

Abstract
BACKGROUND:
Little is understood about neonatal pharmacokinetics immediately after delivery and during the first days of life following intrauterine exposure to maternal medications. Our objective was to develop and evaluate a novel physiological-based pharmacokinetics (PBPK) modeling workflow for predicting perinatal and postnatal disposition of commonly used antiretroviral drugs administered prenatally to pregnant women living with human immunodefciency virus (HIV).
METHODS:
Using previously published maternal-fetal PBPK models for emtricitabine, dolutegravir and raltegravir built with PK-Sim/MoBi®, placental drug transfer was predicted in late pregnancy. The total drug amount in fetal compartments at term delivery was estimated and subsequently integrated as initial conditions in different tissues of a whole-body neonatal PBPK model to predict drug concentrations in the neonatal elimination phase after birth. Neonatal elimination processes were parameterized according to published data. Model performance was assessed by clinical data.
RESULTS:
Neonatal PBPK models generally captured the initial plasma concentrations after delivery but underestimated concentrations in the terminal phase. The mean percentage error for predicted plasma concentrations was −71.5%, −33.8% and 76.7% for emtricitabine, dolutegravir and raltegravir, respectively. Sensitivity analysis suggested that the activity of organic cation transporter (OCT) 2 and uridine diphosphate glucuronosyltransferase (UGT) 1A1 during the first postnatal days in term newborns is ~11% and ~30% of that in adults, respectively.
CONCLUSION:
These findings demonstrate the general feasibility of applying PBPK models to predict washout concentrations of transplacentally acquired drugs in newborns. These models can increase the understanding of pharmacokinetics during the first postnatal days and allow prediction of drug exposure in this vulnerable population.
Keywords: physiologically-based pharmacokinetics, antiretroviral therapy, maternal-fetal, trans-placenta, neonate
Introduction
While more data describing maternal drug pharmacokinetics in pregnancy is becoming available, relatively little is known about pharmacokinetics of transplacentally acquired drugs in neonates.[1] Conducting clinical trials in neonates is challenging and pharmacokinetic data in this vulnerable population are scarce.[2] Extrapolation of pharmacokinetic parameters from adults to newborns is complicated by the rapid and often poorly understood anatomical and physiological changes after birth associated with neonatal adaptation to the extrauterine environment.[3] Consequently, determining optimal drug regimens for newborns is extremely challenging.[4] One approach to filling these knowledge gaps is through application of physiologically based pharmacokinetic (PBPK) models that integrate neonatal anatomical and physiological changes in a mechanistic modeling framework with the aim to predict neonatal pharmacokinetics as a function of the physiological system and physicochemical properties of the drug.[5–7] However, to date, there are no published PBPK models that can be applied to mechanistically characterize neonatal pharmacokinetics of drugs transferred across the placenta to the fetus after prenatal maternal administration. In this article, a novel PBPK modeling framework was developed and used to predict the placental transfer of three widely used antiretroviral (ARV) drugs (emtricitabine (FTC), dolutegravir (DTG) and raltegravir (RAL)) administered to pregnant women prior to delivery and their subsequent disposition in the neonate after birth.
Pregnant women living with HIV receive ARVs during pregnancy to maintain their own health and to prevent vertical HIV transmission (through suppressing viral load). Emtricitabine, dolutegravir and raltegravir are ARVs used for the prevention and treatment of human immunodeficiency virus (HIV) infection. Maternal pharmacokinetics of these ARVs have been well characterized and while reduced plasma exposures during the third trimester compared to postpartum were observed they were deemed not clinically significant.[8–13] Emtricitabine is mainly eliminated unchanged in urine via a combination of glomerular filtration and active tubular secretion which is predominantly mediated by organic cation transporter (OCT)2, while dolutegravir and raltegravir are predominantly metabolized via hepatic uridine diphosphate glucuronosyltransferase (UGT) 1A1. All three drugs readily cross the placenta leading to fetal drug exposure in utero.[14, 15] Washout kinetics of these ARVs in neonates immediately following delivery have previously been reported.[10, 16–18] Building on these efforts, the objective of the current study was to develop a novel PBPK framework capable of reproducing the observed neonatal washout kinetics of emtricitabine, dolutegravir and raltegravir in silico. Given the paucity of quantitative information on relevant transporters and enzymes involved in neonatal drug elimination (e.g. OCT2 and UGT1A1), sensitivity analyses were additionally conducted to identify the apparent activity during the first postnatal days.
Materials and Methods
Software
PBPK models were developed using the open source software tool Open Systems Pharmacology (OSP) version 8.0 (http://www.open-systems-pharmacology.org/) which makes formerly commercial software PK-Sim® and MoBi® available as freeware under the GPLv2 License.[19] WebPlotDigitizer (http://automeris.io/WebPlotDigiti-zer/) was used to extract data from published figures and convert them into digital format. The free software R (version 3.5.1, R Foundation for Statistical Computing, Vienna, Austria; http://www-.r-project.org) was used for non-compartmental analysis and graphics creation.
Clinical data
All clinical data shown herein were from published studies. For emtricitabine, clinical data were obtained from the TEmAA ANRS 12109.[17] Emtricitabine was administered orally as a 400 mg single dose (2 × 200 mg tablets) in pregnant women during labor. For dolutegravir, clinical data were obtained from the IMPAACT P1026s study.[10] As part of ARV therapy, 50 mg of dolutegravir single tablet was orally administered once daily to pregnant women; data from two women were excluded: one because her electronic record was not available for the day before delivery (and hence it was not clear whether dolutegravir concentrations at delivery were at steady state) and another because the dolutegravir dose prior to delivery was apparently not taken. For raltegravir, clinical data were obtained from the IMPAACT P1097 study.[16] Raltegravir was dosed at 400 mg twice daily as an orally administered single tablet in pregnant women. Characteristics of these clinical studies and the patients included therein are listed in Table 1.
Table 1:
Characteristics of the clinical pharmacokinetic studies and studied patients
| Emtricitabine | Dolutegravir | Raltegravir | |
|---|---|---|---|
|
| |||
| Name of pharmacokinetic study | TEmAA ANRS 12109 trial, step 1[17] | IMPAACT P1026s | IMPAACT P1097[16] |
| No. of mother-fetus pairs studied at delivery | 38 | 20 | 19 |
| No. of neonates studied after delivery | 32 | 10 | 19 |
| Maternal posology | 400 mg single dose | 50 mg QD | 400 mg BID |
| Time to delivery since last dose [h] | 4.9 (0.78 – 17.4)a | 11.8 (5.1 – 18.8)a | 4.6 (1.1 – 21)a |
| Gestational age at birth [weeks] | 39 (33 – 42)a | 38 (36 – 40)a | 38 (37 – 40)a |
| Birth weight [g] | 2700 (2300 – 3600)a | 3030 (2670 – 3950)a | 3080 (2200 – 4100)a |
| Gender (males/females) | NA | 3/7 | 13/6 |
PBPK model development
General workflow
Figure 1 schematically illustrates the workflow of this study. Maternal-fetal PBPK models for emtricitabine, dolutegravir and raltegravir have been developed according to a best practice workflow[20] and reported recently[14, 15] and these models were used to estimate the total amount drug of present in the fetal compartments at the time of delivery following the last oral dose administered to the mother prior to delivery. A direct coupling of the fetal to neonatal PBPK model was structurally not possible because the pregnancy PBPK model contains five fetal compartments, but no whole-body fetal sub-model. To allow a better description of the tissue distribution in the newborn, a preliminary PBPK model for neonates (interim model) was built in a second step. In contrast to the final neonatal PBPK model, this interim model did not contain elimination processes. It was used to simulate the distribution of each drug into all organs (and its sub-compartments, namely plasma, blood cells, interstitial and intracellular space) following intravenous (IV) bolus administration. The dose of this administration was set to the total amount of drug that was estimated to be present in the fetal compartments at delivery by the maternal-fetal PBPK models. In the next step, the organ concentrations simulated by the interim model were then integrated as initial values in a final neonatal PBPK model (where elimination processes were activated) and pharmacokinetics were predicted during the washout phase.
Figure 1. Workflow for predicting neonatal pharmacokinetics after maternal dosing prior to birth.
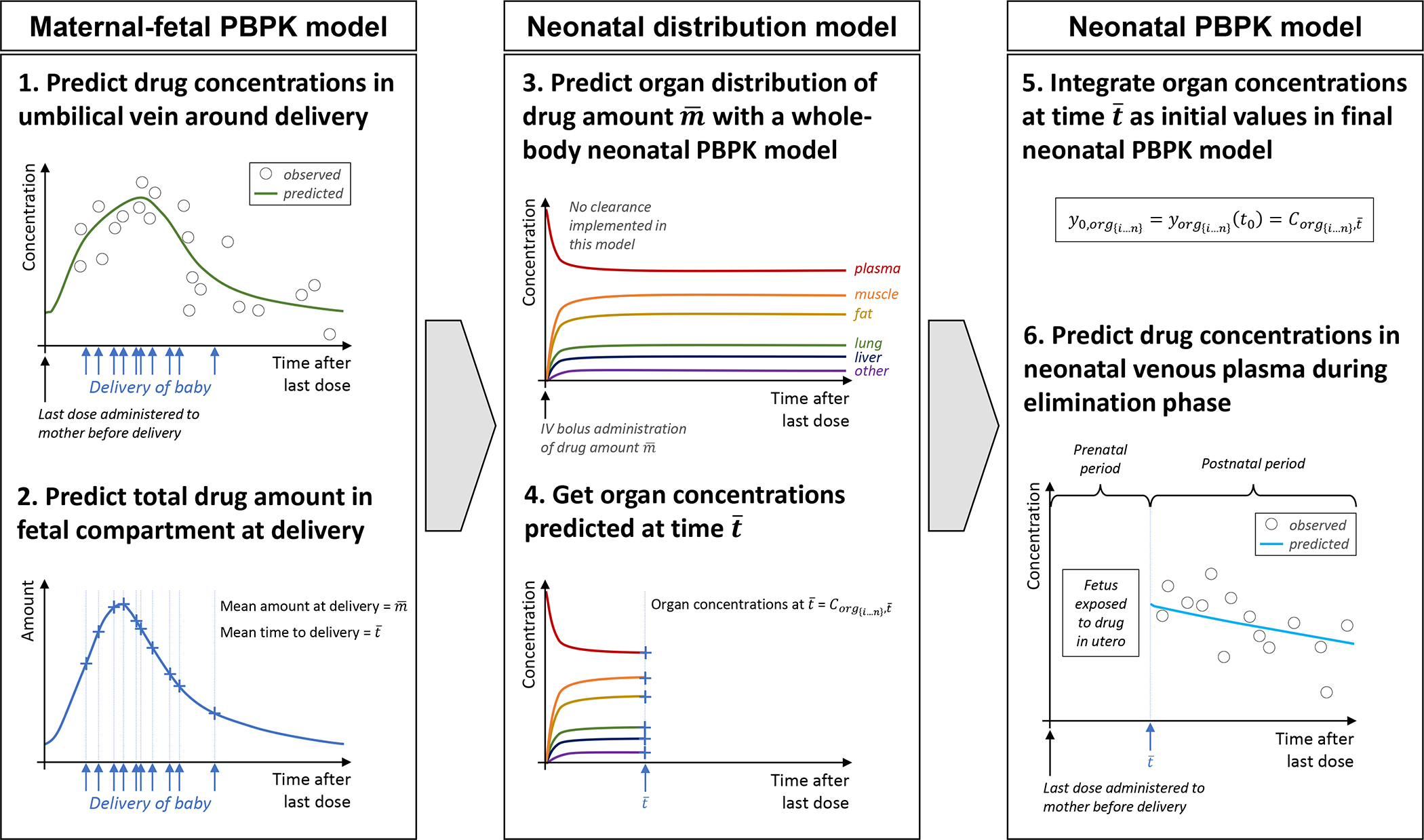
Abbreviations: PBPK: physiologically based pharmacokinetic
Non-pregnant and maternal-fetal PBPK models
Emtricitabine
In non-pregnant adults, about 86% of an emtricitabine dose is eliminated unchanged by renal glomerular filtration and tubular secretion through various transporters. A small portion of emtricitabine (~13%) is eliminated as sulfide and glucuronide metabolites.[21] The model hence includes separate elimination pathways via glomerular filtration, tubular secretion and a hepatic plasma clearance via an unspecified enzyme. Details of the development and evaluation of the non-pregnant and pregnant PBPK models can be found in a previous publication.[14]
Dolutegravir
In adults, dolutegravir is primarily eliminated by hepatic metabolism through various enzymes including UGT1A1, and to a minor extent UGT1A3, UGT1A9 and CYP3A4 (in male adults approximately 51%, 2.8%, 5.5% and 21% of the radioactive dose, respectively[22]). In the models described herein, the contribution of UGT1A3 and 1A9 to total glucuronidation was added to the biotransformation pathway mediated by UGT1A1. The model hence includes separate elimination pathways via UGT1A1 and CYP3A4. Details of the development and evaluation of the non-pregnant and pregnant PBPK models can be found in a previous publication.[15]
Raltegravir
In adults, raltegravir is primarily eliminated by hepatic metabolism via UGT1A1 and UGT1A9 (79% and 11% of the dose in non-pregnant adults, respectively). Additionally, approximately 9% is eliminated unchanged through the kidney.[23] The model hence includes separate elimination pathways via UGT1A1, UGT1A9 and glomerular filtration. Details of the development and evaluation of the non-pregnant and pregnant PBPK models for raltegravir can be found in a previous publication[15] and additionally on GitHub (https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/tree/master/Raltegravir).
Neonatal PBPK models
Transfer of prenatal drug concentrations in the fetus to the neonatal PBPK models
Using our previously developed maternal-fetal PBPK models described above, the total amount of emtricitabine, dolutegravir and raltegravir was predicted in the fetus at the time of delivery. In these models, the fetus is structurally represented as a single compartment consisting of four sub-compartments (plasma, blood cells, interstitial and intracellular space), but a whole-body fetal model is lacking. Therefore, a direct, mechanistic coupling of the pharmacokinetics predicted in the one-compartment fetal model prior to birth with a whole-body newborn model was technically not possible. However, these maternal-fetal PBPK models allow for an estimation of the total drug amount present in the fetus at the time of birth which was transferred to the whole-body newborn PBPK model as described in the following. In an interim step, a neonatal PBPK model was used to approximate the distribution kinetics of the drug into all organs occurring in utero, i.e. the tissue distribution prior to delivery which could not be estimated with the one-compartmental fetal structure of the maternal-fetal PBPK model. The drug amount predicted in the fetus at delivery was administered as IV bolus dose in an interim neonatal PBPK model without any clearance pathways, thereby allowing the drug to distribute to all organs. The simulation duration was set to the average time between drug administration and delivery reported in the clinical study. The concentrations predicted in each organ of this preliminary neonatal PBPK model were then extracted and integrated as initial values in the final neonatal PBPK model (which had clearance processes activated). It should be noted that it was technically not possible to integrate variability in organ drug concentrations as initial values in the final neonatal model and thus mean concentrations were used. Technically, the integration of organ drug concentrations at time zero in the neonatal PBPK model was carried out by importing them as molecule start values in MoBi®. Thereafter, the pharmacokinetics were predicted in the neonates using the latter model.
Parametrization of neonatal PBPK models
Three neonatal models – one each for emtricitabine, dolutegravir and raltegravir – were constructed according to the approach described above. The models were parametrized for the 1st day of postnatal life using the “preterm” population implemented in PK-Sim®[5] with a postnatal age of 0 days and the reported gestational age and anthropometry. No further manual changes in model parameters were made except for clearance parameters described below. The model parameters are listed in Table 2. Variability on anatomical and physiological parameters in the PBPK models was automatically accounted for in PK-Sim®; details on the implemented variability in the pregnant and neonatal populations have been published previously [5, 24, 25].
Table 2:
Summary of input data for PBPK models in non-pregnant/ pregnant subjects
| Parameter [unit] | Emtricitabine | Dolutegravir | Raltegravir | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Value | Reference | Value | Reference | Value | Reference | |
| Molecular weight [g/mol] | 247.3 | Drugbank.ca | 419.38 | Drugbank.ca | 444.42 | Drugbank.ca |
| Lipophilicity [log units] | −0.43 | [21] | 0.98 | fitted a | 0.58 | [39] |
| pKa (acid) | 10.1 | Drugbank.ca | 6.67 | [39] | ||
| pKa (base) | 2.65 | Drugbank.ca | ||||
| Fraction unbound: | ||||||
| Non-pregnant | 0.96 | Drugbank.ca | 0.0070 | [40] | 0.170 | [41] |
| 3rd trimester | 0.97 | calculated | 0.0088 | calculated | 0.206 | calculated |
| Neonates | 0.96 | calculated | 0.0081 | calculated | 0.190 | calculated |
| Major binding protein | Albumin | Drugbank.ca | Albumin | Drugbank.ca | Albumin | [41] |
| Solubility (at pH 7) [mg/L] | 112 | Drugbank.ca | 0.172b | fitted a | 8900c | [42] |
| Intestinal permeability (transcellular) [cm/min] | 3.98E-6 | fitted a | 0.05 | fitted a | 1.71 ∙ 10−5 | fitted a |
| Model for estimating organ-to-plasma partition coefficients | Rogers & Rowland | [43, 44] | Rogers & Rowland | [43, 44] | Rogers & Rowland | [43, 44] |
| GFR fraction | 1.0 | 1.0 | [23] | |||
| Km-UGT1A1 [μM] | 149 | [22] | 99 | [23] | ||
| Vmax−UGT1A1 [nmol/min/mg] | 7.34 | fitted a | 2.74d | fitted a | ||
| Km-UGT1A9 [μM] | 296 | [23] | ||||
| Vmax−UGT1A9 [nmol/min/mg] | 1.63d | fitted a | ||||
| CLspec/Enzyme [l/μmol/min] | 0.036e | fitted a | 0.050f | fitted a | ||
| TSspec [1/min] | 0.57 | fitted a | ||||
Value simultaneously fitted to in vivo plasma concentration-time profiles of non-pregnant subjects and to the reported dose fractions metabolized. For detailed information see references.[14, 15]
Solubility for the tablet formulation administered in fasted state
Solubility implemented as table: PH=1 to 4, solubility=40mg/L; PH=5, solubility=120mg/L; PH=6, solubility=980mg/L; PH=7, solubility=8900mg/L; PH=8, solubility=37300mg/L.
Value fitted to in vivo pharmacokinetic data of non-pregnant subjects following oral administration.
Describes emtricitabine metabolism via an unknown hepatic enzyme.
Describes dolutegravir metabolism via CYP3A4.
Abbreviations: CLspec/Enzyme: specific enzymatic clearance rate; CYP: cytochrome P450; GFR: glomerular filtration rate; Km: Michaelis-Menten constant; PBPK: physiologically based pharmacokinetic; TSspec: specific tubular secretion rate; UGT: uridine diphosphate glucuronosyltransferase; Vmax: maximum reaction rate
Ontogeny of clearance pathways in the neonatal PBPK models
The neonatal PBPK models qualitatively included the same clearance pathways as the adult models (see above). Information of the ontogeny of these clearance pathways during the first days of postnatal life was incorporated in the neonatal PBPK models, if such information was available in the scientific literature. Variability in relevant clearance parameters, e.g. glomerular filtration rate (GFR), kidney volume, and enzyme or transporter tissue concentrations, was included as implemented per default in PK-Sim® unless noted otherwise. This section also includes the fraction unbound in plasma since it is a highly sensitive parameter for the hepatic clearance of highly albumin-bound drugs (such as dolutegravir and raltegravir). Variability in the drug’s fraction unbound was not incorporated in the PBPK models.
No information could be found in the literature on the enzyme(s) involved in emtricitabine metabolism. Therefore, no ontogeny was assumed for this pathway and the specific clearance value incorporated in the adult model was used in the neonatal emtricitabine model.
Ontogeny of Renal Excretion
The ontogeny of glomerular filtration has been extensively studied. The model used the default setting for the kidney ontogeny in PK-Sim® which gives an absolute GFR of 3.43 mL/min in a neonate with a gestational age (GA) of 38 weeks, postnatal age of 1 day and a height of 49 cm and a kidney volume of 37 mL and blood flow of 117.7 mL/min. [5]
The tubular secretion of emtricitabine is mediated through the MATE1 efflux transporter and OCT2 influx transporters.[26] Studies with postmortem frozen renal cortical tissue samples from adults and newborns indicate that MATE1 has an age-independent expression, while OCT2 has significantly lower expression in newborns than in adults.[27] Specifically, the median OCT2 abundance is approximately 29.6 pmol/mg total membrane protein in adults, whereas in a term neonatal sample, abundance was approximately 8.8 pmol/mg total membrane protein.[27] Applying this fraction (0.30) to the specific first order tubular secretion value used in the adult PBPK model (0.57 min−1) resulted in a value of 0.17 min−1 which was implemented in the neonatal PBPK model. A sensitivity analysis was performed to investigate the effect of quantitative changes in renal clearance on the predicted neonatal pharmacokinetics.
Ontogeny of UGT1A1
Little is known about the expression and activity of UGT1A1 in a neonate shortly after delivery. The default ontogeny for UGT1A1 implemented in PK-Sim® started at 36 weeks of age and therefore did not provide information for the first days of life. Using dolutegravir and raltegravir as probe drugs, Badee et al.[28] measured the glucuronide formation rate in pooled or individual liver microsomes from both neonates and adults. They reported a dolutegravir glucuronidation rate of approximately 5.23 pmol/min in newborns and 8.99 pmol/min in adults and a raltegravir glucuronidation rate of approximately 2.01 pmol/min in newborns and 2.5 pmol/min in adults. Based on this information, neonatal UGT1A1 activity was estimated to be 60% of the adult value. Applying this fraction to the UGT1A1 Vmax value used in the adult PBPK model yielded a Vmax value of 4.55 nmol/min/mg which was implemented in the dolutegravir neonatal model. As no information about the variability of UGT1A1 tissue concentrations could be found in literature, geometric standard deviation of 1.4 was assumed. A sensitivity analysis was performed to investigate how quantitative changes in UGT1A1 activity propagate to the final model output (i.e. predicted dolutegravir and raltegravir pharmacokinetics in neonates).
Ontogeny of CYP3A4
CYP3A4 has a relatively low expression in newborns, but its ontogeny is rapid during early age. According to literature data, CYP3A4 activity in newborns was assumed to be 13% of that in adults[5] and hence, in the neonatal PBPK model, dolutegravir clearance via CYP3A4 (CLspec: specific enzymatic clearance rate) was decreased from 0.05 min−1 (adult value) to 0.0065 min−1. A sensitivity analysis was performed to investigate the effect of changes in CYP3A4 activity on predicted dolutegravir pharmacokinetics in neonates.
Ontogeny of Albumin
Albumin is the major plasma binding protein for dolutegravir and raltegravir and the ontogeny of this protein may affect the fraction unbound of these drugs. The adult unbound fraction values are 0.96, 0.007 and 0.17 for emtricitabine, dolutegravir and raltegravir, respectively. In PK-Sim®, the fraction unbound of albumin-bound drugs is automatically scaled with age resulting in a neonatal fraction unbound of 0.96, 0.0081 and 0.19 for emtricitabine, dolutegravir and raltegravir, respectively. A sensitivity analysis was performed to investigate how quantitative changes in the fraction unbound of each drug influence the predicted pharmacokinetics in neonates.
Evaluation of PBPK models
Visual evaluation of predictive model performance comprised superimposing predicted concentration-time profiles and observed clinical data. Predicted concentrations in the neonates were further evaluated by calculating the mean prediction error (MPE) and mean absolute prediction error (MAPE), and by comparing the observed and predicted elimination rate constant (kel). The latter was calculated from the predicted plasma concentration-time profile via non-compartmental analysis in R using a slightly modified form of the package ncappc.[29] A 2-fold error range of the predicted kel was considered adequate (i.e. 0.5 ≤ predicted/observed ≤ 2). Additionally, for dolutegravir and raltegravir, the neonatal drug concentration at 0 hour, i.e. time of delivery, was back-extrapolated from the last two concentration measurements in the individual and compared with the model-predicted concentration (for emtricitabine, subject-specific concentration information was not available and hence no back-extrapolation was conducted). Back-extrapolation was only conducted if the last two concentration measurements were decreasing. Finally, local sensitivity analyses were conducted to assess quantitatively how variations in relevant model inputs propagated to the model output, i.e. the predicted neonatal concentration-time profiles. Model inputs were included in the sensitivity analyses if the degree of uncertainty in this input was considered high. This was the case for clearance parameter values, fraction unbound and the predicted initial drug amount present in the neonate at delivery.
Results
Neonatal PBPK models for newborns
Emtricitabine
Observed and predicted emtricitabine concentration-time profiles in the maternal plasma and umbilical vein at delivery are shown in Figure 2A and B, respectively. On average, the newborns were delivered 4.9 hours after the last maternal emtricitabine dose. At this time, a total drug mass of 2.37 mg (9.58 μmol) was predicted to be present in all fetal compartments of the pregnancy PBPK model (Figure 2C). Integrating this drug mass into the different tissues of a whole-body neonatal PBPK model and predicting neonatal pharmacokinetics after delivery resulted in the concentration-time profile shown in Figure 3A. Generally, clearance was overestimated in the model. Nine out of 37 observed concentrations (24.3%) fell within the predicted 5th – 95th percentile range. MPE and MAPE were −71.5% and 85.8%, respectively. Table 3 lists the observed and predicted mean kel in neonates following birth. The ratio of predicted to observed mean kel was 2.51. Figure 4A shows the results of the sensitivity analysis where the renal elimination was decreased to 75%, 50% and 25% of the original value. Figure 4B shows the results of the sensitivity analysis where fraction unbound was set to ±5%, −10% and −15% of the original value in the model. Figure 5A shows the results of the sensitivity analysis where the drug amount was set to x0.2, x0.5, x2 and x5 of the original value in the model. The results of this sensitivity analysis suggested that a more precise estimation of the drug amount present in the fetus at delivery does not substantially improve the model performance.
Figure 2. Plasma concentration-time profiles of emtricitabine, dolutegravir and raltegravir for pregnant women and their newborns at delivery.
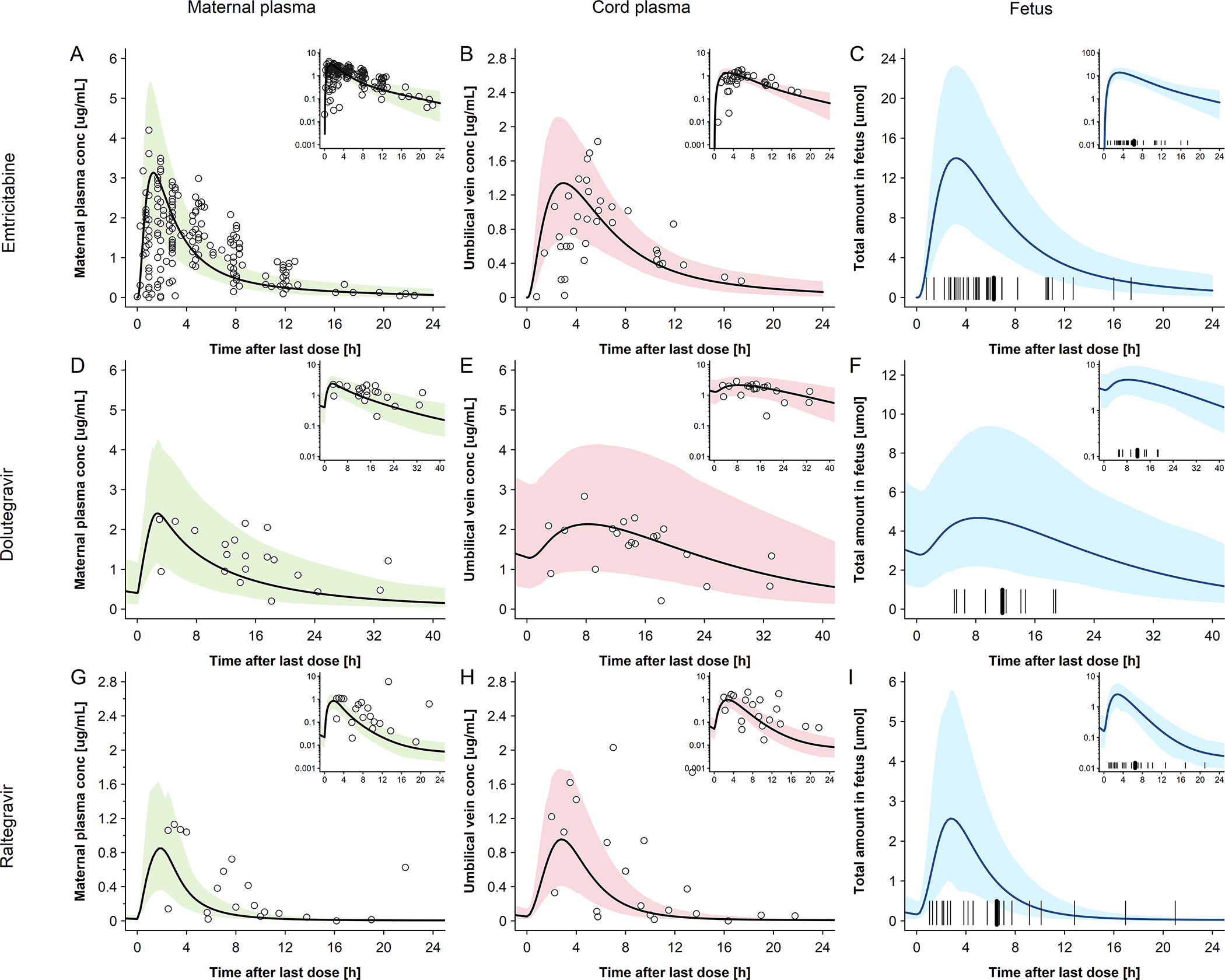
Semi-log scale figures are given as inset figure in the top right corners. Observed steady-state in vivo data were taken from in vivo study of Hirt et al.[17] IMPAACT P1026[10] and Clarke et al.[16] A: emtricitabine 400 mg single dose in pregnant women with an average gestational age of 39 weeks at delivery. Empty circles represent individual concentration data in the maternal plasma taken from in vivo study of Hirt et al.[17]; the line represents the predicted mean concentrations in the maternal plasma; The shaded area represents the predicted 5th – 95th percentile range of the prediction. B: emtricitabine 400 mg single dose in pregnant women with an average gestational age of 39 weeks at delivery. Empty circles represent individual concentration data in the umbilical vein taken from in vivo study of Hirt et al.[17] the line represents the predicted mean concentrations in the umbilical vein. The shaded area represents the predicted 5th – 95th percentile range of the prediction. C: emtricitabine 400 mg single dose in pregnant women with an average gestational age of 39 weeks at delivery. The line represents the predicted mean amount of emtricitabine in fetus. The marks represent the delivery time after last dose. D: dolutegravir 50 mg once a day in pregnant women with an average gestational age of 38 weeks at delivery. Empty circles represent individual concentration data in the maternal plasma taken from in vivo study of IMPAACT P1026;[10] the line represents the predicted mean concentration in the maternal plasma; the shaded area represents the predicted 5th – 95th percentile range of the prediction. E: dolutegravir 50 mg once a day in pregnant women with an average gestational age of 38 weeks at delivery. Empty circles represent individual concentration data in the umbilical vein taken from in vivo study of IMPAACT P1026;[10] the line represents the predicted mean concentration in the umbilical vein. The shaded area presents the predicted 5th – 95th percentile range of the prediction; F: dolutegravir 50 mg once a day in pregnant women with an average gestational age of 38 weeks at delivery; the line represents the predicted mean amount of dolutegravir in fetus. The marks represent the delivery time after last dose. G: raltegravir 400 mg twice a day in pregnant women with an average gestational age of 38 weeks at delivery. Empty circles represent individual concentration data in the maternal plasma taken from in vivo study of Clarke et al.[16]; the line represents the predicted mean concentrations in the maternal plasma; the shaded area represents the predicted 5th – 95th percentile range of the prediction. H: raltegravir 400 mg twice a day in pregnant women with an average gestational age of 38 weeks at delivery. Empty circles represent individual concentration data in the umbilical vein taken from in vivo study of Clarke et al.[16]; the line represents the predicted mean concentrations in the umbilical vein. The shaded area represents the predicted 5th – 95th percentile range of the prediction. I: raltegravir 400 mg twice a day in pregnant women with an average gestational age of 38 weeks at delivery. The line represents the predicted mean amount of raltegravir in fetus. The marks represent the delivery time after last dose.
Figure 3. Plasma concentration-time profiles of dolutegravir, raltegravir and emtricitabine in newborns following oral administration to the mother prior to delivery.
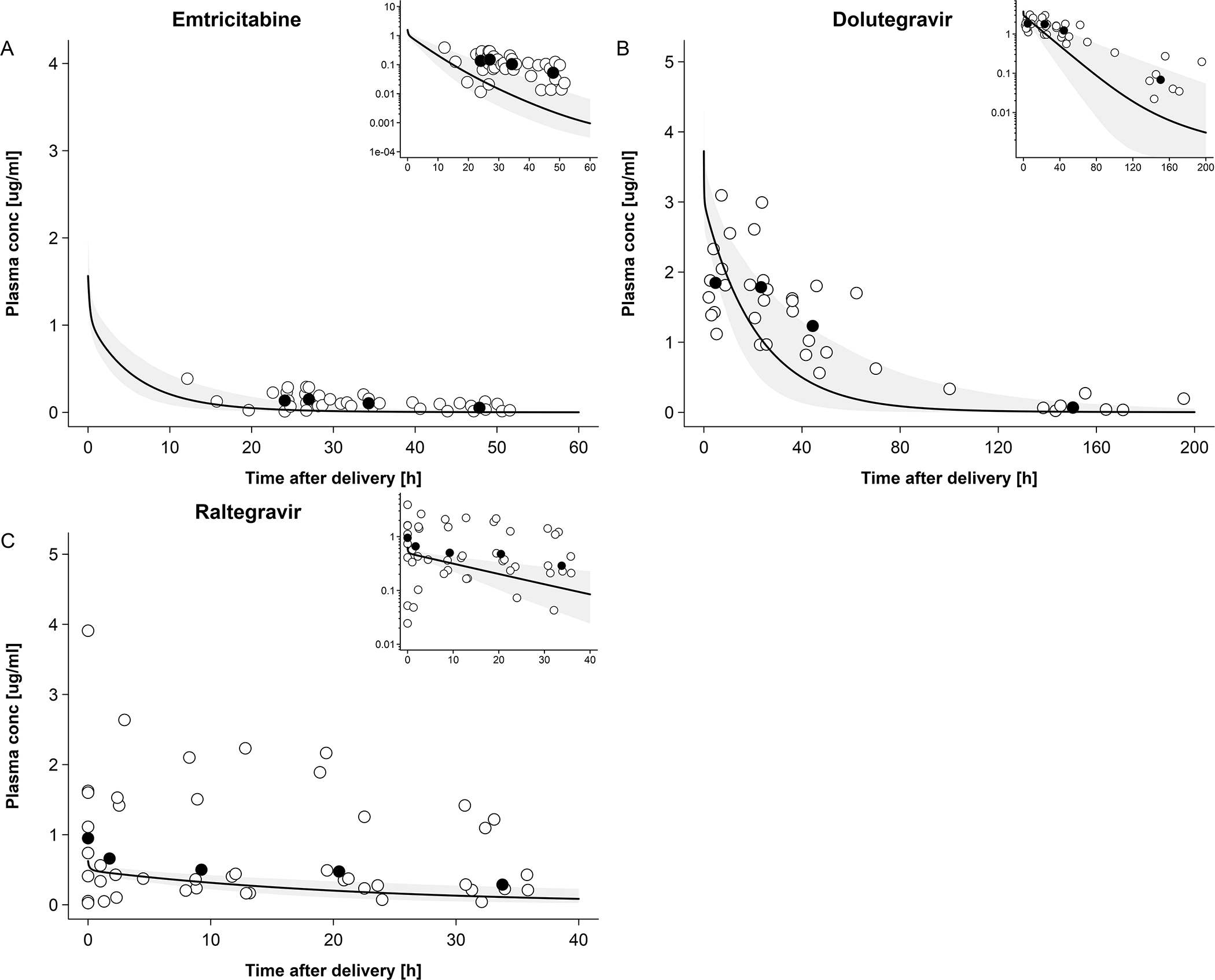
Empty circles represent observed individual concentration data in the newborns plasma and black circles represent median concentration data in the newborns; the solid line represents the predicted median plasma concentration in a population of newborns and the shaded area the predicted 5th – 95th percentile range. Semi-log scale figures are given as inset figure in the top right corners. A: emtricitabine plasma concentration in newborns; maternal dose of 400 mg single dose. Observed data were taken from Hirt et al.[17] B: dolutegravir plasma concentration in newborns; maternal dose of 50 mg once a day. Observed data were taken from IMPAACT P1026.[10] C: raltegravir plasma concentration in newborns; maternal dose of 400 mg twice a day. Observed data were taken from Clarke et al.[16]
Table 3:
Observed vs. predicted elimination rate constant (kel) in neonates after delivery
| Drug | Observed [h−1] | Predicted [h−1] |
|---|---|---|
|
| ||
| Emtricitabine | 0.0653a | 0.164 [0.030 – 0.225]b |
| Dolutegravir | 0.0238 [0.0079 – 0.0320] | 0.0429 [0.0195 – 0.0939] |
| Raltegravir | 0.0261 [0.00377 – 0.0745] | 0.0443 [0.00894 – 0.127] |
mean value
mean value [range]
Data expressed a median [range] unless noted otherwise. For emtricitabine, the observed elimination rate constant was taken from Hirt et al.;[17] for dolutegravir, the observed elimination rate constant was taken from Mulligan et al.;[10] and for raltegravir, the observed elimination rate constant was taken from Clarke et al.[16]
Figure 4. Sensitivity analysis of elimination of emtricitabine, dolutegravir and raltegravir in newborns.
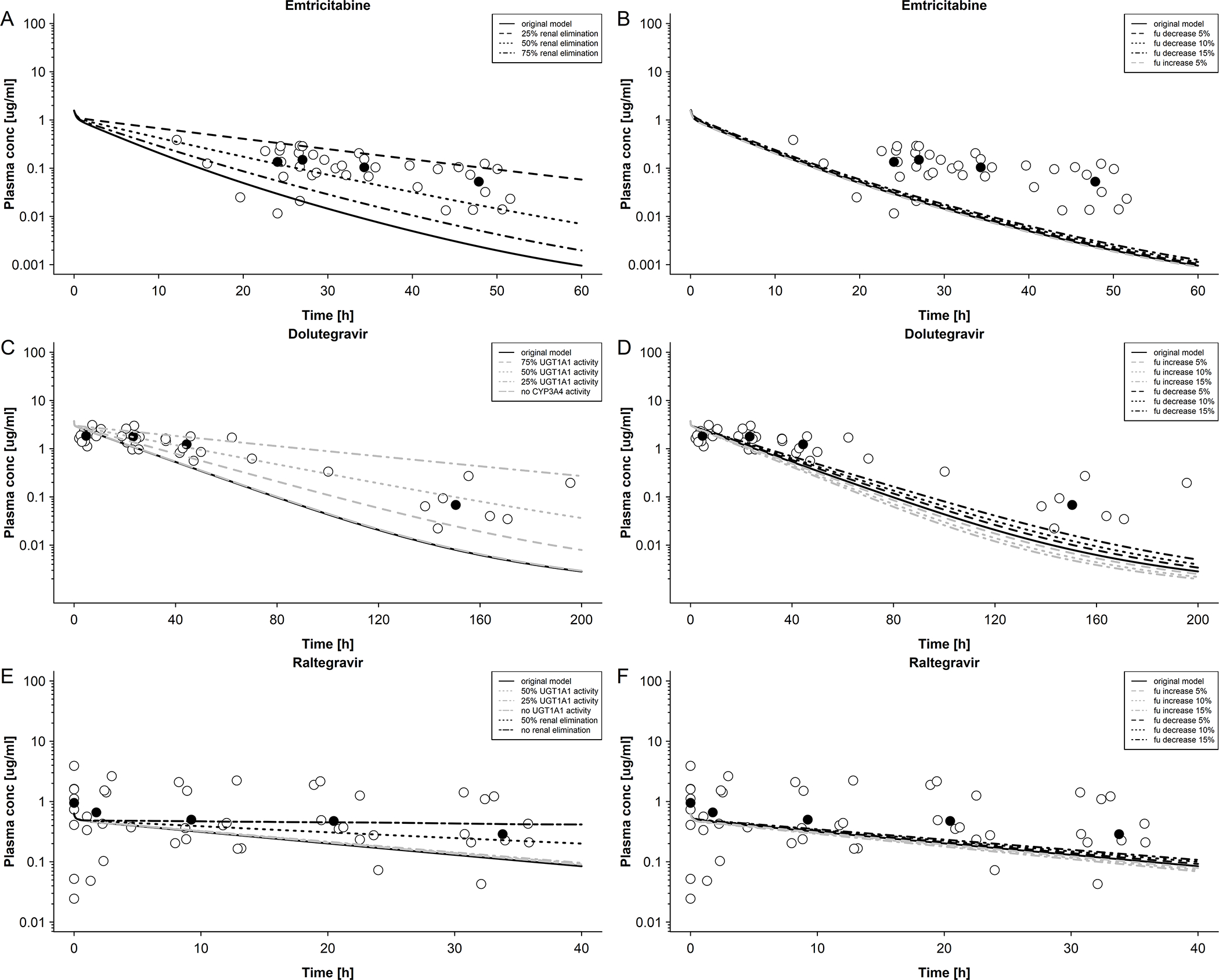
A: Emtricitabine plasma concentration in newborns with differing renal elimination; maternal dose of 400 mg single dose. Empty circles represent observed data were taken from Hirt et al[17] and black circles represent median observed data. B: Emtricitabine plasma concentration in newborns with differing unbound fractions; maternal dose of 400 mg single dose. Empty circles represent observed data were taken from Hirt et al.[17] and black circles represent median observed data. C: Dolutegravir plasma concentration in newborns with differing UGT1A1 and CYP3A4 activities; maternal dose of 50 mg once a day. Empty circles represent observed data were taken from IMPAACT P1026[10] and black circles represent median observed data. D: Dolutegravir plasma concentration in newborns with differing unbound fractions; maternal dose of 50 mg once a day. Empty circles represent observed data were taken from IMPAACT P1026[10] and black circles represent median observed data. E: Raltegravir plasma concentration in newborns with differing UGT1A1 activity and renal elimination; maternal dose of 400 mg twice a day. Empty circles represent observed data were taken from Clarke et al.[16] and black circles represent median observed data. F: Raltegravir plasma concentration in newborns with differing unbound fractions; maternal dose of 50 mg once a day. Empty circles represent observed data were taken from Clarke et al. [16] and black circles represent median observed data.
Figure 5. Sensitivity analysis of dosing of emtricitabine, dolutegravir and raltegravir in newborns.
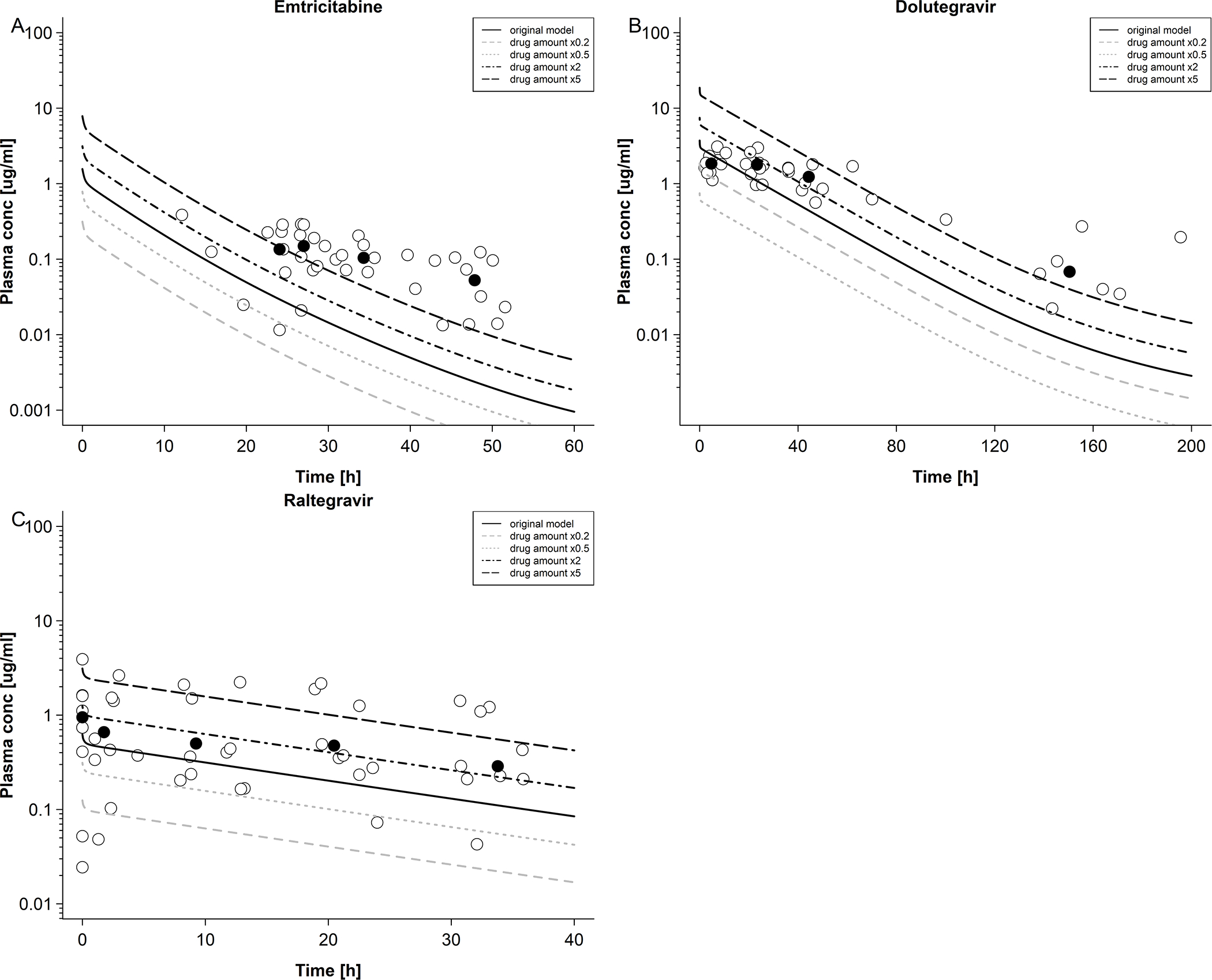
A: Emtricitabine plasma concentration in newborns with differing dosing; maternal dose of 400 mg single dose. Empty circles represent observed data were taken from Hirt et al [17] and black circles represent median observed data. B: dolutegravir plasma concentration in newborns with differing dosing; maternal dose of 50 mg once a day. Empty circles represent observed data were taken from IMPAACT P1026 [10] and black circles represent median observed data. C: raltegravir plasma concentration in newborns with differing dosing; maternal dose of 400 mg twice a day. Empty circles represent observed data were taken from Clarke et al. [16] and black circles represent median observed data.
Dolutegravir
Observed and predicted concentration-time profiles in the maternal plasma and umbilical vein at delivery are shown in Figure 2D and E, respectively. On average, the newborns were delivered 11.8 hours after the last dolutegravir dose had been administered to the mother. At this time, a total drug mass of 2.05 mg (4.89 μmol) was predicted to be present in all fetal compartments of the pregnancy PBPK model (Figure 2F). Integrating this drug mass into the different tissues of a whole-body neonatal PBPK model and predicting neonatal pharmacokinetics after delivery resulted in the concentration-time profile shown in Figure 3B. At the time of delivery (0 hours in Figure 3B), the model predicted a dolutegravir median concentration of 3.60 μg/mL in neonatal plasma; back-extrapolation of the observed plasma concentrations yielded a median concentration of 1.75 μg/mL with a range of 1.10 – 5.13 μg/mL at this time point. The model appeared to capture observed concentrations within the first day of life better than at later time points where, especially in the terminal phase, observed concentrations were underestimated. Twenty-three out of 38 observed concentrations (60.5%) were within the predicted 5th – 95th percentile range. The MPE and MAPE were −33.8% and 56.2%, respectively. Table 3 compares the observed and predicted kel in neonates after delivery. The ratio of predicted to observed median kel was 1.80. Figure 4C shows the results of the sensitivity analysis when reducing the apparent activity of UGT1A1 to 75%, 50% or 25% of the original value or that of CYP3A4 to 0% of the original value (i.e. removing CYP3A4 elimination from the model). Figure 4D shows the results of the sensitivity analysis when fraction unbound was set to ±5%, ±10% and ±15% of the original value in the model. Figure 5B shows the results of the sensitivity analysis where the drug amount was set to x0.2, x0.5, x2 and x5 of the original value in the model. Similar to the findings for emtricitabine, the result of the sensitivity analysis indicated that the initial drug amount in the neonate can, at best, only moderately improve the model performance.
Raltegravir
Observed and predicted concentration-time profiles in the maternal plasma and umbilical vein at delivery are shown in Figure 2G and H, respectively. On average, the newborns were delivered 4.6 hours after the last raltegravir dose had been administered to the mother. At this time, a total drug mass of 0.45 mg (1.01 μmol) was predicted to be present in all fetal compartments of the pregnancy PBPK model (Figure 2I). Integrating this drug mass into the different tissues of a whole-body neonatal PBPK model and predicting neonatal pharmacokinetics after delivery resulted in the concentration-time profile shown in Figure 3C. At the time of delivery (0 h in Figure 3C), the model predicted a raltegravir median concentration of 0.60 μg/mL in neonatal plasma; back-extrapolation of the observed plasma concentrations yielded a median concentration of 0.71 μg/mL with a range of 0.35 – 3.39 μg/mL at this time point. The concentrations in the terminal phase were slightly underestimated by the model, as was variability. Twelve out of 46 observed concentrations (26.1%) fell within the predicted 5th – 95th percentile range. The MPE and MAPE were 76.7% and 157.8%, respectively. Table 3 compares the observed and predicted kel in neonates after delivery. The ratio of predicted to observed median kel was 1.70. Figure 4E shows the results of the sensitivity analysis when either reducing the apparent activity of UGT1A1 to 50%, 25% or 0% (i.e. complete inactivation of UGT1A1) of the original value or without renal elimination. Figure 4F shows the results of the sensitivity analysis when fraction unbound was set to ±5%, ±10% and ±15% of the original value in the model. Figure 5C shows the results of the sensitivity analysis where the drug amount was set to x0.2, x0.5, x2 and x5 of the original value in the model in order to cover the range of predicted drug amounts in fetus (Figure 2C,F,I), suggesting that a more precise estimation of the drug amount present in the fetus at delivery may, at best, only slightly improve the model performance.
Discussion
This study aimed at developing a novel PBPK modeling framework for predicting neonatal pharmacokinetics of drugs that crossed the placenta following administration to the pregnant woman prior to delivery. Previously developed maternal-fetal PBPK models[14, 15] were used to estimate the total drug amount present in the fetus at the time of delivery. The usefulness and limitations of different techniques to model placental drug transfer have been discussed previously[30]. Structurally, the herein used maternal-fetal PBPK models do not contain a full-body fetal sub-model precluding a description of the distribution in different fetal organs in utero. To overcome this shortcoming a whole-body neonatal model without clearance processes was used as interim model to simulate drug distribution into all tissues within a time interval spanning the reported time between drug administration to the mother and birth. Simulated organ drug concentrations at the end of this time interval were then integrated as initial values into the final neonatal PBPK model and drug concentrations were predicted in the first hours of postnatal life.
Our current understanding of early neonatal pharmacokinetics is sparse and only few studies can be found in the literature that have attempted to model neonatal pharmacokinetics following prenatal maternal drug administration.[31, 32] Apart from limited clinical data, one reason for the scarcity of models applied to this scenario might be the technical difficulties associated with using a model structure that undergoes dramatic changes (i.e. a fetal-sub-model embedded in a larger maternal model for describing in utero exposure, followed by the structural elimination of the maternal model and some parts of the fetal model, such as placenta, umbilical cord and amniotic fluid, for describing postnatal exposure). There was only one previously published neonatal PBPK model for alprazolam incorporated an intravenous administration of the retrospectively identified dose needed to result in the observed neonatal peak plasma concentration.[32] The modeling framework presented in this paper is the first of its kind that mechanistically links prenatal with postnatal drug pharmacokinetics.
Although novel, the current modeling framework also has technical limitations and the current model needs further confirmation using additional drugs administered to neonates to further qualify the neonatal model. For example, neonatal washout kinetics could only be described for the mean total drug amount present in the fetus immediately before birth. As can be seen in Figure 2, both delivery time and total drug amount in the fetus varied considerably between different individuals. In carrying over only the mean drug amount in the fetus, the neonatal model was biased towards an underestimation of the variability in postnatal drug concentrations as evident in Figure 3.
Understanding variability is very important for population predictions. In the models for emtricitabine and raltegravir, the variability in drug exposure was underestimated (Figure 3A and 3C). The inter-individual variability in the predicted drug exposure can be influenced by multiple factors such as age, weight, height, ontogeny processes relating to enzyme maturation, genetic polymorphisms and other factors. The current model only involved the standard variabilities in anatomical and physiological parameters implemented per default in PK-Sim® [5]. Variability in predicted concentrations in the fetus were not being carried over from the fetus to neonates. In the emtricitabine and raltegravir models, the sample may not be large enough to adequately represent the variability of the above factors which could have led to the current low variability in the prediction. Additional information in newborns is unavailable or is very limited in the literature, such as the fact that the OCT2 ontogeny factor comes from one neonate’s data. Further in vitro and in vivo investigation is needed in these areas. Additionally, the PBPK model did not account for host genetic polymorphism of UGT1A1. It is currently unclear whether UGT1A1 polymorphisms correlate with raltegravir metabolism.[16] If future studies can establish a significant correlation, this factor should also be incorporated in PBPK models to adequately reflect variability in population predictions. The unspecified enzyme contributing to the clearance of emtricitabine also could affect the prediction of neonatal PK.
The prediction results in neonates suggested that, overall, predictive performance of the neonatal PBPK model could be improved. This could principally be accomplished by a more accurate estimation of the transplacentally acquired drug amount at birth and/or the elimination processes. The neonatal plasma concentrations of dolutegravir and raltegravir predicted at 0 h after delivery fell within the range of the back-extrapolated measured concentrations, suggesting that the models were generally able to capture the drug amount present in the fetus at delivery. Furthermore, the sensitivity analysis on the initial drug amount in the neonate at 0 hour after delivery (Figure 5) suggested that the mismatch between predicted and observed kinetics can, at best, only partially improved by refining this factor. In contrast, sensitivity analysis on elimination processes (Figure 4) identified the extent of elimination as explanatory variable. This indicates that the quantitative information on relevant elimination pathways that was taken from the literature and integrated in the neonatal models resulted in an overestimation of clearance.
Specifically, the sensitivity analysis for emtricitabine showed that OCT2 activity, which only relates to tubular secretion, significantly influenced its elimination (Figure 4A), whereas changes in the fraction unbound did not appear to significantly affect elimination in newborns as was expected given its low protein binding (Figure 4B). It should be noted that the uncertainty in OCT2 ontogeny applied in the model (30% of the adult value) is relatively high because the renal cortical tissue sample was obtained from one term newborn only.[27] Furthermore, samples from infants reported by the same authors showed a high variability with an approximately 6-fold range.[27] Results of the sensitivity analysis suggested that OCT2 activity in term newborns is only about 7.5% – 15% of the adult activity (i.e. 25% – 50% of the value implemented in the model). Additional in vitro and clinical studies are needed to investigate the ontogeny of OCT2 in newborns.
Results of the sensitivity analysis for dolutegravir demonstrated that UGT1A1 plays a major role in dolutegravir elimination in newborns, while the role of CYP3A4 appeared to be insignificant (Figure 4C), probably because of the low expression of this enzyme in the neonate where CYP3A7 has a similar role than CYP3A4 in adults.[33] Although the metabolism of DTG involves UGT1A3 and UGT1A9, there was no UGT1A3 and UGT1A9 involvement in the dolutegravir model. In adults, relative contribution of these enzymes to total clearance is small [22], but in neonates this could change. No information on UGT1A3 activity at birth could be found in the literature. Miyagi et al.[34] found that UGT1A9 has no activity in newborns. The knowledge gap regarding UGT1A3 ontogeny introduces some uncertainty into the prediction and is a limitation of the dolutegravir model. Unfortunately, similar to UGT1A3, there are no data on the potential extent to which CYP3A7 may metabolize dolutegravir thus precluding the incorporation of this enzyme in the model. However, although there was no CYP3A7 involvement in the clearance of dolutegravir, the influence on model performance is minor because the sensitivity analysis demonstrated that an increase of CYP3A4 barely affected the model prediction. Since CYP3A7 has a function similar to CYP3A4, the results suggest that involvement of CYP3A7 would result in a minor effect on predicted neonatal drug concentrations. In addition, the mean delivery time also introduces uncertainty regarding the variability in the predicted fetal drug amount at delivery which was carried over as initial drug amount in the neonatal model. However, the sensitivity analyses of differing initial drug amounts in the neonates were conducted (Figure 5), suggesting that different initial drug amounts did not affect the elimination pathway of the drugs and may, at best, only slightly improve model performance. Alterations in the fraction unbound only moderately affected predicted dolutegravir pharmacokinetics (Figure 4D) which is a surprising finding given that dolutegravir appears to be a highly protein-bound drug with capacity-limited clearance in adults. Taken together, these results suggest that UGT1A1 clearance of dolutegravir in newborns is, on average, about 30% of that in adults.
Interestingly, these findings could not be confirmed for raltegravir where changes in UGT1A1 activity barely affected the predicted plasma concentration-time profile (Figure 4E). However, renal clearance appeared to be a highly sensitive parameter for raltegravir pharmacokinetics. In vitro experiments demonstrated that raltegravir is a substrate for P-glycoprotein,[23] as well as organic anion transporter (OAT) 1 and peptide transporter (PepT) 1,[35] but overall, the contribution of these transporters to total renal clearance seems to be negligible with glomerular filtration likely being the dominant process. This suggests that GFR may be overestimated in the neonatal PBPK model; however, clinical data for drugs predominantly excreted via glomerular filtration in neonates are needed to draw firm conclusions. The picture is further complicated by the fact that raltegravir may undergo enterohepatic recirculation in neonates causing raltegravir concentrations to increase after birth within the first 12 – 24 h in some neonates as was hypothesized by Clarke et al.[16] In fact, for some neonates, raltegravir plasma concentrations were observed to increase during the sampling intervals. It is yet unclear whether this increase is caused by enterohepatic circulation or redistribution processes.[36] Further clinical studies are needed to disentangle these processes and their effect on raltegravir pharmacokinetics.
Breastfeeding may constitute another covariate affecting drug pharmacokinetics. Apart from drug intake via breastfeeding, breast milk may influence neonatal drug concentrations via its effect on UGT1A1 activity. Previous studies have demonstrated that the endogenous compound 5β-pregnane-3α,20β-diol present in breast milk inhibits enzyme activity and transcriptional activity of UGT1A1.[37] Also, various fatty acids present in breast milk have been shown to strongly inhibit UGT1A1 activity.[38] While of general concern for neonatal populations, these findings may be not relevant for the current model predictions since no newborn of the dolutegravir and raltegravir groups was reportedly breastfed. Additionally, the presented findings of the sensitivity analysis suggested that, in contrast to adults, UGT1A1 does not appear to be the major enzyme responsible for raltegravir metabolism with unchanged renal elimination as the predominating clearance pathway (Figure 4E).
Conclusion
Given the challenges of conducting clinical trials in newborns, alternative tools such as PBPK models could help improve our understanding of pharmacokinetics in this vulnerable population. To date, these models have been almost exclusively focused on the pharmacokinetics of drugs administered directly to the neonate, while transplacentally acquired drugs in neonates have been often ignored. This study presents a first attempt on how to model perinatal pharmacokinetics along the temporal axis of maternal drug administration, placental transfer, delivery, and postnatal life. The novel workflow presented is the first of its kind to (semi)mechanistically link prenatal with postnatal drug pharmacokinetics in a PBPK framework. By means of sensitivity analyses, the ontogeny of OCT2 and UGT1A1 was investigated at the first days of life, suggesting that the activity is ~11% and ~30% of that in adults, respectively. However, to verify the presented findings, further confirmation using additional drugs administered to the mother and/or neonate is strictly necessary. Once thoroughly verified, coupled maternal-fetal-neonatal PBPK models could improve the understanding of early neonatal pharmacokinetics. This would support informed decision making concerning the correct neonatal dose of drugs administered directly to the neonate or indirectly to the mother.
Key Points.
The current study provides a physiologically based pharmacokinetic (PBPK) framework to (semi)mechanistically predict neonatal drug exposure after maternal dosing.
During the first postnatal days following term delivery, the activity of organic cation transporter (OCT) 2 and uridine diphosphate glucuronosyltransferase (UGT) 1A1 seems to be approximately 11% and 30% of that in adults, respectively.
Applying this workflow to additional drugs could enhance confidence in the presented findings and improve the mechanistic understanding of pharmacokinetics during the first days of postnatal life.
Funding:
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under Award Numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. The NIH awards numbers 5T32HD087969-03 and 5T32HD087969-02 also support this project.
Footnotes
Conflict of Interest/Disclosure: The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Dr. André Dallmann is an employee of Bayer AG, a company which is part of the Open Systems Pharmacology (OSP) member team and involved in OSP software development used in this study. The results from this study will be presented in part at the American College of Clinical Pharmacology Annual Meeting, September 2020.
Disclaimer: The opinions expressed in this article are those of the authors and should not be interpreted as the position of the U.S. Food and Drug Administration or of the National Institutes of Health. No broader FDA policies or perspectives are intended nor should be inferred. The FDA does not recommend any specific PBPK software.
References
- 1.Pariente G, Leibson T, Carls A, Adams-Webber T, Ito S, Koren G. Pregnancy-Associated Changes in Pharmacokinetics: A Systematic Review. PLoS Med. 2016; 13(11): e1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J, Avant D, Green D, Seo S, Fisher J, Mulberg AE, et al. A Survey of Neonatal Pharmacokinetic and Pharmacodynamic Studies in Pediatric Drug Development. Clin Pharmacol Ther. 2015; 98(3): 328–35. [DOI] [PubMed] [Google Scholar]
- 3.Van Den Anker J, Reed MD, Allegaert K, Kearns GL. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J Clin Pharmacol. 2018; 58(Suppl 10): S10–S25. [DOI] [PubMed] [Google Scholar]
- 4.Ku LC, Smith PB. Dosing in neonates: special considerations in physiology and trial design. Pediatr Res. 2015; 77(1–1): 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claassen K, Thelen K, Coboeken K, Gaub T, Lippert J, Allegaert K, et al. Development of a Physiologically-Based Pharmacokinetic Model for Preterm Neonates: Evaluation with In Vivo Data. Curr Pharm Des. 2015; 21(39): 5688–98. [DOI] [PubMed] [Google Scholar]
- 6.Michelet R, Bocxlaer JV, Vermeulen A. PBPK in Preterm and Term Neonates: A Review. Curr Pharm Des. 2017; 23(38): 5943–5954. [DOI] [PubMed] [Google Scholar]
- 7.Abduljalil K, Pan X, Pansari A, Jamei M, Johnson TN. Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population. Clin Pharmacokinet. 2020; 59(4): 501–518. [DOI] [PubMed] [Google Scholar]
- 8.Stek AM, Best BM, Luo W, Capparelli E, Burchett S, Hu C, et al. Effect of pregnancy on emtricitabine pharmacokinetics. HIV Med. 2012; 13(4): 226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colbers AP, Hawkins DA, Gingelmaier A, Kabeya K, Rockstroh JK, Wyen C, et al. The pharmacokinetics, safety and efficacy of tenofovir and emtricitabine in HIV-1-infected pregnant women. AIDS. 2013; 27(5): 739–48. [DOI] [PubMed] [Google Scholar]
- 10.Mulligan N, Best BM, Wang J, Capparelli EV, Stek A, Barr E, et al. Dolutegravir pharmacokinetics in pregnant and postpartum women living with HIV. AIDS. 2018; 32(6): 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bollen P, Freriksen J, Konopnicki D, Weizsacker K, Hidalgo Tenorio C, Molto J, et al. The Effect of Pregnancy on the Pharmacokinetics of Total and Unbound Dolutegravir and Its Main Metabolite in Women Living With Human Immunodeficiency Virus. Clin Infect Dis. 2020; 10.1093/cid/ciaa006. [DOI] [PubMed] [Google Scholar]
- 12.Watts DH, Stek A, Best BM, Wang J, Capparelli EV, Cressey TR, et al. Raltegravir pharmacokinetics during pregnancy. J Acquir Immune Defic Syndr. 2014; 67(4): 375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blonk MI, Colbers AP, Hidalgo-Tenorio C, Kabeya K, Weizsacker K, Haberl AE, et al. Raltegravir in HIV-1-Infected Pregnant Women: Pharmacokinetics, Safety, and Efficacy. Clin Infect Dis. 2015; 61(5): 809–16. [DOI] [PubMed] [Google Scholar]
- 14.Liu XI, Momper JD, Rakhmanina N, Van Den Anker JN, Green DJ, Burckart GJ, et al. Physiologically Based Pharmacokinetic Models to Predict Maternal Pharmacokinetics and Fetal Exposure to Emtricitabine and Acyclovir. J Clin Pharmacol. 2020; 60(2): 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu XI, Momper JD, Rakhmanina NY, Green DJ, Burckart GJ, Cressey TR, et al. Prediction of Maternal and Fetal Pharmacokinetics of Dolutegravir and Raltegravir Using Physiologically Based Pharmacokinetic Modeling. Clin Pharmacokinet. 2020; 59(11):1433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke DF, Acosta EP, Rizk ML, Bryson YJ, Spector SA, Mofenson LM, et al. Raltegravir pharmacokinetics in neonates following maternal dosing. J Acquir Immune Defic Syndr. 2014; 67(3): 310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirt D, Urien S, Rey E, Arrive E, Ekouevi DK, Coffie P, et al. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1-infected pregnant women and their neonates. Antimicrob Agents Chemother. 2009; 53(3): 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waitt C, Orrell C, Walimbwa S, Singh Y, Kintu K, Simmons B, et al. Safety and pharmacokinetics of dolutegravir in pregnant mothers with HIV infection and their neonates: A randomised trial (DolPHIN-1 study). PLoS Med. 2019; 16(9): e1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lippert J, Burghaus R, Edginton A, Frechen S, Karlsson M, Kovar A, et al. Open Systems Pharmacology Community-An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacometrics Syst Pharmacol. 2019; 8(12): 878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dallmann A, Solodenko J, Ince I, Eissing T. Applied Concepts in PBPK Modeling: How to Extend an Open Systems Pharmacology Model to the Special Population of Pregnant Women. CPT Pharmacometrics Syst Pharmacol. 2018; 7(7): 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Food and Drug Administration: Emtriva™ (emtricitabine) Capsules Access at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/21500_emtriva_lbl.pdf (accessed on June 10, 2020). 2003;
- 22.Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013; 41(2): 353–61. [DOI] [PubMed] [Google Scholar]
- 23.Kassahun K, Mcintosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, et al. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug metabolism and disposition: the biological fate of chemicals. 2007; 35(9): 1657–63. [DOI] [PubMed] [Google Scholar]
- 24.Dallmann A, Ince I, Meyer M, Willmann S, Eissing T, Hempel G. Gestation-Specific Changes in the Anatomy and Physiology of Healthy Pregnant Women: An Extended Repository of Model Parameters for Physiologically Based Pharmacokinetic Modeling in Pregnancy. Clin Pharmacokinet. 2017; 56(11): 1303–1330. [DOI] [PubMed] [Google Scholar]
- 25.Dallmann A, Ince I, Solodenko J, Meyer M, Willmann S, Eissing T, et al. Physiologically Based Pharmacokinetic Modeling of Renally Cleared Drugs in Pregnant Women. Clin Pharmacokinet. 2017; 56(12): 1525–1541. [DOI] [PubMed] [Google Scholar]
- 26.Reznicek J, Ceckova M, Cerveny L, Muller F, Staud F. Emtricitabine is a substrate of MATE1 but not of OCT1, OCT2, P-gp, BCRP or MRP2 transporters. Xenobiotica. 2017; 47(1): 77–85. [DOI] [PubMed] [Google Scholar]
- 27.Cheung KWK, Van Groen BD, Spaans E, Van Borselen MD, De Bruijn A, Simons-Oosterhuis Y, et al. A Comprehensive Analysis of Ontogeny of Renal Drug Transporters: mRNA Analyses, Quantitative Proteomics, and Localization. Clin Pharmacol Ther. 2019; 106(5): 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badee J, Qiu N, Collier AC, Takahashi RH, Forrest WF, Parrott N, et al. Characterization of the Ontogeny of Hepatic UDP-Glucuronosyltransferase Enzymes Based on Glucuronidation Activity Measured in Human Liver Microsomes. J Clin Pharmacol. 2019; 59(Suppl 1):S42–S55. [DOI] [PubMed] [Google Scholar]
- 29.Acharya C, Hooker AC, Turkyilmaz GY, Jonsson S, Karlsson MO. A diagnostic tool for population models using non-compartmental analysis: The ncappc package for R. Comput Methods Programs Biomed. 2016; 127:83–93. [DOI] [PubMed] [Google Scholar]
- 30.Codaccioni M, Brochot C. Assessing the impacts on fetal dosimetry of the modelling of the placental transfers of xenobiotics in a pregnancy physiologically based pharmacokinetic model. Toxicol Appl Pharmacol. 2020; 409:115318. [DOI] [PubMed] [Google Scholar]
- 31.Lommerse J, Clarke D, Kerbusch T, Merdjan H, Witjes H, Teppler H, et al. Maternal-Neonatal Raltegravir Population Pharmacokinetics Modeling: Implications for Initial Neonatal Dosing. CPT Pharmacometrics Syst Pharmacol. 2019; 8(9): 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto K, Fukushima S, Mishima Y, Hashimoto M, Yamakawa K, Fujioka K, et al. Pharmacokinetic assessment of alprazolam-induced neonatal abstinence syndrome using physiologically based pharmacokinetic model. Drug Metab Pharmacokinet. 2019; 34(6): 400–402. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Lampe JN. Neonatal cytochrome P450 CYP3A7: A comprehensive review of its role in development, disease, and xenobiotic metabolism. Arch Biochem Biophys. 2019; 673:108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyagi SJ, Milne AM, Coughtrie MW, Collier AC. Neonatal development of hepatic UGT1A9: implications of pediatric pharmacokinetics. Drug Metab Dispos. 2012; 40(7): 1321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moss DM, Kwan WS, Liptrott NJ, Smith DL, Siccardi M, Khoo SH, et al. Raltegravir is a substrate for SLC22A6: a putative mechanism for the interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2011; 55(2): 879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bunglawala F, Rajoli RKR, Mirochnick M, Owen A, Siccardi M. Prediction of dolutegravir pharmacokinetics and dose optimization in neonates via physiologically based pharmacokinetic (PBPK) modelling. J Antimicrob Chemother. 2020; 75(3): 640–647. [DOI] [PubMed] [Google Scholar]
- 37.Ota Y, Maruo Y, Matsui K, Mimura Y, Sato H, Takeuchi Y. Inhibitory effect of 5beta-pregnane-3alpha,20beta-diol on transcriptional activity and enzyme activity of human bilirubin UDP-glucuronosyltransferase. Pediatr Res. 2011; 70(5): 453–7. [DOI] [PubMed] [Google Scholar]
- 38.Shibuya A, Itoh T, Tukey RH, Fujiwara R. Impact of fatty acids on human UDP-glucuronosyltransferase 1A1 activity and its expression in neonatal hyperbilirubinemia. Sci Rep. 2013; 3:2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moss DM, Siccardi M, Murphy M, Piperakis MM, Khoo SH, Back DJ, et al. Divalent metals and pH alter raltegravir disposition in vitro. Antimicrob Agents Chemother. 2012; 56(6): 3020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.US Food and Drug Administration: Clinical Pharmacology Review: Dolutegravir, GSK1349572. Access at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204790Orig1s000ClinPharmR.pdf (accessed on June 29, 2020). 2012;
- 41.Laufer R, Paz OG, Di Marco A, Bonelli F, Monteagudo E, Summa V, et al. Quantitative prediction of human clearance guiding the development of Raltegravir (MK-0518, isentress) and related HIV integrase inhibitors. Drug Metab Dispos. 2009; 37(4): 873–83. [DOI] [PubMed] [Google Scholar]
- 42.Moss DM, Siccardi M, Back DJ, Owen A. Predicting intestinal absorption of raltegravir using a population-based ADME simulation. J Antimicrob Chemother. 2013; 68(7): 1627–34. [DOI] [PubMed] [Google Scholar]
- 43.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005; 94(6): 1259–76. [DOI] [PubMed] [Google Scholar]
- 44.Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006; 95(6): 1238–57. [DOI] [PubMed] [Google Scholar]