
Fig. 3.
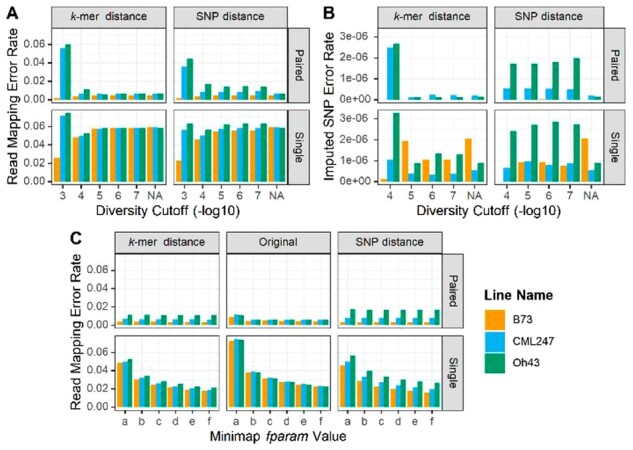
Effect of parameters on accuracy. (A) Read mapping error rate for the whole genome as a function of the maximum diversity (mxDiv) parameter for determining consensus haplotypes, read type (paired-end, single) and distance matrix method (kmer, SNP). Read mapping error rate is the number of reads not mapping to the target haplotype divided by the total number of reads. NA labels the result of mapping against the original, non-consensus haplotypes. (B) Imputed SNP error rate for paired and single-end reads for different values of the diversity cutoff and consensus method. Error rate equals the number of wrong SNP calls divided by the number of base pairs of sequence. Where the B73 bar is absent, the error rate was zero. (C) Read mapping error as a function of minimizer redundancy controlled by the minimap2 f-parameter. f parameter values are (a) f1000,5000 [default]; (b) f5000,6000; (c) f10000,11000; (d)15000,16000; (e) f20000,21000; (f) f25000,26000. mxDiv = 1e-4