
Figure 4. In vivo models and pathology of ocular calpainopathies.
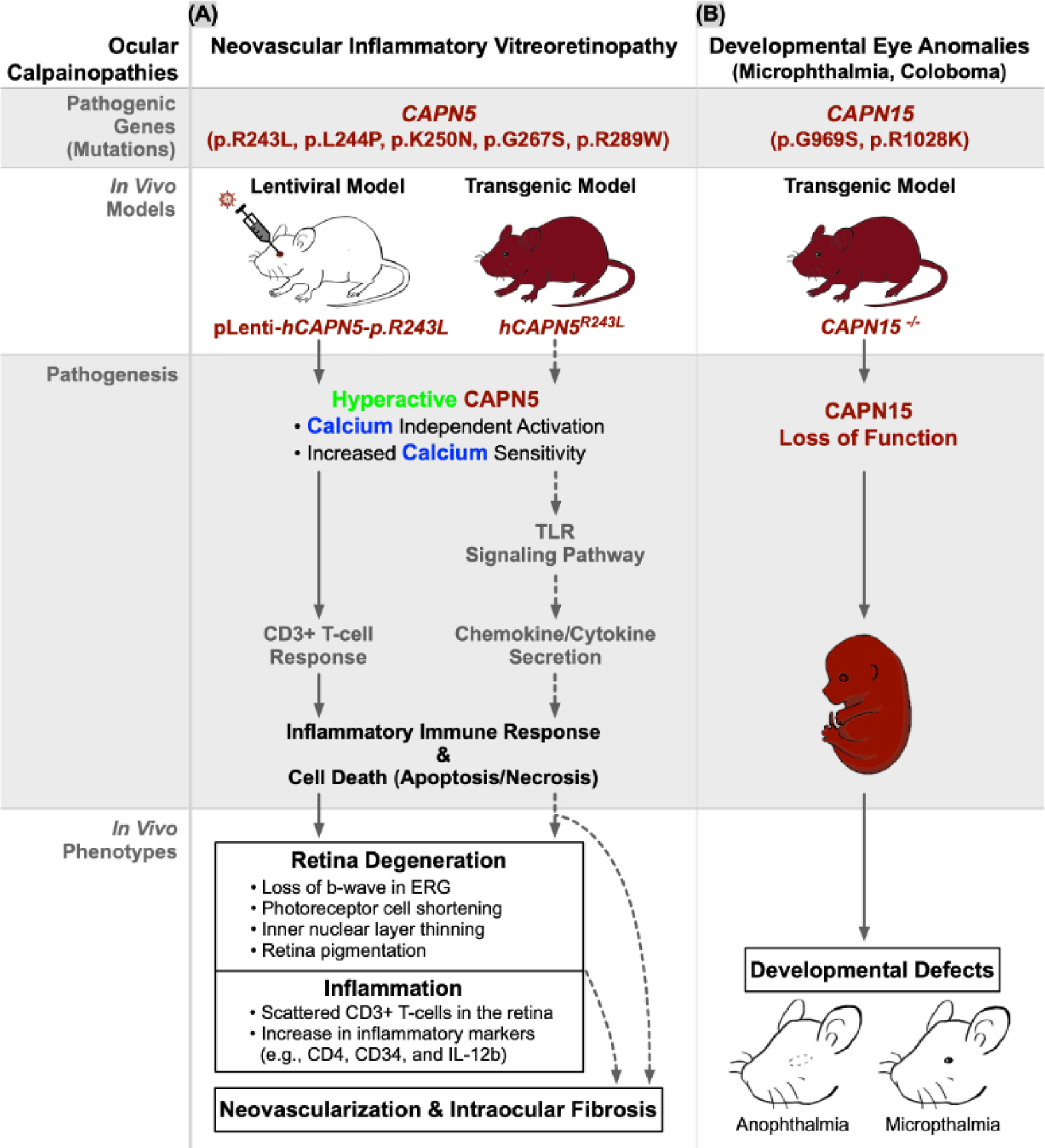
Calpains are involved in a large variety of ocular diseases called calpainopathies that lead to manifestations in (A) neovascular inflammatory vitreoretinopathy (NIV) and (B) developmental eye anomalies. CAPN5 mutations cause CAPN5 hyperactivity either by calcium independent activation or increasing its sensitivity to intracellular calcium concentrations. There are two reported mouse models of NIV to model CAPN5 mutations—a lentiviral model and a transgenic model. NIV models were created based on biochemical findings of five known NIV mutations in the catalytic PC domains that increase CAPN5 catalytic activity: three mutations (p.R243L, p.L244P, and p.K250N) located on the G1 gating loop of the PC2 subdomain (which may impede the G1-loop’s ability to gate substrate binding thus making it constantly available for target proteolysis [8,28,29]); a fourth, p.G267S, on the PC2L2 loop (distantly located from the active site suggesting the mutation works through allosteric effects [55]) and the fifth, p.R289W, located on the G2 gating loop (which disrupts a key conserved calpain structural regulatory motif—the tryptophan “wedge” leading to calcium-independent calpain hyperactivity[149]). A transgenic CAPN15 knockout model to model developmental eye anomalies resulted in developmental defects such as anophthalmia and microphthalmia.