incubated with either 10 nM Cy5-U40 or 10 nM Cy5-PAR16 to achieve complete binding. Increasing concentrations of unlabeled PAR16 or U40 were added to each reaction, respectively, and a decrease in anisotropy was interpreted as a successful displacement of the labeled polymer from FUS.
Turbidity Measurements
Turbidity assays were performed essentially as described previously (Rhine et al., 2020a). Briefly, FUS protein was exchanged into 20 mM Na3PO4, pH 8, as described above (see In Vitro Phase Separation Reactions). FUS (1 μM) and RNA (1 μM) were combined in 1X Cleavage Buffer with TEV protease. PAR was added to 500 nM in applicable reactions. Reactions (100 μL) were prepared in Thermo Scientific Nunc MicroWell 96-well plates, and the A400 value was measured over 2 h in a Tecan Spark 10M plate reader. Turbidity values were normalized to the maximal turbidity for each reaction. The t1/2 value was determined by fitting the turbidity values to a sigmoid curve in Matlab.
Single-Molecule Nucleation
Single-molecule nucleation experiments were performed as described previously (Rhine et al., 2020a; Rhine et al., 2021). Briefly, low-density biotin PEG-passivated slides were purchased from the Slide Production Core for Microscopy at Johns Hopkins Medical Institute. Slides were assembled with 5 lanes using double-sided tape and epoxy, which glued the coverslip and slide together. Reagents were flowed onto the slide in the following order: 1 mg/mL NeutrAvidin, T50 buffer, ~50 pM pdPAR or pdRNA (prepared above), T50 buffer, Imaging Buffer (20 mM Tris-Hcl, pH 7.4, 100 mM KCl, 0.5% (w/v) glucose, 1 mg/mL glucose oxidase, 1.8 U/mL catalase, 10 mM Trolox, 2 U TEV, and 4 U RNase inhibitor), and Imaging Buffer with Cy3-labeled FUS. Long videos of the single-molecule surface (300 s) were obtained to plot Cy3-FUS intensity over time. A short burst of Cy5 exposure at the beginning and end of each video was used to filter out nonspecific binding spots.
Dynamic Light Scattering
FUS was transferred into 20 mM sodium phosphate buffer (pH 7.4). 1 μM of wildtype FUS was used to form droplets with U40 RNA (1μM) or PAR16 (100nM) in 1X Cleavage Buffer. All the components, except RNA and PAR, were filtered by 0.22μM syringe filters before mixing. Samples were incubated in a 96-well plate (Corning®, Product Number 3880). DLS results were acquired immediately after adding TEV protease to the 4-hour time point with an interval of 10 minutes by Wyatt DynaPro Plate Reader II (Wyatt Technology), and the particle sizes were transformed into size distribution data by the Dynamic 7 software (Wyatt Technology). After extracting the distributions into a set of .csv files, a MATLAB script was applied to visualize the data.
Stable Cell Line Production
SH-SY5Y cells cultured without antibiotic were transfected with 800 ng of FUS-Halo and 800 ng of the Super Piggybac Transposase plasmid using Lipofectamine 3000. Two days after transfection, G418 was added to a final concentration of 400 ng/μL. The media was refreshed every 4–7 days until the cells reached confluency and were then cultured as described above.
Live Fluorescence Confocal Microscopy of FUS-Halo Cells
Fibronectin (5 μg/mL) was used to pre-treat 4-well Nunc chambers for 15 min. FUS-Halo cells were passaged into the fibronectin-treated chambers with 500 μL DMEM mixture 2–3 days before imaging so that they would be 60–80% confluent for imaging. Cells were transfected 1 day prior to imaging (see below), if applicable. Sodium arsenite (0.5 mM for all conditions except controls) was added 1 h prior to imaging, and JF549 (25 nM for all stressed conditions except controls) and Hoechst (1 μg/mL) were added 30 min prior to imaging. Cells were washed three times with 1X pre-warmed distilled PBS before adding fresh DMEM supplemented with sodium arsenite for applicable conditions. Cells were imaged in the 408 nm (Hoechst), 488 nm (GFP, when applicable), 550 nm (JF549) channels using an LSM-700 confocal microscope. Digital gain and laser exposure were optimized for each image, and cells were maintained at 37 °C throughout imaging.
shRNA and Plasmid Transfections
Lipofectamine 3000 was used following the manufacturer’s directions. Short-hairpin RNAs (10 nM) and GFP-tagged PARP or PARG mammalian expression plasmids (800 ng) were transfected ~16–24 h prior to imaging.
PARP Inhibitor Treatment
Inhibitors of PARP activity were added 1, 4, or 24 h prior to imaging. To limit intracellular loss of inhibitors, these cells were not washed with PBS prior to imaging. The inhibitors were added at the following final concentrations: Olaparib (10 μM); IWR-1 (5 μM); and G007-LK (1 μM) (Evers et al., 2008; Huang et al., 2009; Voronkov et al., 2013).
Western Blot of FUS Expression
FUS-Halo cells were cultured in 12-well plates to confluency at 37 °C. Cells were treated with shRNA as described above ~16 h prior to harvesting, if applicable. For lysis, the cells were transferred to 4 °C, and the media was removed from the plate. Cells were washed three times with 1X ice-cold dPBS and resuspended in 165 μL NP-40 Lysis Buffer. Lysis occurred on ice for ~10 min. The samples were then transferred to Eppendorf tubes and centrifuged at 21000 x g for 10 min at 4 °C. Following centrifugation, the supernatant fraction from each tube was transferred to a new tube, and the lysate was mixed 1:1 with 2X Laemmli Sample Buffer for electrophoresis. These samples were heated to 100 °C and centrifuged again at 21000 x g for 1 min at room temperature. After loading onto an Any kD TGX Precast Protein Gel, the gel was electrophoresed at 100 V for ~1.5 h. The gel was then transferred to a nitrocellulose membrane in Western Transfer Buffer (10% (v/v) methanol, 25 mM Tris pH 8.3, 192 mM glycine) for 1.5 h at 100 A. The nitrocellulose membrane was treated with primary antibodies at 1:500 dilution in Blocking Buffer (5% (w/v) milk powder, 0.1% (v/v) Tween in 1X dPBS) overnight at 4 °C, secondary antibodies at 1:5000 dilution in Blocking Buffer at room temperature for 2 h, and ECL Blotting Reagent for ~2 min at room temperature with three 1X dPBST (1X dPBS with 0.1% (v/v) Tween-20) washes between each treatment. The membrane was imaged using the “Chemiluminescence” function with high dynamic range on an Amersham Imager 600 RGB.
FUS-Halo IP and PARylation Western Blotting
SH-SY5Y cell lines with endogenous FUS-Halo were passaged onto 100 mm cell culture dishes that were treated with poly-D-lysine (R&D Systems, Minneapolis, MN). All steps were performed on ice or at 4 °C unless otherwise indicated. For each IP, 107 cells were seeded onto five dishes in the DMEM mixture and grown at 37 °C + 5% CO2 for 48 h. The cells were washed with 1X dPBS and then DMEM mixture containing PARG inhibitor, PDD 00017273 (10 μM), sodium arsenite (0.5 mM) was added to a final volume of 10 mL per dish. The cells were incubated at 37 °C + 5% CO2 for 1 hr, then the DMEM mixture was aspirated and cold 1X dPBS containing PDD 00017273 (10 μM), G7-L00K (1 μM) and Olaparib (10 μM) was added (1.5 mL per dish). Cells were harvested with a cell scraper and cells from the five dishes corresponding to each condition were combined in the same tube and pelleted at 400 x g for 3 min. The supernatant was aspirated then 400 μL lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM NaF, 0.5% NP-40, 50 μM Olaparib, 50 μM G7-L00K, 100 μM PDD 00017273, 1X cOmplete protease inhibitor cocktail) was added. The cells were resuspended gently with a pipet then incubated for 30 min. Insoluble material was pelleted at 13,800 x g for 12 min, then the supernatant was transferred to new tubes. Cell extracts were diluted to 1 mL with dilution buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT) then 4 μL was mixed with SDS sample buffer for the input. Remaining extract was incubated with 50 μL of HaloTrap agarose (Chromotek, Planegg-Martinsried, Germany) for 1 h with end-over-end rotation. The samples were transferred to spin columns (Chromotek, Planegg-Martinsried, Germany) and washed three times with 500 μL wash buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.05% NP-40), then 30 μL of SDS sample buffer was added and the columns were incubated at 95 °C for 10 min. IP samples were eluted from the agarose by spinning the columns at 2,500 g for 2 min. Samples were separated with 4–8% Bis-Tris gels in MOPS-Tris running buffer, then transferred to PVDF membranes. Membranes were dried, stained with Revert 700 (Li-Cor Biosciences), then blocked with 5% non-fat dry milk in TBS-T overnight. Membranes were probed with pan-ADP-ribose (Millipore MABE1016) and anti-Halo (Promega G9211) diluted 1:1000 in 5% non-fat dry milk in TBS-T) for 1 hr at ambient temperature, then anti-Rabbit IRDye680 RD (Li-Cor Biosciences) and anti-Mouse Irdye800 CW (Li-Cor Biosciences) diluted 1:10,000 in 5% non-fat dry milk in TBS-T for 1 h at ambient temperature. Data were acquired with an Odyssey (Li-Cor Biosciences).
Immunofluorescence and Super Resolution Imaging of Fixed Cells
SH-SY5Y cell lines with endogenous FUS-Halo were passaged onto Lab-Tek II 4-well chambers. Transfection and arsenite stress was performed as described above (see Live Fluorescence Confocal Microscopy of FUS-Halo Cells). Cells were washed three times with 1X dPBS and fixed with 4% paraformaldehyde for 15 minutes at room temperature. Cells were permeabilized with 1X dPBS supplemented with 0.3% Triton-X100 and 1% BSA. For antibody staining, cells were washed three times with 1X dPBS and antibody was added at the appropriate dilution in permeabilization buffer; fixed cells were incubated overnight at 4 °C with gentle rotation. Following antibody treatment, JF549 and Hoechst were added; the fixed cells were then mounted with Prolong Gold and sealed for imaging. Super resolution imaging was performed on an LSM-800 with an Airyscan attachment. Airyscan processing was performed by Zen Blue (Zeiss), and cell images were further processed as described below (FUS Granule Counting and Cell Image Processing).
QUANTIFICATION AND STATISTICAL ANALYSIS
Processing and Quantification of Droplet Images and Videos
For presentation in the manuscript, Nikon images were converted into TIFF images using Fiji. The brightness and contrast were adjusted in Adobe Photoshop, and the same adjustment was generally applied to all conditions in a figure when appropriate. Certain images with lower concentrations of PAR had increased contrast to better visualize droplets. For multi-channel images, each channel was processed separately before being overlaid.
Droplet statistics were quantified by intensity thresholding using a custom Matlab script or the NIS-Element AR cell counting software (Nikon). Both software packages calculated several droplet characteristics, including droplet count, average area, average circularity, and overall area coverage. Droplet colocalization was calculated by employing a separate Matlab script described previously (Rhine et al., 2020a). Each condition was given a colocalization score by dividing the number of overlapping droplets by the total number of droplets in both fluorescence channels.
FRAP videos were analyzed by a custom Matlab script which was employed in our previous work (Niaki et al., 2020). Regions of interest (ROIs) were manually defined by the user, including background and reference ROIs that corrected for basal photobleaching caused by the Cy3 or Cy5 laser. The average fluorescence intensity of each ROI was calculated, corrected, and normalized. A similar script was used to analyze dissolution and spike videos, in which intensity changes in individual droplets were tracked over time before being normalized as a percentage of their initial value.
Analysis of Optical Trapping Data
The fusion time was calculated essentially as described previously with Matlab scripts (Rhine et al., 2021). Briefly, we converted each frame into a binary image and modeled the fusing droplets as a single ellipse. We then calculated the aspect ratio (the ratio of major axis length to minor axis length) for each frame. The aspect ratio as a function of time was fit to an exponential decay function, starting at the initial aspect ratio before fusion and asymptotically approaching the final aspect ratio of the fully relaxed droplet, to obtain the relaxation time constant for that fusion event. Initial and final aspect ratios were also included as fit parameters.
EMSA and Western Blot Image Processing
Images were processed as described above (Analysis of Droplet Images and Videos). The intensities of individual bands were quantified using ImageJ’s gel analysis function and were normalized to the highest intensity band. For Western Blots, intensities were generally normalized to the tubulin band. Intensity plots were generated using Fiji’s plot gel function, and intensity values were normalized to the highest intensity in each channel. Line plots of multichannel fluorescence intensity were made using Fiji’s plot profile function, and the intensities were normalized to the highest intensity in each fluorescence channel.
Binding Isotherm Plots
Binding isotherms were constructed by plotting anisotropy over concentration of FUS. Kd_app values were determined by fitting each isotherm in Prism 8 with a four-component total binding fit where the ‘NS’ (nonspecific binding) component was constrained to 0. Three independent Kd_app fits were used to generate an average and calculate error.
Analysis of Single-Molecule Data
Single-molecule videos were processed using IDL as described previously (Roy et al., 2008). Processed trace data were visualized with Matlab (Sarkar and Myong, 2018). Nucleation data was analyzed as described previously (Rhine et al., 2020a; Rhine et al., 2021), except that binding events were manually binned into “short” (<1 s) and “long” (>2 s) FUS interactions and the intensity of each binding interaction was also recorded.
FUS Granule Counting and Cell Image Processing
Images of cells were processed for publication as described above (see Processing and Quantification of Droplet Images and Videos). FUS granule counts were determined using a Matlab script. Briefly, this script identified cells by masking the nuclear Hoechst stain and granules through dynamic masking of higher-intensity granules within the cytoplasm. Granules were assigned to the nearest nuclear signal. Nuclei with weak FUS signal, granules that were too distant to be mapped to a nuclear mask, and granules that were too large were all filtered. The number of granules per identified nucleus and the size of each of these granules were calculated by the Matlab script. Intensity profiles of antibody and FUS-Halo intensities were generated as described above in EMSA Image Processing using the plot profile function in Fiji.
Supplementary Material
Supplemental Video 1 (related to Figure 7): Karyopherin-β2 preferentially removes longer polymers from FUS condensates.
Supplemental Video 2 (related to Figure 7): RNase treatment reverts droplets to FUS-PAR-only morphology.
Supplemental Video 3 (related to Figure 7): RNA ejects PAR from pre-formed FUS-PAR condensates.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal FUS/TLS antibody | Santa Cruz Biotech | sc-47711 |
| eIF3b antibody | Santa Cruz Biotech | sc-16377 |
| Mouse monoclonal G3BP1 antibody | Santa Cruz Biotech | sc-365338 |
| Rabbit monoclonal MAR/PAR antibody | Cell Signaling Tech | 83732 |
| Rabbit polyclonal TNKS antibody | Santa Cruz Biotech | sc-8337 |
| Monoclonal Halo-Tag antibody | Promega | G9211 |
| Pan-ADP-ribose binding reagent | Millipore | MABE1016 |
| Mouse anti-alpha Tubulin antibody [DM1α] | Abcam | ab7291 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A21202 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) | Abcam | ab150077 |
| Rabbit anti-Goat IgG (H+L), Superclonal™ Recombinant Secondary Antibody, Alexa Fluor 488 | Invitrogen | A27012 |
| Goat anti-Mouse IgG (H+L) Irdye 800 CW Secondary Antibody | Li-Cor | 926-32210 |
| Goat anti-Rabbit IgG (H+L) Irdye 680 RD Secondary Antibody | Li-Cor | 926-68071 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Scientific | 31430 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Scientific | 31460 |
| Revert 700 Total Protein Stain | Li-Cor | 926-11011 |
| Bacterial and Virus Strains | ||
| E. Coli BL21(DE3) Chemically Competent Cells | Millipore Sigma | CMC0014-20X40UL |
| BL21-CodonPlus (DE3)-RIL Competent Cells | Agilent | 230245 |
| NEB® Turbo Competent E. coli (High Efficiency) | New England Biolabs | C2984H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AcTEV Protease | Fisher Scientific | 12-575-015 |
| Ribonuclease A from bovine pancreas | Millipore Sigma | R6513 |
| cOmplete™ Protease Inhibitor Cocktail | Roche | 11697498001 |
| Glucose Oxidase from Aspergillus niger | Sigma-Aldrich | G2133 |
| Catalase from bovine liver | Sigma-Aldrich | C3155 |
| IGEPAL® CA-630 | Sigma-Aldrich | I8896 |
| Alexa Fluor™ 488 TFP ester | Thermo Fisher | A37563 |
| Cy3 NHS Ester | Cytiva | PA13101 |
| Cy5 NHS Ester | Cytiva | PA15100 |
| Janelia Fluor® 549 HaloTag® Ligand | Promega | GA1110 |
| Rnase Inhibitor, Murine | New England Biolabs | M0314L |
| 1,6-Hexanediol,99% | Sigma-Aldrich | 240117-50G |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Scientific | L3000001 |
| Sodium (meta)arsenite (>90%) | Millipore Sigma | S7400-100G |
| Fibronectin bovine plasma | Millipore Sigma | F1141-1MG |
| Poly-D-lysine | R&D Systems | 3439-200-01 |
| Geneticin™ Selective Antibiotic (G418 Sulfate) | Thermo Scientific | 10131035 |
| NP40 Cell Lysis Buffer | Thermo Scientific | FNN0021 |
| Pierce™ ECL Western Blotting Substrate | Thermo Scientific | 32109 |
| Olaparib | Selleckchem | S1060 |
| G007-LK | Sigma-Aldrich | 504907 |
| IWR-1 | Cayman Chemical | 13659 |
| PDD 00017273 | MedChemExpress | HY-108360 |
| Alkyne linker: (2-[2-(2-Propynyloxy)ethoxy]ethylamine) CAS# 944561-44-8 | TCI | P2225 |
| dATP-Cy3= N6-(6-Aminohexyl)-dATP-Cy3 | Jena Bioscience | NU-835-CY3 |
| EDAC, Hydrochloride | Sigma-Aldrich | 25952-53-8 |
| Tris(3-hydroxypropyltriazolylmethyl)amine | Sigma-Aldrich | 760952-88-3 |
| Experimental Models: Cell Lines | ||
| Human: SH-SY5Y | ATCC | CRL-2266 |
| Human: WT-FUS-Halo SH-SY5Y (GenR) | This paper | N/A |
| Oligonucleotides | ||
| Biotin-18mer: 5’- /biotin/rUrGrG rCrGrA rCrGrG rCrArG rCrGrA rGrGrC/3AmMO/ −3’ | Niaki et al., 2020; IDT | N/A |
| U50-18mer: 5’- /5AmMC6/rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrG rCrCrU rCrGrC rUrGrC rCrGrU rCrGrC rCrA −3’ | Niaki et al., 2020; IDT | N/A |
| U40: 5’- rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rU/3AmMO/ −3’ | Niaki et al., 2020; IDT | N/A |
| Bio-U40: 5’- rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rU/Bio/ −3’ | IDT | N/A |
| DNA-18-mer: 5’- TGG CGA CGG CAG CGA GGC −3’ | IDT | N/A |
| Bio-DNA-18-mer: 5’- /Cy5/GCC TCG CTG CCG TCG CCA/biotin/ −3’ | IDT | N/A |
| PARP1-shRNA-1-F: 5’-rUrGrArCrUrUrGrGrArArGrUrCrArUrCrGrArUrArUrCrUTT-3’ | IDT | hs.Ri.PARP1.13.1 |
| PARP1-shRNA-1-R: 5’-rArArArGrArUrArUrCrGrArUrGrArCrUrUrCrCrArArGrUrCrArUrA-3’ | IDT | hs.Ri.PARP1.13.1 |
| PARP1-shRNA-2-F: 5’-rCrUrGrArArGrGrArGrCrUrArCrUrCrArUrCrUrUrCrArACA-3’ | IDT | hs.Ri.PARP1.13.2 |
| PARP1-shRNA-2-R: 5’-rUrGrUrUrGrArArGrArUrGrArGrUrArGrCrUrCrCrUrUrCrArGrGrU-3’ | IDT | hs.Ri.PARP1.13.2 |
| PARP1-shRNA-3-F: 5’-rGrGrArArGrGrUrArUrCrArArCrArArArUrCrUrGrArArAAG | IDT | hs.Ri.PARP1.13.6 |
| PARP1-shRNA-3-R: 5’-rCrUrUrUrUrCrArGrArUrUrUrGrUrUrGrArUrArCrCrUrUrCrCrUrC-3’ | IDT | hs.Ri.PARP1.13.6 |
| PARP5a-shRNA-1-F: 5’-rGrUrCrArCrArGrArArCrUrGrCrUrArCrUrArArArGrCrATG-3’ | IDT | hs.Ri.TNKS.13.1 |
| PARP5a-shRNA-1-R: 5’-rrArUrGrCrUrUrUrArGrUrArGrCrArGrUrUrCrUrGrUrGrArCrUrU-3’ | IDT | hs.Ri.TNKS.13.1 |
| PARP5a-shRNA-2-F: 5’- rArUrGrArArUrArUrCrArGrCrCrArArUrUrUrCrUrArArAAA-3’ | IDT | hs.Ri.TNKS.13.2 |
| PARP5a-shRNA-2-R: 5’-rUrUrUrUrUrArGrArArArUrUrGrGrCrUrGrArUrArUrUrCrArUrGrU-3’ | IDT | hs.Ri.TNKS.13.2 |
| PARP5a-shRNA-3-F: 5’-rGrArCrArGrArArUrUrGUrUrArCrUrUrArGrArArArArGGA-3’ | IDT | hs.Ri.TNKS.13.5 |
| PARP5a-shRNA-3-R: 5’-rUrCrCrUrUrUrUrCrUrArArGrUrArArCrArArUrUrCrUrGrUrCrArC-3’ | IDT | hs.Ri.TNKS.13.5 |
| Recombinant DNA | ||
| pTHMT/FUSWT (encoding 6xHis-MBP-FUS-WT) | Niaki et al., 2020 | N/A |
| pTHMT/FUSR244C (encoding 6xHis-MBP-FUS-R244C) | Niaki et al., 2020 | N/A |
| pTHMT/FUSG156E (encoding 6xHis-MBP-FUS-G156E) | Niaki et al., 2020 | N/A |
| pTHMT/FUSY>S (encoding 6xHis-MBP-FUS(Y>S)) | Genscript | N/A |
| pTHMT/FUSR>G (encoding 6xHis-MBP-FUS(R>G)) | Genscript | N/A |
| Super piggyBac Transposase expression vector | Systems Biosciences | PB210PA-1 |
| pFUSWT-Halo_piggybac-EF1-Halo | Rhine et al., 2020a | N/A |
| pET24a-PARP5a (1093-1327) | Tan et al., 2012 | N/A |
| pGFP-cPARG102 | Leung et al., 2011 | N/A |
| pSAT1-PARG (318-976) | This paper | N/A |
| GST-TEV-Kapβ2 | Guo et al., 2018 | N/A |
| Software and Algorithms | ||
| Matlab | Mathworks | N/A |
| NIS-Elements Ar Package | Nikon Inc. | N/A |
| Adobe Photoshop CC | Adobe (https://www.adobe.com/products/photoshop.html ) | N/A |
| Prism 8 | GraphPad (https://www.graphpad.com/scientific-software/prism/ ) | N/A |
| ImageJ (Fiji) | NIH (https://imagej.nih.gov/ij/ ) | N/A |
| ImageStudio 5.2 | Li-Cor Biosciences | N/A |
| Zen Blue | Zeiss | N/A |
| Matlab code | This paper | DOI: 10.5281/zenodo.5866686 |
| Other | ||
| Nunc™ Lab-Tek™ Chambered Coverglass | Thermo Scientific | 155361 |
| Nunc™ Lab-Tek™ II Chamber Slide™ System | Thermo Scientific | 154526PK |
| Superdex™ 200 Increase 10/300 GL | Cytiva | 28990944 |
| HisTrap™ Fast Flow Crude | Cytiva | 17528601 |
| HisPur™ Ni-NTA Resin | Thermo Scientific | 88221 |
| Glutathione Sepharose 4 Fast Flow, 100 mL | Cytiva | 17513202 |
| HiTrap™ Q HP | Cytiva | 17115401 |
| Streptavidin Coated Polystyrene Particles | Spherotech | SVP-40-5 |
| DNA Retardation Gels (6%) | Invitrogen | EC63655BOX |
| Any kD™ Mini-PROTEAN® TGX™ Precast Protein Gels, 15-well, 15 μL | Bio Rad | 4569036 |
| Nitrocellulose membranes, roll, pore size 0.45 μm | VWR | 10-6000-02 |
| Amicon™ Ultra-0.5 Centrifugal Filter Unit (10K MWCO) | Millipore Sigma | UFC501096 |
| Amicon™ Ultra-15 Centrifugal Filter Unit (10K MWCO) | Millipore Sigma | UFC901024 |
| Zeba™ Spin Desalting Columns, 7K MWCO, 0.5 mL | Thermo Scientific | 89883 |
| Corning™ 96-Well, Non-Treated, Flat-Bottom, Half-Area Microplate | Fisher Scientific | 07-200-840 |
| High Pure miRNA Isolation Kit | Roche | 5080576001 |
| HiTrap Desalting | Cytiva | 17140801 |
| Monarch PCR & DNA Cleanup Kit | New England Biolabs | T1030S |
Highlights.
Low concentrations (1 nM) of PAR trigger liquid-liquid phase separation of FUS.
Transient FUS–PAR interactions promote FUS–FUS contacts that induce condensation.
Knockdown or catalytic inhibition of PARP5a reduces cellular FUS condensation.
PAR and RNA co-condense with FUS.
Acknowledgements
We thank Drs. Steven Boeynaems and Phil Sharp for feedback on the manuscript. The authors acknowledge technical support from Nathalie Djaja and Shang-Jung Cheng. The LSM microscopes were purchased and maintained by the Johns Hopkins University Integrated Imaging Center, and we thank Erin Pryce, Gabby Vidaurre, and Jonathan Snedeker for assistance with training. Anti-mouse and anti-rabbit secondary antibodies were gifted by Dr. Xin Chen. The super piggybac transposase vector was gifted by Dr. Zhe Liu. We acknowledge funding support from the National Institutes of Health grants F31-NS113439 (K.R.), T32-GM007231 (K.R.), T32-GM080189 (M.D.), F31-NS111870 (C.M.F.), T32-GM008275 (C.M.F), R21-NS090205 (J.S.), R01-GM104135 (A.K.L.L.), RF1-AG071326 (A.K.L.L. and S.M.), and RF1-NS113636 (S.M.), the National Science Foundation grant PHY-1430124 via the Center for Physics of Living Cells (S.M.), the Johns Hopkins Provost’s Undergraduate Research Award (J.Y.), The G. Harold and Leila Y. Mathers Charitable Foundation (J.S.), Target ALS (J.S.), ALSA (J.S.), the Packard Center for ALS Research (J.S.), and the Johns Hopkins Discovery Award (A.K.L.L. and S.M.).
Footnotes
Declaration of Interests
J.S. is a consultant for Dewpoint Therapeutics, Maze Therapeutics, Vivid Sciences, Korro, and ADRx. S.M. is an advisory board member for Molecular Cell. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham R, McPherson RL, Dasovich M, Badiee M, Leung AKL, and Griffin DE (2020). Both ADP-Ribosyl-Binding and Hydrolase Activities of the Alphavirus nsP3 Macrodomain Affect Neurovirulence in Mice. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, and Mittag T. (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MD, Streicher W, Jungmichel S, Nielsen ML, et al. (2015). Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun 6, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando Y, Elkayam E, McPherson RL, Dasovich M, Cheng SJ, Voorneveld J, Filippov DV, Ong SE, Joshua-Tor L, and Leung AKL (2019). ELTA: Enzymatic Labeling of Terminal ADP-Ribose. Mol Cell 73, 845–856 e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103, 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyappan V, Wat R, Barber C, Vivelo CA, Gauch K, Visanpattanasin P, Cook G, Sazeides C, and Leung AKL (2021). ADPriboDB 2.0: an updated database of ADP-ribosylated proteins. Nucleic Acids Res 49, D261–D265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert E, Boeynaems S, Kato M, Guo L, Caulfield TR, Steyaert J, Scheveneels W, Wilmans N, Haeck W, Hersmus N, et al. (2018). Molecular Dissection of FUS Points at Synergistic Effect of Low-Complexity Domains in Toxicity. Cell Rep 24, 529–537 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JM, Holehouse AS, and Pappu RV (2020). Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu Rev Biophys 49, 107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Lanson NA Jr., Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, and Pandey UB (2013). RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet 22, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasovich M, Beckett MQ, Bailey S, Ong SE, Greenberg MM, and Leung AKL (2021). Identifying Poly(ADP-ribose)-Binding Proteins with Photoaffinity-Based Proteomics. J Am Chem Soc 143, 3037–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, and Jankovic J. (2014). The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10, 337–348. [DOI] [PubMed] [Google Scholar]
- Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, and Bozzoni I. (2014). An ALS-associated mutation in the FUS 3’-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun 5, 4335. [DOI] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, Ma Z, Qian B, Deng X, Zhang K, et al. (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res 29, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S. (2019). Matter over mind: Liquid phase separation and neurodegeneration. J Biol Chem 294, 7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers B, Drost R, Schut E, de Bruin M, van der Burg E, Derksen PW, Holstege H, Liu X, van Drunen E, Beverloo HB, et al. (2008). Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 14, 3916–3925. [DOI] [PubMed] [Google Scholar]
- Fahrer J, Kranaster R, Altmeyer M, Marx A, and Burkle A. (2007). Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res 35, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrer J, Popp O, Malanga M, Beneke S, Markovitz DM, Ferrando-May E, Burkle A, and Kappes F. (2010). High-affinity interaction of poly(ADP-ribose) and the human DEK oncoprotein depends upon chain length. Biochemistry 49, 7119–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y, Alam JM, Noshiro D, Mouri K, Ando T, Okada Y, May AI, Knorr RL, Suzuki K, Ohsumi Y, et al. (2020). Phase separation organizes the site of autophagosome formation. Nature. [DOI] [PubMed] [Google Scholar]
- Ghosh SG, Becker K, Huang H, Dixon-Salazar T, Chai G, Salpietro V, Al-Gazali L, Waisfisz Q, Wang H, Vaux KK, et al. (2018). Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. Am J Hum Genet 103, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, and Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, et al. (2018). Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173, 677–692 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, and Miwa M. (2004). Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A 101, 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon TS, Holehouse AS, Rosen MK, and Pappu RV (2017). Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AF, and Shorter J. (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem J 474, 1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama K, Nemoto Y, Ueda K, and Hayaishi O. (1986). Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J Biol Chem 261, 14902–14911. [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, et al. (2018). Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 173, 706–719 e713. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Julicher F. (2014). Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, and Simon A. (2017). Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. [Google Scholar]
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Leung AK (2014). Poly(ADP-ribose): an organizer of cellular architecture. J Cell Biol 205, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, and Chang P. (2011). Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKL (2020). Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol 30, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin FE, Lukavsky PJ, Kazeeva T, Reber S, Hock EM, Colombo M, Von Schroetter C, Pauli P, Clery A, Muhlemann O, et al. (2019). The Solution Structure of FUS Bound to RNA Reveals a Bipartite Mode of RNA Recognition with Both Sequence and Shape Specificity. Mol Cell 73, 490–504 e496. [DOI] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillen-Boixet J, Franzmann TM, et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, Grace CR, Soranno A, Pappu RV, and Mittag T. (2020). Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo M, Bu X, Aoyama K, Kato J, Ishiwata-Endo H, Stevens LA, Kasamatsu A, Wolfe LA, Toro C, Adams D, et al. (2019). PARP1 inhibition alleviates injury in ARH3-deficient mice and human cells. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo M, Kato J, and Moss J. (2013). ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci U S A 110, 18964–18969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG, Shorter J, and Bonini NM (2018a). Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol Cell 71, 703–717 e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Mojsilovic-Petrovic J, Van Deerlin VM, Shorter J, Kalb RG, Lee VM, Trojanowski JQ, Lee EB, and Bonini NM (2018b). Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Rifai OM, and Bonini NM (2019). Poly(ADP-Ribosylation) in Age-Related Neurological Disease. Trends Genet 35, 601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Rifai OM, and Bonini NM (2020). TDP-43, a protein central to amyotrophic lateral sclerosis, is destabilized by tankyrase-1 and −2. J Cell Sci 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min A, Im SA, Yoon YK, Song SH, Nam HJ, Hur HS, Kim HP, Lee KH, Han SW, Oh DY, et al. (2013). RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther 12, 865–877. [DOI] [PubMed] [Google Scholar]
- Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, and Kriwacki RW (2016). Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, and Tycko R. (2017). Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 171, 615–627 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, Hurst M, Lee JC, Koh HR, Guo L, et al. (2020). Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Portz B, Lee BL, and Shorter J. (2021). FUS and TDP-43 Phases in Health and Disease. Trends Biochem Sci 46, 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourfarjam Y, Kasson S, Tran L, Ho C, Lim S, and Kim IK (2020). PARG has a robust endo-glycohydrolase activity that releases protein-free poly(ADP-ribose) chains. Biochem Biophys Res Commun 527, 818–823. [DOI] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EC, Miyashita A, Williams D, Strohl F, et al. (2018). FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 173, 720–734 e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhine K, Makurath MA, Liu J, Skanchy S, Lopez C, Catalan KF, Ma Y, Fare CM, Shorter J, Ha T, et al. (2020a). ALS/FTLD-Linked Mutations in FUS Glycine Residues Cause Accelerated Gelation and Reduced Interactions with Wild-Type FUS. Mol Cell 80, 666–681 e668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhine K, Skanchy S, and Myong S. (2021). Single-molecule and ensemble methods to probe RNP nucleation and condensate properties. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhine K, Vidaurre V, and Myong S. (2020b). RNA Droplets. Annu Rev Biophys 49, 247–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, and Ha T. (2008). A practical guide to single-molecule FRET. Nat Methods 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gomez-Herreros F, Hafezparast M, and Caldecott KW (2014). PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res 42, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar J, and Myong S. (2018). Single-Molecule and Ensemble Methods to Probe Initial Stages of RNP Granule Assembly. Methods Mol Biol 1814, 325–338. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, and Cech TR (2013). RNA seeds higher-order assembly of FUS protein. Cell Rep 5, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, et al. (2013). Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J 32, 1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, and Buchman VL (2014). Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum Mol Genet 23, 5211–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singatulina AS, Hamon L, Sukhanova MV, Desforges B, Joshi V, Bouhss A, Lavrik OI, and Pastre D. (2019). PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Rep 27, 1809–1821 e1805. [DOI] [PubMed] [Google Scholar]
- Svetoni F, Frisone P, and Paronetto MP (2016). Role of FET proteins in neurodegenerative disorders. RNA Biol 13, 1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ES, Krukenberg KA, and Mitchison TJ (2012). Large-scale preparation and characterization of poly(ADP-ribose) and defined length polymers. Anal Biochem 428, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, Taroni F, Kwiatkowski TJ Jr., McKenna-Yasek DM, Sapp PC, et al. (2009). Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73, 1180–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronkov A, Holsworth DD, Waaler J, Wilson SR, Ekblad B, Perdreau-Dahl H, Dinh H, Drewes G, Hopf C, Morth JP, et al. (2013). Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J Med Chem 56, 3012–3023. [DOI] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa T, Ali R, Jiou J, Fung HYJ, Burke KA, Kim SJ, Lin Y, Peeples WB, Saltzberg D, Soniat M, et al. (2018). Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 173, 693–705 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wu YC, Mullane P, Ji YJ, Liu H, He L, Arora A, Hwang HY, Alessi AF, Niaki AG, et al. (2018). FUS Regulates Activity of MicroRNA-Mediated Gene Silencing. Mol Cell 69, 787–801 e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1 (related to Figure 7): Karyopherin-β2 preferentially removes longer polymers from FUS condensates.
Supplemental Video 2 (related to Figure 7): RNase treatment reverts droplets to FUS-PAR-only morphology.
Supplemental Video 3 (related to Figure 7): RNA ejects PAR from pre-formed FUS-PAR condensates.
Data Availability Statement
Microscopy, single-molecule, and other raw data materials are available from the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication at the following DOI: https://doi.org/10.5281/zenodo.5866686.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.