

Abstract
Recent data suggests that (pre)diabetes onset is preceded by a period of hyperinsulinemia. Consumption of the “modern” Western diet, over-nutrition, genetic background, decreased hepatic insulin clearance, and fetal/metabolic programming may increase insulin secretion, thereby causing chronic hyperinsulinemia. Hyperinsulinemia is an important etiological factor in the development of metabolic syndrome, type 2 diabetes, cardiovascular disease, polycystic ovarian syndrome, and Alzheimer’s disease. Recent data suggests that the onset of prediabetes and diabetes are preceded by a variable period of hyperinsulinemia. Emerging data suggest that chromic hyperinsulinemia is also a driving force for increased activation of the hypothalamic-adrenal-pituitary (HPA) axis in subjects with the metabolic syndrome, leading to a state of “functional hypercortisolism”. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia. It also disturbs energy balance by shifting energy fluxes away from muscles toward abdominal fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote abdominal visceral obesity and insulin resistance which are core pathophysiological components of the metabolic syndrome. It is hypothesized that hyperinsulinemia-induced increased activation of the HPA axis plays an important etiological role in the development of the metabolic syndrome and its consequences. Numerous studies have demonstrated reversibility of hyperinsulinemia with lifestyle, surgical, and pharmaceutical-based therapies. Longitudinal studies should be performed to investigate whether strategies that reduce hyperinsulinemia at an early stage are successfully in preventing increased activation of the HPA axis and the metabolic syndrome.
Keywords: hyperinsulinemia, relative hypoinsulinemia, insulin, insulin resistance, insulin receptor, westernized diet, over-nutrition, metabolic syndrome, Cushing syndrome, cortisoluria, CBG, 11β-HSD1
1. Introduction
In the Western populations there is a cluster of metabolic risk factors which include hypertension, glucose intolerance, abdominal obesity, and hyperlipidemia. Since these factors are observed together more frequently than by chance alone, the concept was developed that these factors are interrelated and together produce the metabolic syndrome [1]. The metabolic syndrome increases risks for developing cardiovascular diseases (such as heart attacks and strokes), type 2 diabetes mellitus, and cancer. The metabolic syndrome has its own set of underlying risk factors of which hyperinsulinemia and insulin resistance, defined as a subnormal response (of blood glucose levels) to insulin, are considered the most important. Other risk factors for the metabolic syndrome are abdominal adiposity, physical inactivity, aging, stress, sleep disturbance, circadian disruption, and the Western diet.
For many years, the dogma has been that insulin resistance was the primary etiological factor in the development of obesity, the metabolic syndrome, and type 2 diabetes, and preceded hyperinsulinemia [2,3]: hyperinsulinemia was considered secondary representing a compensatory mechanism to overcome systemic (peripheral) insulin resistance. However, at present there is no satisfactory explanation for how insulin resistance might stimulate insulin secretion, while there are only a few naturally occurring or genetic models of primary insulin resistance, and few diabetes genes are implicated in insulin resistance [3].
The direct contributions of insulin itself in causing or sustaining insulin resistance have received little sustained attention [2]. Recent data place hyperinsulinemia mechanistically upstream of insulin resistance and suggest that insulin hypersecretion, rather than beta cell dysfunction, identifies otherwise normal individuals at risk for type 2 diabetes [4]. After a period of 3 years, Ferrannini et al. found in a prospective study that subjects with higher insulin secretion at baseline were more likely to progress to impaired glucose tolerance or T2D than those with lower insulin secretion [5]. This result, which is consistent with other human studies [6,7,8,9,10], suggests thus that primary insulin hypersecretion is a triggering event in T2D pathogenesis [11,12] (Figure 1). In this alternate concept, hyperinsulinemia precedes impaired insulin secretory function during the conversion from normoglycemia to (pre)diabetes onset: hyperinsulinemia-induced insulin resistance is considered a physiological protective defense mechanism of the body that tries to prevent hypoglycemia and to protect critical tissues from metabolic stress and nutrient-induced injury [12,13,14]. Thus, hyperinsulinemia may be both a result and a driver of insulin resistance [2]. In this latter case, hyperinsulinemia may be the direct consequence of consumption of the “modern” Western diet, overnutrition, decreased hepatic insulin clearance, genetic factors, fetal/metabolic programming, defects in pancreatic β-cells, and loss of pulsatile insulin secretion [12] (Figure 2).
Figure 1.

(A) The traditional model vs. the alternate model of the pathogenesis of insulin resistance. The traditional model (which is a widely held view) posits that insulin resistance leads to hyperinsulinemia, which in time is followed by β-cell dysfunction (left). In the traditional model, insulin resistance (1) precedes hyperinsulinemia (2), which may be followed by β-cell exhaustion and finally frank type 2 diabetes. (B) In the alternate model, hypersecretion of insulin and the resulting hyperinsulinemia (1) primarily cause insulin resistance (2), which may be followed by β-cell exhaustion and finally frank type 2 diabetes (right). Note that in the alternate model hyperinsulinemia is already present when there is still a normal glucose tolerance. Reproduced from [12].
Figure 2.

Factors involved in primary hyperinsulinemia. Consumption of the “modern” Western diet, over-nutrition, genes, defects in pancreatic β-cells, decreased hepatic insulin clearance, loss of pulsatile insulin secretion, and fetal/metabolic programming may increase insulin secretion, thereby causing chronic hyperinsulinemia and hyperinsulinemia-induced insulin resistance. In the alternate concept, hyperinsulinemia precedes impaired insulin secretory function during the conversion from normoglycemia to (pre) diabetes onset: hyperinsulinemia-induced insulin resistance is considered a physiological protective defense mechanism of the body that tries to prevent hypoglycemia and to protect critical tissues from metabolic stress and nutrient-induced injury.
The metabolic syndrome and Cushing syndrome share several features, including abdominal visceral obesity, insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus, hypertension, and hypertriglyceridemia (Table 1). The overlap in clinical characteristics between the metabolic syndrome and Cushing syndrome may be caused by common underlying mechanisms. Earlier studies did not see consistent relationships between plasma cortisol levels and the presence of the metabolic syndrome [15]. However, new data suggest (a mild) hyperactivity of the HPA axis inducing a state of “functional hypercortisolism” in subjects with the metabolic syndrome [16,17,18]. Despite the hyperactivity of the HPA axis, in most subjects with the metabolic syndrome, plasma cortisol concentrations are usually low/normal, while simultaneously urinary free cortisol clearance is increased [16,17,18] (Table 1). It has been suggested that increased cortisol clearance is responsible for the observed low plasma cortisol concentrations in subjects with the metabolic syndrome. With decreased negative feedback at the hypothalamus and the pituitary, low plasma cortisol might in turn result in an enhanced adrenocorticotropic hormone (ACTH)-induced cortisol response [19]. An alternative explanation for the low/normal plasma cortisol concentrations in subjects with the metabolic syndrome might be hyperreactivity of the target cells to cortisol [20].
Table 1.
Overlap and differences in clinical characteristics between the metabolic syndrome and Cushing syndrome.
| Metabolic Syndrome | Cushing Syndrome |
|
|---|---|---|
| Plasma Insulin levels | ↑↑ | ↑ * or ↓ ** |
| insulin resistance | + | + |
| AbdominalObesity | + | + |
| impaired glucose tolerance | + | + |
| Hypertriglyceridemia | + | + |
| Hypertension | + | + |
| HPA Activity | ↑ | ↑ |
| Plasma Cortisol | ↓/N | ↑ |
| 24 h-Free Cortisoluria | ↑ | ↑ |
+ = present; ↑ or ↑↑ = elevated; N = normal; ↓ = decreased; * “relative hypoinsulinemia”: insulin levels are increased but lower than would be expected for the actual level of glucose; ** about 20% of patients with Cushing syndrome will finally develop frank diabetes.
Subjects with endogenous Cushing syndrome and the metabolic syndrome differ significantly with respect to insulin secretion. While hyperinsulinemia in the metabolic syndrome is primary, hyperinsulinemia in endogenous Cushing syndrome is secondary, representing a compensatory mechanism to overcome insulin resistance. Moreover, while hyperinsulinemia is an obligatory finding in subjects with the metabolic syndrome, subjects with endogenous hypercortisolemia (Cushing syndrome) and impaired glucose tolerance show a relative hypoinsulinemia, wherein insulin levels have increased, but less than would be expected for the level of plasma glucose [21]. In subjects with Cushing syndrome as the endogenous hypercortisolemia exacerbates, the relative insulinopenia becomes more paramount, suggesting that relative insulinopenia is caused by cortisol-mediated direct or indirect “toxic” effects on the pancreatic β-cells, which suppresses endogenous insulin secretion [21,22]. As a direct consequence, insulin secretion in subjects with Cushing syndrome will not suffice to adequately control plasma glucose in response to a glucose load [21,22,23] (Table 1). Excess cortisol has negative effects on insulin secretion, insulin sensitivity, and glucose tolerance but does not primarily induce hyperinsulinemia (see below paragraph Effects of glucocorticoids on insulin secretion for more details).
In this paper, data are presented showing that the hyperinsulinemia-mediated increased activation of the HPA axis may play an etiological role in the development of the metabolic syndrome. It is postulated that hyperinsulinemia-induced “functional hypercortisolism” disturbs the energy homeostasis of the body and functions as a physiological defense mechanism, preventing hypoglycemia and protecting muscles and other critical tissues from metabolic stress and nutrient-induced injury by shifting energy from the muscles to visceral adipocytes [12]. Before going into more detail, background information about the HPA axis and insulin will be first provided in the next paragraphs.
2. Regulation of the Hypothalamic-Pituitary-Adrenal (HPA)-Axis in Healthy Subjects
Hypothalamus and pituitary regulate cortisol synthesis and release in healthy subjects (Figure 3). Corticotropin-releasing hormone (CRH) is released by the hypothalamus and stimulates the anterior pituitary to release ACTH. ACTH then acts on the adrenal cortex which promotes the secretion of cortisol from the zona fasciculata. The secretion of cortisol provides a negative feedback loop by inhibiting release of CRH and ACTH from the hypothalamus and anterior pituitary, respectively. The activity of 11 beta-hydroxysteroid dehydrogenase (11b-HSD) plays an important role in extra-adrenal cortisol metabolism [24]. At least two isozymes of 11 beta-HSD exist, which catalyze the interconversion of hormonally active glucocorticoids (cortisol, corticosterone) and their inactive metabolites (cortisone, 11-dehydrocorticosterone) [25] 11β-hydroxysteroid dehydrogenase-2 (11β-HSD2), is mainly expressed in the kidneys and protects the mineralocorticoid receptor from glucocorticoid excess by converting cortisol to cortisone [25]. It thereby promotes the access of aldosterone to the mineralocorticoid receptors in the kidney. The other isoform, 11β-hydroxysteroid dehydrogenase-1 (11β-HSD1), is widely expressed in classic insulin target tissues as the liver, muscle, and adipose tissue.
Figure 3.
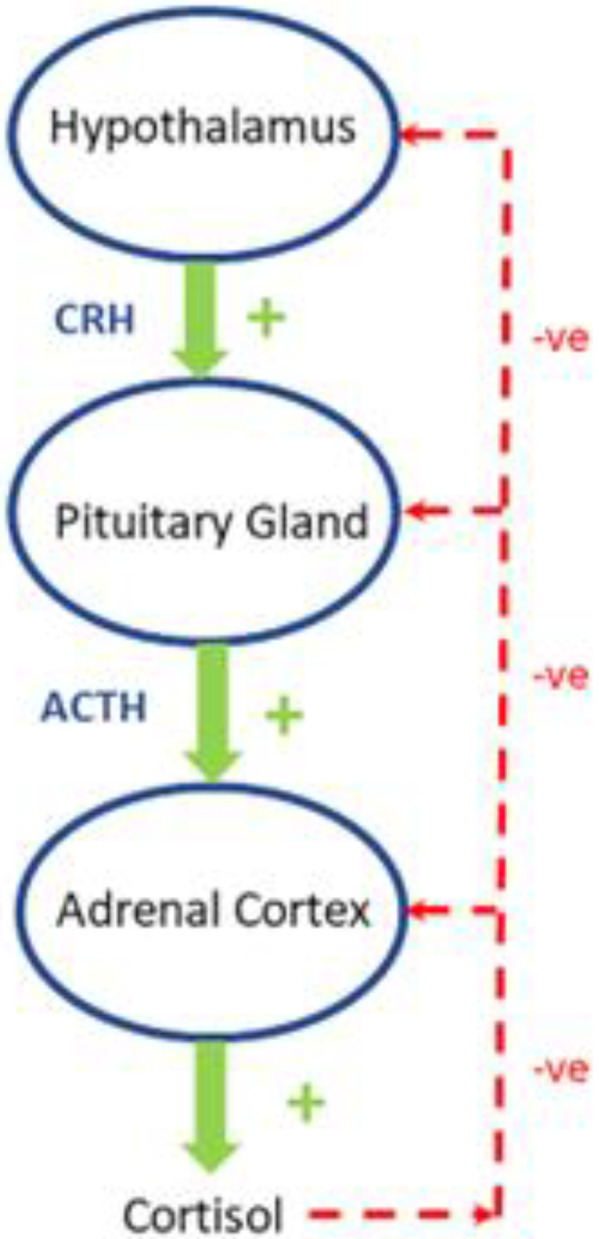
In healthy subjects, corticotropin-releasing hormone (CRH) stimulates the secretion of adrenocorticotropic hormone (ACTH) in the anterior lobe of the pituitary gland. ACTH in turn acts on the adrenal cortex which produces glucocorticoid hormones (mainly cortisol in humans) in response to stimulation by ACTH. Cortisol in turn acts back on the hypothalamus and the pituitary (to suppress CRH and ACTH) production in a negative feedback cycle. Stimulatory effects are shown in green and inhibitory effects are shown in red. See text for more details.
Circulating levels of cortisol are regulated through a balance between synthesis in the adrenal cortex and clearance via metabolic pathways in the liver. An increase in the level of circulating cortisol may be related to stimulation of 11β-HSD type 1 in the liver and/or reduced cortisol clearance by inhibition of 5α- reductase or 5β-reductase [24].
Cortisol metabolism is not only regulated centrally, but is also regulated peripherally. A rise of 11β-HSD1 activity (locally) at the tissue level stimulates conversion of cortisone to cortisol, and in this way cortisol receptors are locally exposed to increased cortisol concentrations [26,27]. Cortisol itself stimulates 11β-HSD1 expression in hepatocytes, adipocytes, and myoblasts [18]. Moreover, other factors which may increase 11β-HSD1 expression in a tissue-specific manner are tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and interleukin-6 (IL-6), whereas insulin, insulin-like growth factor-I (IGF-I), and growth hormone reduce 11β-HSD1 activity [18].
Visceral and omental adipocytes show a higher number of cortisol receptors than subcutaneous adipocytes [28]. In addition, 11β-HSD1 in visceral and omental adipocytes converts locally more cortisone to (active) cortisol than subcutaneous adipocytes [29]. As an immediate result, visceral and omental adipocytes are exposed to higher cortisol concentrations than are present in the circulation and this may contribute to the preferential deposition of fat at intra-abdominal rather than subcutaneous sites [28,30].
3. How Does Cortisol Affect Intermediary Metabolism?
Cortisol is one of the main hormones that regulate fuel homeostasis. After uptake of free cortisol from the circulation, cortisol exerts its effects by binding to intracellular cortisol receptors [31]. Cortisol increases availability of potential fuel substrates by mobilization of glucose, free fatty acids, and amino acids from the body energy stores (adipose tissue, muscle protein, and glycogen) and thereby stimulates energy expenditure [32].
The peripheral effects of cortisol (i.e., outside the central nervous system (CNS)) are primarily catabolic [9]. Cortisol enhances hepatic gluconeogenesis, inhibits glucose uptake in muscles, and increases circulating free fatty acid levels by increasing adipose tissue lipolysis [33].
Under experimental conditions, the amplitude of the diurnal variations in insulin was significantly related to amplitude of cortisol rhythm, suggesting that cortisol day rhythm may mediate, at least partially, the diurnal variation in carbohydrate tolerance [34]. It has been suggested that cortisol induces increased insulin resistance and in this way may contribute to development of impaired glucose tolerance and the metabolic syndrome [35].
Control of food intake is complex and involves numerous brain neurotransmitters and central and peripheral neural structures. Centrally, the HPA axis is believed to interact with the hypothalamic neurotransmitters mediating effects on nutrient intake [36]. In particular, central effects mediated by CRH and cortisol are primarily anabolic and increase appetite and caloric and food intake [36,37,38]. Elevated cortisol in situations of chronic stress increases appetite and intake of especially nutrient dense food and this makes it possible to quickly replace nutrients that are lost during a fight or flight acute stress response [39].
4. How Does Insulin Affect Intermediary Metabolism?
Insulin is considered the master regulator of metabolism in the body [40]. Peripheral actions of insulin (i.e., outside the CNS) are primarily anabolic and stimulate energy storage [29]. In the liver, insulin stimulates glucose utilization, glycolysis, and glycogenesis, and it downregulates glucose production by suppressing gluconeogenesis and glycogenolysis [41]. In the skeletal muscles, insulin facilitates uptake of glucose and amino acids from the bloodstream [42]. Subsequently, glucose is mostly utilized in glycolysis in order to produce energy in the form of ATP which is thereafter used for functional protein synthesis [42]. Insulin converts glucose in muscles to glycogen and further stimulates lipogenesis by promoting uptake of fatty acids into adipocytes. These fatty acids are subsequently converted into triglycerides, which function as long-term energy sources [42]. Simultaneously, insulin inhibits protein degradation (proteolysis) and suppresses lipolysis and ketogenesis.
The circumventricular organs (which include the median eminence and adjacent neurohypophysis) are structures that permit substances that do not cross the blood brain barrier to trigger changes in brain function [43]. Uptake of plasma insulin into the central nervous system (CNS) is mediated by a saturable mechanism consistent with insulin binding to blood-brain barrier and/or blood-cerebrospinal fluid (CSF) barrier insulin receptors and subsequent transcytosis into the CNS [44]. This transport process may be subject to regulation by endogenous factors and interventions that reduce efficiency of insulin uptake in in the CNS [45]. Because the bulk of brain glucose uptake is not affected by insulin in humans, the brain had long been considered “insulin insensitive” [46,47,48]. While there is evidence for glucose transport with the insulin-sensitive GLUT4 in a few selected nuclei in the brain (see below), glucose transport into most neurons is GLUT3-dependent, while the glia and brain endothelial cells depend on GLUT1 activity for glucose uptake from brain interstitial fluid (ISF) and plasma, respectively [49]. Thus insulin does not play a major role in glucose transport into most brain cells [46]. However, insulin functions as a key afferent signal to the CNS for the control of energy balance [50]. In the CNS, insulin has primarily catabolic effects by inhibiting food intake and thereby energy acquisition. In addition, insulin actions in the hypothalamus increase outflow of the sympathetic nervous system to brown adipose tissue and this increases energy expenditure and production of heat by stimulating fatty acid oxidation [50]. In addition, when insulin is infused into the third cerebral ventricle, this induces a sharp decline of hepatic glucose production (HGP) without any changes in circulating insulin levels or the so called counterregulatory hormones [51]. Central insulin signaling suppresses HGP by activating Kupffer cells in the liver to release interleukin-6 (IL-6). IL-6 in turn acts via phosphorylation of signal transducer and activator of transcription-3 (STAT-3) in the liver leading to the suppression of hepatic glucose production [52,53].
The insulin-regulatable glucose transporter, GLUT-4, is expressed in the hypothalamus [54]. The hypothalamus lays outside the blood brain barrier and is therefore sensitive to circulating insulin [54]. It has been further reported that hyperinsulinemia under normoglycemic conditions increases local glucose utilization significantly in the ventromedial, dorsomedial, and anterior hypothalamic nuclei, suggesting that these nuclei are physiologically activated in response to hyperinsulinemia [55]. It has been further proposed that insulin-stimulated uptake of glucose into areas of the hypothalamus represents a form of metabolic feedback regulation, allowing circulating insulin and glucose (with other mechanisms) to maintain blood glucose homeostasis [54].
5. The Bidirectional Interactions between Insulin and the HPA Axis
In healthy subjects, insulin normally shows a reciprocal relationship with cortisol: insulin inhibits food intake while cortisol stimulates food intake [56]. Insulin and cortisol are major antagonistic regulators of energy balance. Effects of cortisol and insulin on food intake may be mediated through regulation of hypothalamic neuropeptide-Y (NPY) synthesis and secretion [56]. In the arcuate nuclei, insulin inhibits and cortisol stimulates the expression of NPY mRNA, which may explain in part the reciprocal actions of insulin and the HPA axis on energy acquisition during the day. It has been further suggested that inhibition of insulin transport across the blood brain barrier by glucocorticoids could be the basis for the enhanced appetite seen with glucocorticoid treatments [57].
6. Effects of Glucocorticoids on Insulin Secretion
Healthy subjects in the Japanese general population with serum cortisol levels within the normal range showed a significant negative relationship between serum cortisol levels and insulin secretion [58]. When healthy subjects were divided in three equal tertiles based on serum cortisol levels, subjects in the highest tertile were at greater risk of decreased insulin secretion than subjects in the lowest tertile [58]. Further adjustments for insulin resistance confirmed this negative relationship [58]. Overall, these findings suggest that cortisol levels (even within the normal range) suppress insulin secretion [46].
Decreased insulin release by pancreatic beta cells is observed following prolonged exposure (days) to glucocorticoids [59,60,61]. Chronic exposure to high circulating glucocorticoid levels inhibits insulin release by binding to glucocorticoid receptors present on pancreatic beta cells [23]. Glucocorticoids reduce the uptake and metabolism of glucose in pancreatic β-cells by genomic actions (i.e., modulation of gene expression by binding of glucocorticoids to the nuclear glucocorticoid receptors). As an immediate consequence, efficacy of cytoplasmic Ca2+ on the exocytosis of insulin granules in the pancreatic β-cells will decrease [59]. Glucocorticoids may further impair pancreatic β-cells glucose metabolism by reducing expression of glucose transporter GLUT-2 and glucokinase thereby decreasing glucose uptake and phosphorylation of pancreatic β-cells [61]. As a direct result, both ATP synthesis and calcium influx, considered essential for a normal pancreatic insulin secretion, decrease and insulin levels after a glucose load will become insufficient to maintain normal plasma glucose levels. Chronic exposure to glucocorticoids may further reduce the insulinotropic effects of glucagon-like peptide-1 (GLP-1) [62]. Thus, with several mechanisms, glucocorticoids may inhibit pancreatic β-cell mediated insulin secretion and thereby induce a “relative hypoinsulinemia”. In addition, glucocorticoids may induce insulin resistance (see next paragraph Glucocorticoids, insulin resistance and metabolism). As long as pancreatic insulin secretion is sufficient, cortisol-mediated increase of insulin resistance and hepatic glucose production will not materially affect glucose tolerance. However, failure of the pancreas to mount an adequate compensatory insulinemic response (due to cortisol-mediated suppression of pancreatic insulin release) may lead to hyperglycemia and impaired glucose tolerance (Figure 4).
Figure 4.

Effects of (high) glucocorticoids on insulin, insulin resistance, and glucose metabolism. Cortisol directly suppresses pancreatic insulin release. It also reduces glucagon-like peptide-1 (GLP-1) production, which further decreases insulin secretion. Increased cortisol also induces glycogenolysis and the expression of key gluconeogenic enzymes, which will increase hepatic glucose production and release. In addition, cortisol may impair insulin receptor signaling (at the receptor and post-receptor level) and thereby induce insulin resistance. Failure of the pancreas to mount an adequate compensatory insulinemic response (“relative hypoinsulinemia” due to cortisol-mediated suppression of pancreatic insulin release) with cortisol-induced insulin resistance may lead to hyperglycemia and impaired glucose tolerance. However, if insulin secretion is sufficient to overcome cortisol-mediated insulin resistance, cortisol will not materially affect glucose tolerance.
7. Glucocorticoids, Insulin Resistance and Metabolism
Glucocorticoids are associated with insulin resistance and antagonize (counteract) insulin-mediated uptake and utilization of glucose in adipose tissue and muscles [63,64]. These latter effects are mediated by multiple mechanisms: glucocorticoids translocate glucose transporters from the plasma membrane thereby reducing insulin-mediated glucose uptake [64]. In addition, glucocorticoids decrease insulin receptor affinity, induce post-insulin receptor defects by decreasing key mediators of insulin action in peripheral tissues as insulin receptor substrate-1 (IRS-1), phosphatidylkinaseinositol-3 kinase, and protein kinase B [59,65,66]. The liver seems particularly vulnerable to the negative effects of glucocorticoids on insulin actions. In the liver glucocorticoids promote gluconeogenesis by inducing phosphoenolpyruvate carboxykinase [18]. Glucocorticoids further increase gluconeogenesis by potentiating actions of insulin-antagonistic hormones like glucagon [65].
Skeletal muscles play an important role in glucose metabolism. Immediately after a meal, skeletal muscles are responsible for approximately 80% of glucose uptake from the circulation. The skeletal muscles contain the body’s largest glycogen stores. Peripheral skeletal muscle atrophy is a classic sign of prolonged (chronic) exposure to relative high levels of glucocorticoids. When there is peripheral skeletal muscle atrophy due to chronic exposure to relative high levels of glucocorticoids, size of glycogen stores as well as glucose uptake from the circulation will be reduced. In addition, excess glucocorticoids decrease insulin-stimulated glycogen synthase kinase-3 phosphorylation in skeletal muscles which may result in decreased glycogen synthesis [67].
Glucocorticoids further influence insulin sensitivity by modifying lipid metabolism. Adipose tissues from various sites of the body show marked differences in metabolism and sensitivity to glucocorticoids. Glucocorticoids induce hormone sensitive lipase and inhibit tissue lipoprotein lipase activity in peripheral subcutaneous fat cells [68]. This induces an increased fat mobilization from the subcutaneous fat depots. Despite these glucocorticoid-mediated effects on lipolysis, the net effect of glucocorticoid excess is a relocation of fat depots since glucocorticoids simultaneously enhance fat deposition in the visceral compartment [69]. Synergistic effects of glucocorticoids and insulin in visceral adipose tissue induce differentiation and proliferation of fat precursor stem cells and increase tissue lipoprotein lipase [70]. These synergistic effects may contribute to a selective increase of abdominal fat cells and central adiposity, which—as discussed previously—are classical characteristics of both the metabolic syndrome and Cushing syndrome [70]. Thus, contrary to the prevailing opinion that cortisol causes insulin resistance (in all tissues of the body), increased visceral adiposity may be in fact due to synergistic effects of insulin and cortisol on fat cells [35]. Visceral adipose tissue is less sensitive to antilipolytic actions of insulin than subcutaneous adipose tissue. Reduced insulin sensitivity may lead to the release of free fatty acids from visceral fat depots and thereby increase plasma free fatty acid levels. This will further exacerbate insulin resistance and contribute to decreased insulin-mediated glucose uptake and disposal in muscles.
8. The Biologically Defended Level of Glycemia in Health and Disease
The biologically defended level of glycemia (BDLG) is determined by the balance between rates of glucose appearance into and disappearance from the circulation, and imbalance in these rates contributes to an elevated BDLG [4]. In health, acute deviations from the BDLG are counteracted by both insulin-dependent and insulin-independent mechanisms that restore blood glucose levels into the normal range [4]. A rise in blood glucose levels stimulate an increased glucose-stimulated insulin secretion, while conversely a fall in blood glucose levels triggers adaptive neuroendocrine and autonomic counterregulatory responses, including an increased HPA activity and cortisol secretion [4]. However, a gradual and pathological increase in the BDLG may occur during the development of type 2 diabetes [4]. As a consequence, the lower boundary of the BDLG is increased and when glucose falls induction of adaptive neuroendocrine and autonomic counterregulatory responses (including an increased HPA activity), occurs already at a higher plasma glucose level. In healthy subjects a rise in plasma cortisol was observed when blood glucose concentrations decreased from 5.5 to 3.5 mmol/L, while in diabetic subjects a rise in plasma cortisol was already observed when plasma glucose decreased from 13 to 5.8 mmol/L [71,72]. It has been further found that the glycemic threshold for triggering counterregulatory responses including the HPA activity (presumably, the level at which the brain perceives a glucose deficit) is elevated by 40% or more in people with T2D compared to nondiabetic individuals [4,73,74]. This may also explain why in routine clinical practice T2D patients frequently report feelings of hypoglycemia when measured plasma glucose levels are within the normal range [73]. Although the underlying mechanism(s) for the increase in glycemic threshold in T2D remains to be elucidated, we hypothesize that hyperinsulinemia may be involved in the gradual and pathological increase in the BDLG during the development of type 2 diabetes (see next paragraphs below for more details).
9. Hyperinsulinemia Induces a State of “Functional Hypercortisolism”
As previously discussed, numerous recent data are supportive of the concept that hyperinsulinemia per se is primary and causes insulin resistance [3,11] (Figure 1). In this alternate concept, insulin resistance is proposed to be a physiological defense mechanism of the body preventing hyperinsulinemia-induced hypoglycemia and protecting against overstimulation of target tissues from metabolic stress and nutrient-induced injury [12,13,14]. This concept is even more interesting against data suggesting that hyperinsulinemia per se directly stimulates activation of the HPA axis and thereby induces a state of “functional hypercortisolism”. This “functional hypercortisolism” may help to antagonize insulin actions and in this way prevent both hypoglycemia and overstimulation of target tissues from nutrient-induced injury (see next paragraph entitled Evidence for insulin regulation of the HPA activity below for more details).
10. Evidence for Insulin Regulation of the HPA Activity
Most people are familiar with the central interactions between insulin and the HPA axis during hypoglycemia. From a physiological perspective, activation of the HPA axis during hyperinsulinemia may develop to prevent an impending fall in blood glucose. Increased release and actions of cortisol after hyperinsulinemia may prevent hypoglycemia by cortisol-mediated effects and enlarging the body’s sensitivity to other counterregulatory hormones [75]. In addition, it was further shown that epinephrine and norepinephrine levels progressively increased with increasing doses of insulin, suggesting that insulin also plays a direct role in stimulating release and effects of catecholamines to prevent hypoglycemia in dogs [76]. The insulin-mediated increase of epinephrine and norepinephrine cause—like cortisol— an elevation of plasma glucose levels in normal humans due to an epinephrine- and norepinephrine-induced suppression of endogenous insulin secretion and a direct inhibitory effect on insulin-stimulated glucose utilization [77].
Hyperinsulinemia influences many aspects of cortisol metabolism at both a central and a peripheral level (Figure 5). A rise in plasma insulin during euglycemia (i.e., during normal plasma glucose levels) appears to moderately stimulate activation of the HPA axis in both normal and diabetic animals [78]. During hyperinsulinemic-euglycemic glucose clamps, insulin alone increased HPA activity in normal and diabetic rats by stimulating CRH, vasopressin mRNA, plasma ACTH, and corticosterone [78]. This response was further accompanied by increased glucocorticoid receptor mRNA expression in the anterior pituitary and paraventricular nucleus of the hypothalamus [78]. However, these insulin-mediated effects appeared dose-dependent since the HPA axis was only clearly stimulated during hyperinsulinemia but not when insulin was administered in physiological concentrations [78].
Figure 5.

Hyperinsulinemia influences many aspects of cortisol metabolism at both the central and the peripheral level. Interactions are shown between hyperinsulinemia, HPA axis activity, 11β-HSD, sensitivity of adrenals to ACTH, SF-I activity (adrenals), CBG (liver), and adrenal cellular proliferation (See text for more details). Stimulatory effects are shown in green (dark green: central effects; light green: peripheral effects) and inhibitory effects are shown in red. ↑ = increased; ↓ = decreased. List of abbreviations. HPA = hypothalamus-pituitary-adrenal axis activity; CBG = cortisol binding globulin; ACTH = adrenocorticotropic hormone; SF-1 = steroidogenic factor 1; 11β HSD = 11 beta-hydroxysteroid dehydrogenase.
During hyperinsulinemic-euglycemic-clamp studies in animal studies, glucose utilization in the medial basal hypothalamus, locus coeruleus, and motor cortex was found to be decreased (despite normal plasma glucose levels) [79]. The decreased glucose utilization in these brain regions induced marked elevations of serum corticosterone levels [79].
Only a few numbers of studies have investigated effects of insulin on the HPA axis in humans. Fruehwald-Schultes et al. reported that in human subjects acute hyperinsulinemia under euglycemic conditions induced an increase in plasma ACTH and cortisol concentrations in a similar way as previously reported in animal studies [80]. Comparable to animal studies, stimulatory effects of insulin on the HPA axis activity were also only found when relatively high (supra-physiological) doses of insulin were administered, but were absent during administration of relatively low insulin doses [80].
As mentioned previously, circulating insulin can cross the blood brain barrier and thereby exert direct effects at the hippocampus, the hypothalamus, and the pituitary [80]. In normoinsulinemic conditions, the hippocampus inhibits the HPA axis and thereby prevents excess cortisol release [81] (Figure 6A). However, in hyperinsulinemic conditions the inhibitory activity of the hippocampus on the HPA axis is decreased (releases “the brake”). Consequently, the homeostatic setpoint (the predetermined level) of the HPA axis activity is set to a higher level: cortisol secretion per 24 h increases (compared to normoinsulinemic conditions), thereby inducing a state of “functional hypercortisolism” [81,82] (Figure 6B,C). The exaggerated cortisol secretion per 24 h during hyperinsulinemia can be explained by increased forward drive to the HPA axis, and/or reduced sensitivity of the HPA axis to negative feedback by cortisol [16,83,84,85] (Figure 6C).
Figure 6.


Insulin can cross the blood brain barrier and exert direct effects at the hippocampus. In normoinsulinemic conditions, the hippocampus has inhibitory control over the HPA axis preventing excess cortisol release. Cortisol in turn has negative feedback on the HPA activity and positive feedback on the hippocampus (A). In hyperinsulinemic conditions the inhibitory activity of the hippocampus on the HPA axis is decreased (releases “the brake”). Consequently, the homeostatic setpoint (the predetermined level) of the HPA axis activity will change: cortisol secretion per 24 h increases (compared to normoinsulinemic conditions) and thereby induces a state of “functional hypercortisolism” (B). The exaggerated cortisol secretion per 24 h during hyperinsulinemia can be explained by increased forward drive to the HPA axis, and/or reduced sensitivity of the HPA axis to negative feedback by cortisol (C). Stimulatory effects are shown in green and inhibitory effects are shown in red. See text for more details.
Both birth and infant weights have been found to be inversely associated with the prevalence of impaired glucose tolerance, the metabolic syndrome, type 2 diabetes, and polycystic ovary syndrome later in life [86]. In addition, it has become clear that intra-uterine environment may also have long-lasting and permanent effects on the activity of the HPA axis [87,88,89]. Urinary excretion of total glucocorticoid metabolites in a population sample of 9-year-old children was higher in children who had a low birthweight [87]. In a study in 60- to 70-year-old men, fasting plasma cortisol concentrations were inversely related to birth weight and body mass index [88]. Furthermore, it was shown that pituitary and adrenal progenitor cells exposed to hyperinsulinemia are metabolically primed to a hyper-functional state of the HPA axis and enhanced cortisol production later in life [89]. This latter mechanism might explain how hyperinsulinemia early in life increases the risk for developing the metabolic syndrome in adulthood [89].
11. HPA Activity in Obesity and the Metabolic Syndrome
Earlier studies proved that the total daily cortisol production rate is somewhat enhanced in obesity and the metabolic syndrome [90,91,92,93]. This is further supported by data showing that the HPA axis responsiveness in obesity, particularly when there was visceral obesity, was enhanced to stimuli such as acute stress, hypoglycemia, a standard meal, or CRH/arginine vasopressin [83,91,94]. In addition, ACTH hyper-responsiveness after CRH/AVP stimulation also appeared to be related to hyperinsulinemia, although the exact underlying mechanism(s) of this relationship is unclear [94].
Increased adipose tissue 11b-HSD-1 expression and activity is also found in human obesity [83]. In subjects with (central) obesity, contrary to those with Cushing syndrome, basal plasma cortisol levels are often not elevated [83]. Despite hyperinsulinemia-mediated increase in cortisol production in overweight and obese subjects, plasma cortisol levels are either similar to or lower than those in non-obese individuals, suggesting that there is also an increased plasma cortisol clearance [95,96]. Insulin-mediated changes in cortisol binding globulin (CBG) concentrations may play a role in the increased plasma cortisol clearance [95,96]. It has been found that plasma CBG levels were negatively related with the insulin response to a glucose load and this relationship was independent of insulin sensitivity [97]. Hyperinsulinemia might decrease CBG liver production and/or increase CBG plasma clearance rate or may have both effects [97]. A decrease in CBG plasma levels may increase plasma cortisol clearance and act to amplify the availability of metabolically free cortisol at peripheral target organs at the tissue level [98]. The reduction in plasma levels of cortisol caused by CBG might decrease feedback inhibition on the hypothalamus and/or pituitary. This in turn would cause upregulation of the HPA axis with increased cortisol synthesis in the adrenals. Thus hyperinsulinemia-induced decrease in plasma CBG levels may help to explain why in subjects with hyperinsulinemia and (central) obesity, lowered plasma total cortisol levels are found despite the presence of an increased HPA activity and an increased total daily cortisol production [95]. However, there are likely to be several other mechanisms involved in the increased HPA activity, such as the negative feedback effects of cortisol on the HPA axis and the tissue selective pleotropic effects of hyperinsulinemia (see below).
12. Direct Effects of Insulin on the Adrenals
Penhoat and colleagues showed insulin receptors in bovine adrenal fasciculata cells [99]. Furthermore, they demonstrated in a series of in vitro experiments that insulin enhances the steroidogenic and cAMP response to ACTH [99]. The enhanced steroidogenic responsiveness of insulin-treated cells was related to an enhanced capacity to produce pregnenolone and an increased activity of several steroid hydroxylases [99]. Thus, by increasing local sensitivity of the adrenals to ACTH, insulin may contribute in the adrenals to enhanced adrenocorticotropic effects (Figure 5). It has been further postulated that insulin may directly influence adrenal steroidogenesis independent of ACTH actions. It was demonstrated that insulin regulates steroidogenesis in the adrenals by activating steroidogenic factor 1 (SF-1) and its steroidogenic target genes important for steroid hormone synthesis through a mechanism independent of the canonical CRH/ACTH/MC2R/PKA pathway and its adrenocorticotropic effects [100] (Figure 5). Thus, insulin may be directly involved, at least in part, in increased cortisol production rate by the adrenal glands (see also below, paragraph Effects of insulin on local cortisol metabolism).
Cross-sectional studies suggest a significant association between hyperinsulinemia and the rising number of adrenal incidentalomas/tumors [101]. Hyperinsulinemia may be responsible for adrenal cellular proliferation and tissue proliferation and exerts its mitogenic effects directly by acting on the insulin receptors or, indirectly, by reducing the production of IGFBP-1 in the liver and thereby increasing the bioavailability of free IGF-I [102] (Figure 5). Interestingly, adrenal gland volume has been suggested as a marker of HPA axis activity reflecting chronic cortisol burden and it was recently reported in a population-based cohort study that mean adrenal gland volume measured by MRI was significantly greater in subjects with prediabetes and diabetes than in healthy controls [103,104].
13. Effects of Insulin on Local Cortisol Metabolism
Cortisol metabolism is not only centrally regulated. As discussed previously, in target tissues local cortisol concentrations are tissue-specific controlled at the prereceptor level through 11b-HSD1, which interconverts hormonally inactive cortisone to active cortisol. In this way local tissue cortisol concentrations are regulated independent of circulating cortisol concentrations [105]. Insulin is probably one of the main factors that exert effects on local cortisol metabolism at the prereceptor level by dynamically regulating 11β-HSD1 [106,107]. Data from transgenic mice selectively over-expressing 11βHSD-1 in adipocytes provide evidence for a causal role of 11βHSD-1 in the development of visceral obesity and the metabolic syndrome [108]. Conversely, 11βHSD-1 knockout mice have lower glucocorticoid levels in adipose tissue and are protected from dietary-induced obesity and show an improved lipid profile, hepatic insulin sensitization, and a potentially atheroprotective phenotype [109].
Through a posttranscriptional mechanism that involves activation of the p38 MAPK signaling pathway, insulin stimulates adipocyte 11β-HSD1 activity and expression in concentration-dependent manner [107]. Hyperinsulinemia-induced expression of 11β-HSD1 may stimulate local cortisol secretion in adipocytes through enhanced conversion of cortisone to cortisol and in this way may contribute to increased cortisol concentrations in vivo [110] (Figure 5). The increased cortisol secretion will stimulate glucocorticoid receptor activation and promote central adiposity [106,107,111,112].
In contrast, in the liver, hyperinsulinemia suppresses 11β-HSD1 expression and activity, leading to a decreased hepatic conversion of cortisone into cortisol [111]. It has been hypothesized that decreased hepatic conversion of cortisone into cortisol due to hyperinsulinemia-suppressed 11β-HSD1 activity in the liver represents a physiological mechanism to improve hepatic insulin sensitivity and to lower fasting plasma glucose levels [111].
14. Can Hyperinsulinemia-Induced Activation of the HPA Axis Be Modified?
A number of different treatment approaches can prevent, reduce, and reverse hyperinsulinemia and insulin resistance, obesity, and metabolic syndrome, regardless of whether hyperinsulinemia or insulin resistance “comes first” (i.e., calorie restricted, low carbohydrate diets and gastric bypass surgery). What needs to be further investigated is the effect of these interventions on the HPA axis and “functional hypercortisolism” hypothesis as outlined in this manuscript. This would help elucidate the role of hyperinsulinemia and cortisol in the pathophysiology of the metabolic syndrome.
At this moment it is unclear whether there is a successful strategy to modify hyperinsulinemia-induced “functional hypercortisolism” in subjects prone to develop the metabolic syndrome. Most studies in humans have not shown consistent effects of energy restriction on activation of the HPA axis. It has been reported that long-term moderate energy restriction in (obese) humans resulted in increases, decreases, as well as no change in HPA axis function [113,114,115,116,117,118].
Sodium–glucose cotransporter 2 (SGLT2) inhibitors may also indirectly reduce hyperinsulinemia [12]. Sodium–glucose cotransporter-2 inhibition by SGLT2 inhibitors leads to glycosuria and the lowering of plasma glucose [119]. Another consequence of using SGLT2 inhibitors is the development of a relative hypoinsulinemia, which is part of the first line of defense against hypoglycemia [120]. Of note in this context are findings of a beneficial effects of sodium–glucose transport protein 2 (SGLT-2) inhibitors on central nervous (hypothalamic?) actions [121]. It was recently reported that the SGLT-2 inhibitor tofogliflozin decreased serum ACTH and cortisol levels in subjects with type 2 diabetes, indicating that tofogliflozin also influences the HPA axis [122]. Moreover, metformin, an insulin-sensitizer used as the first-line drug for treating type 2 diabetes, does not only lower plasma insulin and glucose levels, but also can reduce ACTH levels and cortisol [123,124]. For a long time, it has been unclear how the glucose-lowering effect of metformin was related to AMPK activation. Recently, it was found that metformin in rat pituitary cells induced AMPK/liver X receptor a (LXRa) phosphorylation which was followed by pro-opiomelanocortin (POMC) suppression [124]. The authors suggested that part of the anti-hyperglycemic effect of metformin could be attributed to reduced POMC/adrenocorticotropic hormone (ACTH)/cortisol levels following AMPK phosphorylation in the pituitary gland [124]. However, whether reduced activity of the HPA axis was secondary to metformin-mediated effects on hyperinsulinemia was not investigated in this study.
To date, there is no evidence that normalizing hyperinsulinemia in subjects with the metabolic syndrome can reduce (increased) activation of the HPA axis. It is also unknown whether this strategy would exert long-term beneficial effects on health. Therefore additional (longitudinal) studies are required to determine the importance of chronic hyperinsulinemia as driving force for increased HPA axis activity in subjects with the metabolic syndrome. Moreover, when these studies provide evidence for hyperinsulinemia as driving force for increased HPA axis activity in subjects with the metabolic syndrome, future research should focus on developing (new) strategies/drugs that can successfully prevent/reduce hyperinsulinemia-induced “functional hypercortisolism” in subjects with the metabolic syndrome.
In conclusion, emerging data suggest that chromic hyperinsulinemia is the driving force for increased HPA axis activity in subjects with the metabolic syndrome. Despite an increased HPA activity, plasma cortisol levels in subjects with the metabolic syndrome are often not elevated, due an increased plasma cortisol clearance. Nevertheless, subjects with the metabolic syndrome show a state of “functional hypercortisolism”. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia, disturb energy homeostasis, and shift energy fluxes away from muscle toward fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote fat accumulation in visceral fat cells and so contribute to abdominal obesity. Chronic hyperinsulinemia-induced activation of the HPA axis may play an important etiological role in the development of the metabolic syndrome and all its consequences.
Acknowledgments
The author is grateful to Michael P. Brugts and Aan Kharagjitsing for comments and proofreading the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Reaven G. Insulin resistance, type 2 diabetes mellitus, and cardiovascular disease: The end of the beginning. Circulation. 2005;112:3030–3032. doi: 10.1161/CIRCULATIONAHA.105.504670. [DOI] [PubMed] [Google Scholar]
- 2.Shanik M.H., Xu Y., Skrha J., Dankner R., Zick Y., Roth J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care. 2008;31((Suppl. 2)):S262–S268. doi: 10.2337/dc08-s264. [DOI] [PubMed] [Google Scholar]
- 3.Corkey B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care. 2012;35:2432–2437. doi: 10.2337/dc12-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirzadeh Z., Faber C.L., Schwartz M.W. Central Nervous System Control of Glucose Homeostasis: A Therapeutic Target for Type 2 Diabetes? Annu. Rev. Pharmacol. Toxicol. 2022;62:55–84. doi: 10.1146/annurev-pharmtox-052220-010446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trico D., Natali A., Arslanian S., Mari A., Ferrannini E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight. 2018;3:e124912. doi: 10.1172/jci.insight.124912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brons C., Jensen C.B., Storgaard H., Hiscock N.J., White A., Appel J.S., Jacobsen S., Nilsson E., Larsen C.M., Astrup A., et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009;587:2387–2397. doi: 10.1113/jphysiol.2009.169078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrannini E., Natali A., Bell P., Cavallo-Perin P., Lalic N., Mingrone G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR) J. Clin. Investig. 1997;100:1166–1173. doi: 10.1172/JCI119628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loves S., van Groningen L., Filius M., Mekking M., Brandon T., Tack C.J., Hermus A., de Boer H. High-Dose, Diazoxide-Mediated Insulin Suppression Boosts Weight Loss Induced by Lifestyle Intervention. J. Clin. Endocrinol. Metab. 2018;103:4014–4022. doi: 10.1210/jc.2018-01147. [DOI] [PubMed] [Google Scholar]
- 9.Dankner R., Chetrit A., Shanik M.H., Raz I., Roth J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care. 2009;32:1464–1466. doi: 10.2337/dc09-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizza R.A., Mandarino L.J., Genest J., Baker B.A., Gerich J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia. 1985;28:70–75. doi: 10.1007/BF00279918. [DOI] [PubMed] [Google Scholar]
- 11.Corkey B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes. 2012;61:4–13. doi: 10.2337/db11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021;22:7797. doi: 10.3390/ijms22157797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nolan C.J., Ruderman N.B., Kahn S.E., Pedersen O., Prentki M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64:673–686. doi: 10.2337/db14-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schofield C.J., Sutherland C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012;29:972–979. doi: 10.1111/j.1464-5491.2012.03655.x. [DOI] [PubMed] [Google Scholar]
- 15.Abraham S.B., Rubino D., Sinaii N., Ramsey S., Nieman L.K. Cortisol, obesity, and the metabolic syndrome: A cross-sectional study of obese subjects and review of the literature. Obesity. 2013;21:E105–E117. doi: 10.1002/oby.20083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosmond R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology. 2005;30:1–10. doi: 10.1016/j.psyneuen.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Anagnostis P., Athyros V.G., Tziomalos K., Karagiannis A., Mikhailidis D.P. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009;94:2692–2701. doi: 10.1210/jc.2009-0370. [DOI] [PubMed] [Google Scholar]
- 18.Stulnig T.M., Waldhausl W. 11beta-Hydroxysteroid dehydrogenase Type 1 in obesity and Type 2 diabetes. Diabetologia. 2004;47:1–11. doi: 10.1007/s00125-003-1284-4. [DOI] [PubMed] [Google Scholar]
- 19.Duclos M., Marquez Pereira P., Barat P., Gatta B., Roger P. Increased cortisol bioavailability, abdominal obesity, and the metabolic syndrome in obese women. Obes. Res. 2005;13:1157–1166. doi: 10.1038/oby.2005.137. [DOI] [PubMed] [Google Scholar]
- 20.Iida S., Nakamura Y., Fujii H., Nishimura J., Tsugawa M., Gomi M., Fukata J., Tarui S., Moriwaki K., Kitani T. A patient with hypocortisolism and Cushing’s syndrome-like manifestations: Cortisol hyperreactive syndrome. J. Clin. Endocrinol. Metab. 1990;70:729–737. doi: 10.1210/jcem-70-3-729. [DOI] [PubMed] [Google Scholar]
- 21.Friedman T.C., Mastorakos G., Newman T.D., Mullen N.M., Horton E.G., Costello R., Papadopoulos N.M., Chrousos G.P. Carbohydrate and lipid metabolism in endogenous hypercortisolism: Shared features with metabolic syndrome X and NIDDM. Endocr. J. 1996;43:645–655. doi: 10.1507/endocrj.43.645. [DOI] [PubMed] [Google Scholar]
- 22.Beaupere C., Liboz A., Feve B., Blondeau B., Guillemain G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021;22:623. doi: 10.3390/ijms22020623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scaroni C., Zilio M., Foti M., Boscaro M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017;38:189–219. doi: 10.1210/er.2016-1105. [DOI] [PubMed] [Google Scholar]
- 24.Traish A.M., Guay A.T., Zitzmann M. 5alpha-Reductase inhibitors alter steroid metabolism and may contribute to insulin resistance, diabetes, metabolic syndrome and vascular disease: A medical hypothesis. Horm. Mol. Biol. Clin. Investig. 2014;20:73–80. doi: 10.1515/hmbci-2014-0025. [DOI] [PubMed] [Google Scholar]
- 25.Stewart P.M., Krozowski Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999;57:249–324. [PubMed] [Google Scholar]
- 26.Tomlinson J.W., Stewart P.M. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract. Res. Clin. Endocrinol. Metab. 2001;15:61–78. doi: 10.1053/beem.2000.0119. [DOI] [PubMed] [Google Scholar]
- 27.Napolitano A., Voice M.W., Edwards C.R., Seckl J.R., Chapman K.E. 11Beta-hydroxysteroid dehydrogenase 1 in adipocytes: Expression is differentiation-dependent and hormonally regulated. J. Steroid Biochem. Mol. Biol. 1998;64:251–260. doi: 10.1016/S0960-0760(97)00200-8. [DOI] [PubMed] [Google Scholar]
- 28.Janssen J., Lamberts S. Diabetes associated with glucocorticoid excess. In: Ghigo E., Porta M., editors. Diabetes Secondary to Endocrine and Pancreatic Disorders. Karger; Basel, Switzerland: 2014. pp. 22–33. [Google Scholar]
- 29.Dallman M.F., Akana S.F., Strack A.M., Hanson E.S., Sebastian R.J. The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann. N. Y. Acad. Sci. 1995;771:730–742. doi: 10.1111/j.1749-6632.1995.tb44724.x. [DOI] [PubMed] [Google Scholar]
- 30.Goodman H.M. Basic Medical Endocrinology. 4th ed. Elsevier; San Diego, CA, USA: 2009. [Google Scholar]
- 31.Arlt W., Stewart P.M. Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol. Metab. Clin. N. Am. 2005;34:293–313. doi: 10.1016/j.ecl.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Christiansen J.J., Djurhuus C.B., Gravholt C.H., Iversen P., Christiansen J.S., Schmitz O., Weeke J., Jorgensen J.O., Moller N. Effects of cortisol on carbohydrate, lipid, and protein metabolism: Studies of acute cortisol withdrawal in adrenocortical failure. J. Clin. Endocrinol. Metab. 2007;92:3553–3559. doi: 10.1210/jc.2007-0445. [DOI] [PubMed] [Google Scholar]
- 33.Geer E.B., Islam J., Buettner C. Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol. Metab. Clin. N. Am. 2014;43:75–102. doi: 10.1016/j.ecl.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Cauter E., Blackman J.D., Roland D., Spire J.P., Refetoff S., Polonsky K.S. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J. Clin. Investig. 1991;88:934–942. doi: 10.1172/JCI115396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews R.C., Walker B.R. Glucocorticoids and insulin resistance: Old hormones, new targets. Clin. Sci. 1999;96:513–523. doi: 10.1042/CS19980388. [DOI] [PubMed] [Google Scholar]
- 36.Leal A.M., Moreira A.C. Food and the circadian activity of the hypothalamic-pituitary-adrenal axis. Braz. J. Med. Biol. Res. 1997;30:1391–1405. doi: 10.1590/S0100-879X1997001200003. [DOI] [PubMed] [Google Scholar]
- 37.Tataranni P.A., Larson D.E., Snitker S., Young J.B., Flatt J.P., Ravussin E. Effects of glucocorticoids on energy metabolism and food intake in humans. Am. J. Physiol. 1996;271:E317–E325. doi: 10.1152/ajpendo.1996.271.2.E317. [DOI] [PubMed] [Google Scholar]
- 38.Burt M.G., Gibney J., Ho K.K. Characterization of the metabolic phenotypes of Cushing’s syndrome and growth hormone deficiency: A study of body composition and energy metabolism. Clin. Endocrinol. 2006;64:436–443. doi: 10.1111/j.1365-2265.2006.02488.x. [DOI] [PubMed] [Google Scholar]
- 39.Torres S.J., Nowson C.A. Relationship between stress, eating behavior, and obesity. Nutrition. 2007;23:887–894. doi: 10.1016/j.nut.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Norton L., Shannon C., Gastaldelli A., DeFronzo R.A. Insulin: The master regulator of glucose metabolism. Metabolism. 2022;129:155142. doi: 10.1016/j.metabol.2022.155142. [DOI] [PubMed] [Google Scholar]
- 41.Hatting M., Tavares C.D.J., Sharabi K., Rines A.K., Puigserver P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018;1411:21–35. doi: 10.1111/nyas.13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahman M.S., Hossain K.S., Das S., Kundu S., Adegoke E.O., Rahman M.A., Hannan M.A., Uddin M.J., Pang M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021;22:6403. doi: 10.3390/ijms22126403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganong W.F. Circumventricular organs: Definition and role in the regulation of endocrine and autonomic function. Clin. Exp. Pharmacol. Physiol. 2000;27:422–427. doi: 10.1046/j.1440-1681.2000.03259.x. [DOI] [PubMed] [Google Scholar]
- 44.Baura G.D., Foster D.M., Porte D., Jr., Kahn S.E., Bergman R.N., Cobelli C., Schwartz M.W. Saturable transport of insulin from plasma into the central nervous system of dogs In Vivo. A mechanism for regulated insulin delivery to the brain. J. Clin. Investig. 1993;92:1824–1830. doi: 10.1172/JCI116773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baura G.D., Foster D.M., Kaiyala K., Porte D., Jr., Kahn S.E., Schwartz M.W. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes. 1996;45:86–90. doi: 10.2337/diab.45.1.86. [DOI] [PubMed] [Google Scholar]
- 46.Gray S.M., Meijer R.I., Barrett E.J. Insulin regulates brain function, but how does it get there? Diabetes. 2014;63:3992–3997. doi: 10.2337/db14-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hasselbalch S.G., Knudsen G.M., Videbaek C., Pinborg L.H., Schmidt J.F., Holm S., Paulson O.B. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes. 1999;48:1915–1921. doi: 10.2337/diabetes.48.10.1915. [DOI] [PubMed] [Google Scholar]
- 48.Seaquist E.R., Damberg G.S., Tkac I., Gruetter R. The effect of insulin on In Vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes. 2001;50:2203–2209. doi: 10.2337/diabetes.50.10.2203. [DOI] [PubMed] [Google Scholar]
- 49.McEwen B.S., Reagan L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004;490:13–24. doi: 10.1016/j.ejphar.2004.02.041. [DOI] [PubMed] [Google Scholar]
- 50.Porte D., Jr., Baskin D.G., Schwartz M.W. Insulin signaling in the central nervous system: A critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes. 2005;54:1264–1276. doi: 10.2337/diabetes.54.5.1264. [DOI] [PubMed] [Google Scholar]
- 51.Obici S., Zhang B.B., Karkanias G., Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 52.Inoue H., Ogawa W., Asakawa A., Okamoto Y., Nishizawa A., Matsumoto M., Teshigawara K., Matsuki Y., Watanabe E., Hiramatsu R., et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006;3:267–275. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 53.Inoue H., Ogawa W., Ozaki M., Haga S., Matsumoto M., Furukawa K., Hashimoto N., Kido Y., Mori T., Sakaue H., et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism In Vivo. Nat. Med. 2004;10:168–174. doi: 10.1038/nm980. [DOI] [PubMed] [Google Scholar]
- 54.Livingstone C., Lyall H., Gould G.W. Hypothalamic GLUT 4 expression: A glucose- and insulin-sensing mechanism? Mol. Cell Endocrinol. 1995;107:67–70. doi: 10.1016/0303-7207(94)03423-Q. [DOI] [PubMed] [Google Scholar]
- 55.Lucignani G., Namba H., Nehlig A., Porrino L.J., Kennedy C., Sokoloff L. Effects of insulin on local cerebral glucose utilization in the rat. J. Cereb. Blood Flow Metab. 1987;7:309–314. doi: 10.1038/jcbfm.1987.68. [DOI] [PubMed] [Google Scholar]
- 56.Strack A.M., Sebastian R.J., Schwartz M.W., Dallman M.F. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am. J. Physiol. 1995;268:R142–R149. doi: 10.1152/ajpregu.1995.268.1.R142. [DOI] [PubMed] [Google Scholar]
- 57.Banks W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004;490:5–12. doi: 10.1016/j.ejphar.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 58.Kamba A., Daimon M., Murakami H., Otaka H., Matsuki K., Sato E., Tanabe J., Takayasu S., Matsuhashi Y., Yanagimachi M., et al. Association between Higher Serum Cortisol Levels and Decreased Insulin Secretion in a General Population. PLoS ONE. 2016;11:e0166077. doi: 10.1371/journal.pone.0166077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mazziotti G., Gazzaruso C., Giustina A. Diabetes in Cushing syndrome: Basic and clinical aspects. Trends Endocrinol. Metab. 2011;22:499–506. doi: 10.1016/j.tem.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 60.Lambillotte C., Gilon P., Henquin J.C. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J. Clin. Investig. 1997;99:414–423. doi: 10.1172/JCI119175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Raalte D.H., Ouwens D.M., Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Investig. 2009;39:81–93. doi: 10.1111/j.1365-2362.2008.02067.x. [DOI] [PubMed] [Google Scholar]
- 62.Hansen K.B., Vilsboll T., Bagger J.I., Holst J.J., Knop F.K. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J. Clin. Endocrinol. Metab. 2010;95:3309–3317. doi: 10.1210/jc.2010-0119. [DOI] [PubMed] [Google Scholar]
- 63.Olefsky J.M. Effect of dexamethasone on insulin binding, glucose transport, and glucose oxidation of isolated rat adipocytes. J. Clin. Investig. 1975;56:1499–1508. doi: 10.1172/JCI108231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horner H.C., Munck A., Lienhard G.E. Dexamethasone causes translocation of glucose transporters from the plasma membrane to an intracellular site in human fibroblasts. J. Biol. Chem. 1987;262:17696–17702. doi: 10.1016/S0021-9258(18)45435-X. [DOI] [PubMed] [Google Scholar]
- 65.Kahn C.R., Goldfine I.D., Neville D.M., Jr., De Meyts P. Alterations in insulin binding induced by changes In Vivo in the levels of glucocorticoids and growth hormone. Endocrinology. 1978;103:1054–1066. doi: 10.1210/endo-103-4-1054. [DOI] [PubMed] [Google Scholar]
- 66.Yasuda K., Hines E., 3rd, Kitabchi A.E. Hypercortisolism and insulin resistance: Comparative effects of prednisone, hydrocortisone, and dexamethasone on insulin binding of human erythrocytes. J. Clin. Endocrinol. Metab. 1982;55:910–915. doi: 10.1210/jcem-55-5-910. [DOI] [PubMed] [Google Scholar]
- 67.Pivonello R., De Leo M., Vitale P., Cozzolino A., Simeoli C., De Martino M.C., Lombardi G., Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92((Suppl. 1)):77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- 68.Qi D., Rodrigues B. Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am. J. Physiol. Endocrinol. Metab. 2007;292:E654–E667. doi: 10.1152/ajpendo.00453.2006. [DOI] [PubMed] [Google Scholar]
- 69.Kola B., Christ-Crain M., Lolli F., Arnaldi G., Giacchetti G., Boscaro M., Grossman A.B., Korbonits M. Changes in adenosine 5′-monophosphate-activated protein kinase as a mechanism of visceral obesity in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 2008;93:4969–4973. doi: 10.1210/jc.2008-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hauner H., Schmid P., Pfeiffer E.F. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J. Clin. Endocrinol. Metab. 1987;64:832–835. doi: 10.1210/jcem-64-4-832. [DOI] [PubMed] [Google Scholar]
- 71.Santiago J.V., Clarke W.L., Shah S.D., Cryer P.E. Epinephrine, norepinephrine, glucagon, and growth hormone release in association with physiological decrements in the plasma glucose concentration in normal and diabetic man. J. Clin. Endocrinol. Metab. 1980;51:877–883. doi: 10.1210/jcem-51-4-877. [DOI] [PubMed] [Google Scholar]
- 72.DeFronzo R.A., Hendler R., Christensen N. Stimulation of counterregulatory hormonal responses in diabetic man by a fall in glucose concentration. Diabetes. 1980;29:125–131. doi: 10.2337/diab.29.2.125. [DOI] [PubMed] [Google Scholar]
- 73.Spyer G., Hattersley A.T., MacDonald I.A., Amiel S., MacLeod K.M. Hypoglycaemic counter-regulation at normal blood glucose concentrations in patients with well controlled type-2 diabetes. Lancet. 2000;356:1970–1974. doi: 10.1016/S0140-6736(00)03322-5. [DOI] [PubMed] [Google Scholar]
- 74.Chakera A.J., Hurst P.S., Spyer G., Ogunnowo-Bada E.O., Marsh W.J., Riches C.H., Yueh C.Y., Markkula S.P., Dalley J.W., Cox R.D., et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol. Metab. 2018;17:17–27. doi: 10.1016/j.molmet.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dirlewanger M., Schneiter P.H., Paquot N., Jequier E., Rey V., Tappy L. Effects of glucocorticoids on hepatic sensitivity to insulin and glucagon in man. Clin. Nutr. 2000;19:29–34. doi: 10.1054/clnu.1999.0064. [DOI] [PubMed] [Google Scholar]
- 76.Rashid S., Shi Z.Q., Niwa M., Mathoo J.M., Vandelangeryt M.L., Bilinski D., Lewis G.F., Vranic M. Beta-blockade, but not normoglycemia or hyperinsulinemia, markedly diminishes stress-induced hyperglycemia in diabetic dogs. Diabetes. 2000;49:253–262. doi: 10.2337/diabetes.49.2.253. [DOI] [PubMed] [Google Scholar]
- 77.Sherwin R.S., Shamoon H., Hendler R., Sacca L., Eigler N., Walesky M. Epinephrine and the regulation of glucose metabolism: Effect of diabetes and hormonal interactions. Metabolism. 1980;29:1146–1154. doi: 10.1016/0026-0495(80)90024-4. [DOI] [PubMed] [Google Scholar]
- 78.Chan O., Inouye K., Akirav E., Park E., Riddell M.C., Vranic M., Matthews S.G. Insulin alone increases hypothalamo-pituitary-adrenal activity, and diabetes lowers peak stress responses. Endocrinology. 2005;146:1382–1390. doi: 10.1210/en.2004-0607. [DOI] [PubMed] [Google Scholar]
- 79.Grunstein H.S., James D.E., Storlien L.H., Smythe G.A., Kraegen E.W. Hyperinsulinemia suppresses glucose utilization in specific brain regions: In Vivo studies using the euglycemic clamp in the rat. Endocrinology. 1985;116:604–610. doi: 10.1210/endo-116-2-604. [DOI] [PubMed] [Google Scholar]
- 80.Fruehwald-Schultes B., Kern W., Bong W., Wellhoener P., Kerner W., Born J., Fehm H.L., Peters A. Supraphysiological hyperinsulinemia acutely increases hypothalamic-pituitary-adrenal secretory activity in humans. J. Clin. Endocrinol. Metab. 1999;84:3041–3046. doi: 10.1210/jcem.84.9.5953. [DOI] [PubMed] [Google Scholar]
- 81.Jacobson L., Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr. Rev. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- 82.Palovcik R.A., Phillips M.I., Kappy M.S., Raizada M.K. Insulin inhibits pyramidal neurons in hippocampal slices. Brain Res. 1984;309:187–191. doi: 10.1016/0006-8993(84)91028-X. [DOI] [PubMed] [Google Scholar]
- 83.Asensio C., Muzzin P., Rohner-Jeanrenaud F. Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 2004;28((Suppl. 4)):S45–S52. doi: 10.1038/sj.ijo.0802856. [DOI] [PubMed] [Google Scholar]
- 84.Ljung T., Andersson B., Bengtsson B.A., Bjorntorp P., Marin P. Inhibition of cortisol secretion by dexamethasone in relation to body fat distribution: A dose-response study. Obes. Res. 1996;4:277–282. doi: 10.1002/j.1550-8528.1996.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 85.Rosmond R., Dallman M.F., Bjorntorp P. Stress-related cortisol secretion in men: Relationships with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J. Clin. Endocrinol. Metab. 1998;83:1853–1859. doi: 10.1210/jc.83.6.1853. [DOI] [PubMed] [Google Scholar]
- 86.Hales C.N., Barker D.J., Clark P.M., Cox L.J., Fall C., Osmond C., Winter P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Clark P.M., Hindmarsh P.C., Shiell A.W., Law C.M., Honour J.W., Barker D.J. Size at birth and adrenocortical function in childhood. Clin. Endocrinol. 1996;45:721–726. doi: 10.1046/j.1365-2265.1996.8560864.x. [DOI] [PubMed] [Google Scholar]
- 88.Phillips D.I., Barker D.J., Fall C.H., Seckl J.R., Whorwood C.B., Wood P.J., Walker B.R. Elevated plasma cortisol concentrations: A link between low birth weight and the insulin resistance syndrome? J. Clin. Endocrinol. Metab. 1998;83:757–760. doi: 10.1210/jcem.83.3.4634. [DOI] [PubMed] [Google Scholar]
- 89.Werdermann M., Berger I., Scriba L.D., Santambrogio A., Schlinkert P., Brendel H., Morawietz H., Schedl A., Peitzsch M., King A.J.F., et al. Insulin and obesity transform hypothalamic-pituitary-adrenal axis stemness and function in a hyperactive state. Mol. Metab. 2021;43:101112. doi: 10.1016/j.molmet.2020.101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andrew R., Phillips D.I., Walker B.R. Obesity and gender influence cortisol secretion and metabolism in man. J. Clin. Endocrinol. Metab. 1998;83:1806–1809. doi: 10.1210/jcem.83.5.4951. [DOI] [PubMed] [Google Scholar]
- 91.Stewart P.M., Boulton A., Kumar S., Clark P.M., Shackleton C.H. Cortisol metabolism in human obesity: Impaired cortisone → cortisol conversion in subjects with central adiposity. J. Clin. Endocrinol. Metab. 1999;84:1022–1027. doi: 10.1210/jc.84.3.1022. [DOI] [PubMed] [Google Scholar]
- 92.Pasquali R., Vicennati V., Cacciari M., Pagotto U. The hypothalamic-pituitary-adrenal axis activity in obesity and the metabolic syndrome. Ann. N. Y. Acad. Sci. 2006;1083:111–128. doi: 10.1196/annals.1367.009. [DOI] [PubMed] [Google Scholar]
- 93.Vicennati V., Pasquali R. Abnormalities of the hypothalamic-pituitary-adrenal axis in nondepressed women with abdominal obesity and relations with insulin resistance: Evidence for a central and a peripheral alteration. J. Clin. Endocrinol. Metab. 2000;85:4093–4098. doi: 10.1210/jcem.85.11.6946. [DOI] [PubMed] [Google Scholar]
- 94.Pasquali R., Gagliardi L., Vicennati V., Gambineri A., Colitta D., Ceroni L., Casimirri F. ACTH and cortisol response to combined corticotropin releasing hormone-arginine vasopressin stimulation in obese males and its relationship to body weight, fat distribution and parameters of the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 1999;23:419–424. doi: 10.1038/sj.ijo.0800838. [DOI] [PubMed] [Google Scholar]
- 95.Holt H.B., Wild S.H., Postle A.D., Zhang J., Koster G., Umpleby M., Shojaee-Moradie F., Dewbury K., Wood P.J., Phillips D.I., et al. Cortisol clearance and associations with insulin sensitivity, body fat and fatty liver in middle-aged men. Diabetologia. 2007;50:1024–1032. doi: 10.1007/s00125-007-0629-9. [DOI] [PubMed] [Google Scholar]
- 96.Lottenberg S.A., Giannella-Neto D., Derendorf H., Rocha M., Bosco A., Carvalho S.V., Moretti A.E., Lerario A.C., Wajchenberg B.L. Effect of fat distribution on the pharmacokinetics of cortisol in obesity. Int. J. Clin. Pharmacol. Ther. 1998;36:501–505. [PubMed] [Google Scholar]
- 97.Fernandez-Real J.M., Grasa M., Casamitjana R., Pugeat M., Barret C., Ricart W. Plasma total and glycosylated corticosteroid-binding globulin levels are associated with insulin secretion. J. Clin. Endocrinol. Metab. 1999;84:3192–3196. doi: 10.1210/jcem.84.9.5946. [DOI] [PubMed] [Google Scholar]
- 98.Manco M., Fernandez-Real J.M., Valera-Mora M.E., Dechaud H., Nanni G., Tondolo V., Calvani M., Castagneto M., Pugeat M., Mingrone G. Massive weight loss decreases corticosteroid-binding globulin levels and increases free cortisol in healthy obese patients: An adaptive phenomenon? Diabetes Care. 2007;30:1494–1500. doi: 10.2337/dc06-1353. [DOI] [PubMed] [Google Scholar]
- 99.Penhoat A., Chatelain P.G., Jaillard C., Saez J.M. Characterization of insulin-like growth factor I and insulin receptors on cultured bovine adrenal fasciculata cells. Role of these peptides on adrenal cell function. Endocrinology. 1988;122:2518–2526. doi: 10.1210/endo-122-6-2518. [DOI] [PubMed] [Google Scholar]
- 100.Kinyua A.W., Doan K.V., Yang D.J., Huynh M.K.Q., Choi Y.H., Shin D.M., Kim K.W. Insulin Regulates Adrenal Steroidogenesis by Stabilizing SF-1 Activity. Sci. Rep. 2018;8:5025. doi: 10.1038/s41598-018-23298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Altieri B., Tirabassi G., Della Casa S., Ronchi C.L., Balercia G., Orio F., Pontecorvi A., Colao A., Muscogiuri G. Adrenocortical tumors and insulin resistance: What is the first step? Int. J. Cancer. 2016;138:2785–2794. doi: 10.1002/ijc.29950. [DOI] [PubMed] [Google Scholar]
- 102.Tsatsoulis A. The Role of Insulin Resistance/Hyperinsulinism on the Rising Trend of Thyroid and Adrenal Nodular Disease in the Current Environment. J. Clin. Med. 2018;7:37. doi: 10.3390/jcm7030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Golden S.H., Wand G.S., Malhotra S., Kamel I., Horton K. Reliability of hypothalamic-pituitary-adrenal axis assessment methods for use in population-based studies. Eur. J. Epidemiol. 2011;26:511–525. doi: 10.1007/s10654-011-9585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Askani E., Rospleszcz S., Lorbeer R., Kulka C., von Kruchten R., Muller-Peltzer K., Hasic D., Kellner E., Reisert M., Rathmann W., et al. Association of MRI-based adrenal gland volume and impaired glucose metabolism in a population-based cohort study. Diabetes Metab. Res. Rev. 2022;38:e3528. doi: 10.1002/dmrr.3528. [DOI] [PubMed] [Google Scholar]
- 105.Stewart P.M., Toogood A.A., Tomlinson J.W. Growth hormone, insulin-like growth factor-I and the cortisol-cortisone shuttle. Horm. Res. 2001;56((Suppl. 1)):1–6. doi: 10.1159/000048126. [DOI] [PubMed] [Google Scholar]
- 106.Ahmed A., Rabbitt E., Brady T., Brown C., Guest P., Bujalska I.J., Doig C., Newsome P.N., Hubscher S., Elias E., et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS ONE. 2012;7:e29531. doi: 10.1371/journal.pone.0029531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balachandran A., Guan H., Sellan M., van Uum S., Yang K. Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology. 2008;149:4069–4079. doi: 10.1210/en.2008-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Masuzaki H., Paterson J., Shinyama H., Morton N.M., Mullins J.J., Seckl J.R., Flier J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 109.Morton N.M., Holmes M.C., Fievet C., Staels B., Tailleux A., Mullins J.J., Seckl J.R. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J. Biol. Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- 110.Bujalska I.J., Kumar S., Stewart P.M. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet. 1997;349:1210–1213. doi: 10.1016/S0140-6736(96)11222-8. [DOI] [PubMed] [Google Scholar]
- 111.Tomlinson J.W., Walker E.A., Bujalska I.J., Draper N., Lavery G.G., Cooper M.S., Hewison M., Stewart P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- 112.Hazlehurst J.M., Gathercole L.L., Nasiri M., Armstrong M.J., Borrows S., Yu J., Wagenmakers A.J., Stewart P.M., Tomlinson J.W. Glucocorticoids fail to cause insulin resistance in human subcutaneous adipose tissue In Vivo. J. Clin. Endocrinol. Metab. 2013;98:1631–1640. doi: 10.1210/jc.2012-3523. [DOI] [PubMed] [Google Scholar]
- 113.Seimon R.V., Hostland N., Silveira S.L., Gibson A.A., Sainsbury A. Effects of energy restriction on activity of the hypothalamo-pituitary-adrenal axis in obese humans and rodents: Implications for diet-induced changes in body composition. Horm. Mol. Biol. Clin. Investig. 2013;15:71–80. doi: 10.1515/hmbci-2013-0038. [DOI] [PubMed] [Google Scholar]
- 114.Parra M.D., Martinez de Morentin B.E., Alfredo Martinez J. Impact of weight loss on cortisol secretion in obese men with and without metabolic syndrome features. Nutr. Metab. Cardiovasc. Dis. 2006;16:28–34. doi: 10.1016/j.numecd.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 115.Giovannini C., Ciucci E., Clementi R., Cugini P., Facchinetti F., Negri M. Beta-endorphin, insulin, ACTH and cortisol plasma levels during oral glucose tolerance test in obesity after weight loss. Horm. Metab. Res. 1990;22:96–100. doi: 10.1055/s-2007-1004859. [DOI] [PubMed] [Google Scholar]
- 116.Ho J.T., Keogh J.B., Bornstein S.R., Ehrhart-Bornstein M., Lewis J.G., Clifton P.M., Torpy D.J. Moderate weight loss reduces renin and aldosterone but does not influence basal or stimulated pituitary-adrenal axis function. Horm. Metab. Res. 2007;39:694–699. doi: 10.1055/s-2007-985354. [DOI] [PubMed] [Google Scholar]
- 117.Nonino-Borges C.B., Martins Borges R., Bavaresco M., Suen V.M., Moreira A.C., Marchini J.S. Influence of meal time on salivary circadian cortisol rhythms and weight loss in obese women. Nutrition. 2007;23:385–391. doi: 10.1016/j.nut.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 118.Purnell J.Q., Kahn S.E., Samuels M.H., Brandon D., Loriaux D.L., Brunzell J.D. Enhanced cortisol production rates, free cortisol, and 11beta-HSD-1 expression correlate with visceral fat and insulin resistance in men: Effect of weight loss. Am. J. Physiol. Endocrinol. Metab. 2009;296:E351–E357. doi: 10.1152/ajpendo.90769.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kalra S., Gupta Y., Patil S. Sodium-glucose cotransporter-2 inhibition and the insulin: Glucagon ratio: Unexplored dimensions. Indian J. Endocrinol. Metab. 2015;19:426–429. doi: 10.4103/2230-8210.152793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ferrannini E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017;26:27–38. doi: 10.1016/j.cmet.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 121.Schlogl H., Stumvoll M. The Brains behind SGLT2 Inhibition. Diabetes Care. 2022;45:273–275. doi: 10.2337/dci21-0050. [DOI] [PubMed] [Google Scholar]
- 122.Higashikawa T., Ito T., Mizuno T., Ishigami K., Kuroki K., Maekawa N., Usuda D., Morita T., Hamada K., Takagi S., et al. Effects of tofogliflozin on adrenocorticotropic hormone, renin and aldosterone, and cortisol levels in elderly patients with diabetes mellitus: A retrospective study of a patient cohort. Medicine. 2021;100:e27638. doi: 10.1097/MD.0000000000027638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shin A.C., Balasubramanian P., Suryadevara P., Zyskowski J., Herdt T.H., MohanKumar S.M.J., MohanKumar P.S. Metformin effectively restores the HPA axis function in diet-induced obese rats. Int. J. Obes. 2021;45:383–395. doi: 10.1038/s41366-020-00688-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cho K., Chung J.Y., Cho S.K., Shin H.W., Jang I.J., Park J.W., Yu K.S., Cho J.Y. Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRalpha/POMC pathway. Sci. Rep. 2015;5:8145. doi: 10.1038/srep08145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.