

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the causative agent of coronavirus disease 2019 (COVID‐19), has caused millions of deaths in the past two years. Although initially little was understood about this virus, recent research has significantly advanced and landed interferons (IFNs) in the spotlight. While Type I and III IFN have long been known as central to antiviral immunity, in the case of COVID‐19 their role was initially controversial. However, the protective function of IFN is now well supported by the identification of human deficiencies in IFN responses as a predictor of disease severity. Here, we will review the cell types and pathways that lead to IFN production as well as the importance of IFN timing and location for disease outcome. We will further discuss the mechanisms that SARS‐CoV‐2 uses to evade IFN responses, and the current efforts to implement IFNs as therapeutics in the treatment of COVID‐19. It is essential to understand the relationships between SARS‐CoV‐2 and IFN to better inform treatments that exploit IFN functions to alleviate COVID‐19.
Keywords: COVID‐19, immune evasion, interferon, SARS‐CoV‐2
1. INTRODUCTION
A central challenge of the innate immune system is to use hard‐coded sensors to recognize and respond to unknown and evolving challenges. This is especially salient with respect to viral infections, which evolve quickly, and may spill freely between related species. 1 Therefore, over the course of their lifetime, a long‐lived organism can expect to encounter viral infections that did not even exist at the time of their genesis. To this end, organisms have evolved a web of responses which allows for the recognition of diverse‐yet‐conserved microbial patterns (reviewed in 2 ). The millennia‐long coevolution of viruses alongside these antiviral responses has tangled this web into knots. However, if the web can be said to have a center, it could be that of interferons (IFNs). IFNs are ancient cytokines, existing in all jawed vertebrates, 3 where they have undergone multiple rounds of duplication and diversification (reviewed in 4 ). In humans, three major families of IFN are currently recognized (I, II, and III) (reviewed in 5 ). Type I and III IFNs are primarily produced as the first line of alarm and can be generated by most cell types, while Type II IFNs are mostly made secondarily by specialized immune subsets (ie, NK cells or T cells) (reviewed in 6 ). Therefore, Type I and III IFN responses are crucial to the initial control of viral infections. 7 These IFN are induced by an incredible diversity of systems including dozens of pattern recognition receptors (PRRs) (eg, TLRs, RLRs, STING) (reviewed in 2 ). Once produced, IFNs activate intrinsic antiviral state in the cells that sense them via specific IFN receptors, a state that is characterized by the eponymous IFN Stimulated Gene (ISG) signature. 5 Additionally, IFNs activate higher order processes (ie, antigen presentation, NK cell activity, T‐cell expansion, differentiation, and function) which are critical for the control of viral infections (reviewed in 7 , 8 ).
The rise of the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) pandemic in 2019 was an abject lesson in the threatening potential of novel‐virus emergence. The pandemic has also sparked an inundating wealth of studies focused on the SARS‐CoV‐2‐induced responses that protect, or harm, its hosts. Given the central role of IFN in the control of viral infections, it is thus unsurprising that these cytokines have emerged as critical components in the control of SARS‐CoV‐2. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 The effects that IFNs play during SARS‐CoV‐2 infection appear, however, to be complex, nuanced, and highly dependent on context. IFNs are, for example, clearly crucial for the control of SARS‐CoV‐2, 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 but also potential contributors to the severest form of 2019 coronavirus associated disease (COVID‐19). 31 , 32 , 33 , 34 , 35 This is further complicated by the capacity for evasion of human IFN responses clearly present, but perhaps yet untuned, in this novel virus (reviewed in 36 ). In this review, we summarize the burgeoning field at the intersection of SARS‐CoV‐2 infection and its relationship to IFN, and push toward a deeper understanding of how the existing web of IFN regulation has incorporated this novel disease as well as the complexities and seeming paradoxes therein.
2. THE COMPLEX ROLE OF IFN IN SARS‐COV‐2 CONTROL AND COVID‐19
2.1. IFN responses are protective against COVID‐19
While there are certainly complexities in the overarching role of IFNs in COVID‐19 pathology, it is also clear that initial IFN responses are highly protective against SARS‐CoV‐2 infection. This is partly supported by multiple avenues of evidence in people with diverse deficiencies in the IFN response pathway. Indeed, genetic deficiencies related to IFN induction 9 , 10 , 12 , 23 , 30 as well as IFN neutralizing auto‐antibodies 11 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 24 , 25 , 26 , 27 , 28 , 29 have been identified in people with severe COVID‐19. These correlations are epidemiologically significant. For example, 1.5% of severe COVID‐19 cases can be traced to specific deficiencies in TLR7, 12 while ~10% of people with severe COVID‐19 have autoantibodies against one or more Type I or Type III IFNs. 11 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 24 , 25 , 26 , 27 , 28 , 29 Furthermore, allelic variants of the ISG OAS1 are also predictive of COVID‐19 severity. 37 , 38 , 39 , 40 These data definitively show that IFNs are indispensable for the control of SARS‐CoV‐2 and the prevention of severe COVID‐19.
2.2. Untangling correlations of IFN signatures with COVID‐19 severity
Despite the essential role of IFN in preventing severe COVID‐19, several studies have come to seemingly opposite conclusions about the role of IFNs in COVID‐19 disease. Indeed, in addition to the aforementioned mechanistic evidence for a protective IFN role, multiple studies have reported that higher levels of systemic IFN and IFN signatures associate with a milder disease, 41 , 42 , 43 while a nearly equal number of reports have observed the opposite. 31 , 32 , 33 , 34 , 35 The reasons for these differences could be multifarious, and it is critical to acknowledge the specific contexts of these studies, as these details allow a deeper understanding of the factors that could have given rise to seemingly paradoxical results, but still abide by a deeper core logic that reconciles these apparent dichotomies.
Several non‐competing explanations for the apparent contradictory findings of both high‐ and low‐IFN signal correlating with COVID‐19 severity exist. One such explanation is in the timing of these measurements. Humans are rarely studied immediately following infection and prior to the development of symptoms, and to the best of our knowledge, there is no study quantifying initial IFN levels in patients within the first hours/days of SARS‐CoV‐2 infection. This could represent a caveat because IFN production can be highly dynamic throughout the course of a viral infection. In animal models IFN levels dramatically vary at different times throughout a viral infection, reaching peak systemic concentrations within days after inoculation and reduced, or even undetectable, levels thereafter regardless of viral clearance or persistence. 44 , 45 , 46 , 47 Similarly, systemic IFN levels in HIV infected patients peak a few days after exposure before decreasing. 48 As symptoms upon human infection with the early SARS‐CoV‐2 variants start 4–5 days and up to 14 days after exposure, 49 , 50 most human SARS‐CoV‐2 studies likely measured IFN and ISG levels relatively “late” in the innate response. If the comparison is late enough, a positive correlation between IFN levels and disease severity could be a consequence of higher viral RNA levels, which have been reported in severe cases 51 , 52 and would lead to enhanced engagement of PRR and IFN induction. In contrast, in mild cases, higher early IFN induction (and downstream mechanisms of viral control) may have preceded the time of analysis. It cannot be ruled out, however, that the high ISG signature driven by the increased titers in severe cases may in some instances contribute to the gravity of disease (Figure 1).
FIGURE 1.
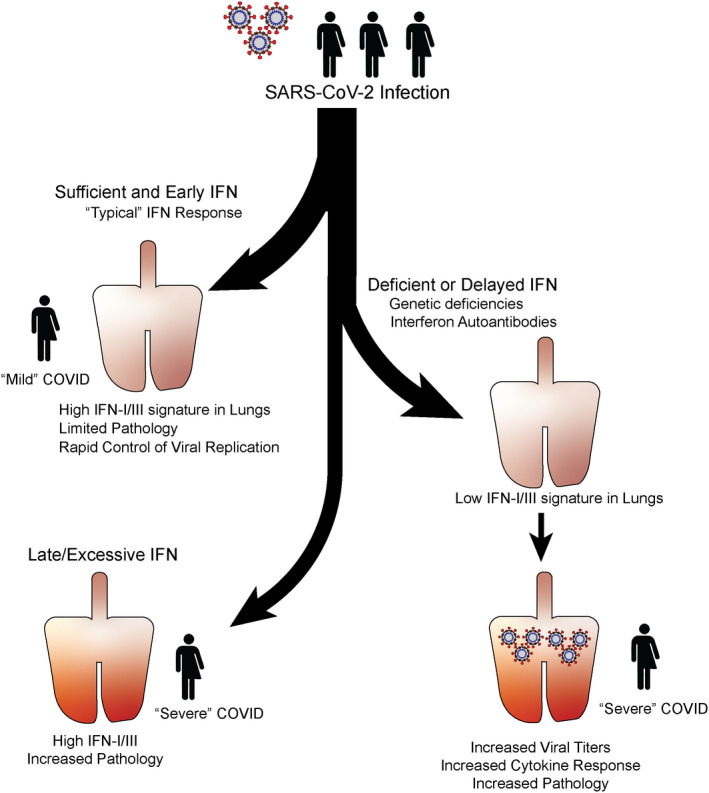
Branching paths of SARS‐CoV‐2 pathology. Model of the heterogeneity of SARS‐CoV‐2 infection outcome as it relates to IFN levels and timing. Early production of sufficient IFN is likely associated with rapid control of infection, limited pathology, and “Mild” disease (Top left). Alternatively, deficiency in IFN response either through genetic deficiency in virus sensing or response, or through the presence of anti‐IFNI autoantibodies associates with reduced control of the virus, increased cytokine response, increased pathology and “Severe” disease (Right). Finally, sustained SARS‐CoV‐2 may lead to late and excessive interferon signature in the lungs, which may contribute to increased pathology and “Severe” disease (Bottom Left). IFN, interferon; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Beside timing of IFN measurements, the type of ISGs induced downstream of IFN signaling may have also confounded the relationship between IFN‐signature and disease progression. Indeed, some ISGs were shown to be more efficient than others at restricting SARS‐CoV‐2 growth and may affect distinct steps in SARS‐CoV‐2 life cycle 53 such as cell entry (Ly6E, UBD, FAM46C), RNA replication (SPATS2L, ZBP1, IFIT3), and viral egress (ERLin1, APOL2, BST2, CNP). 53 Interestingly, some of these ISGs were specifically inhibitory to Coronaviruses, blocking replication of SARS‐CoV‐2 and SARS‐CoV, but not other viruses. 53
In addition to timing and ISG type, IFN location is also a critical variable. Due to its accessibility as an organ, many studies measured IFN and ISG signatures within the peripheral blood, 31 , 32 , 34 , 41 , 42 , 43 but analysis of this compartment does not represent the full picture of the IFN response. Indeed, correlation to disease severity differs depending on whether IFN or IFN signature is detected in the upper or lower airway. 33 Patients who present with severe COVID‐19 are characterized by high levels of IFN‐λ 2 and to a lesser degree IFNα/β in the lower airways. On the contrary, high levels of IFN‐λ 1 and 3 in the upper airway correlate with protection. Finally, correlations between IFN and viral loads were only observed in patients who were <70 years old, suggesting an even more complicated interplay between disease and IFNs that is influenced by aging. 33
3. CELLULAR SOURCES OF IFN AND THEIR SENSING PATHWAYS IN SARS‐COV‐2 INFECTION
3.1. Plasmacytoid Dendritic Cells and TLR7
Plasmacytoid dendritic cells (pDCs) are specialized producers of type I IFN that are critical for the control of many viral infections (reviewed in 54 ). Canonically, pDCs sense viral RNA and DNA through endosomal TLRs, TLR7 and TLR9 respectively, and while often refractory to viral infection, pDCs are able to produce IFN‐I without themselves being infected. 54 This important distinction from other cell types allows pDCs to recognize viruses without exposing themselves to infection‐intrinsic forms of viral evasion. Notably, while human pDCs appear refractory to SARS‐CoV‐2 infection, 55 several studies have now indicated that pDCs are able to produce IFN‐I in response to exposure to SARS‐CoV‐2. 12 , 55 , 56
By analogy to other related viruses, it was suspected from the beginning of the pandemic that SARS‐CoV‐2 would be recognized by TLR7. 57 Indeed, computational analysis of SARS‐CoV‐2 predicted its genome to have even more recognition sequences for TLR7 than the related SARS‐CoV or MERS. 58 This is now supported by several studies that have identified TLR7 deficiency itself, as well as TLR7 signaling regulators, as a critical risk factors for the development of severe COVID‐19. 9 , 10 , 12 , 23 By analyzing patients with loss of function TLR7 variants, it has recently been demonstrated that the IFN‐I response of pDCs to SARS‐CoV‐2 is specifically and highly dependent on TLR7, 12 as well as regulators of TLR7 localization and signaling. 55
Given the established role of pDCs in the control of a variety of infections, and the critical need for TLR7, but not TRL8 (which is expressed in other cells but typically not expressed at high levels in pDCs 59 , 60 ), in preventing severe COVID‐19 9 , 10 , 12 , 23 (Figure 2), it is highly likely that pDC derived IFN‐I is essential for the control of SARS‐CoV‐2 infection. However, it is also important to note that pDCs adapt after viral infection in several ways that reduce the availability of pDC derived IFN‐I (reviewed in 44 , 61 ). Particularly, in the case of SARS‐CoV‐2 infection, pDC numbers are severely reduced, 41 , 43 , 62 , 63 , 64 and the intrinsic capacity of pDCs to produce IFN‐I, which is normally exceptional, is also compromised. 62 Thus, while pDCs are likely crucial components of SARS‐CoV‐2 control early after infection, the aforementioned pDC adaptations may wane their contribution to IFN levels at later stages, likely providing a window of opportunity for the virus to spread and cause severe disease.
FIGURE 2.
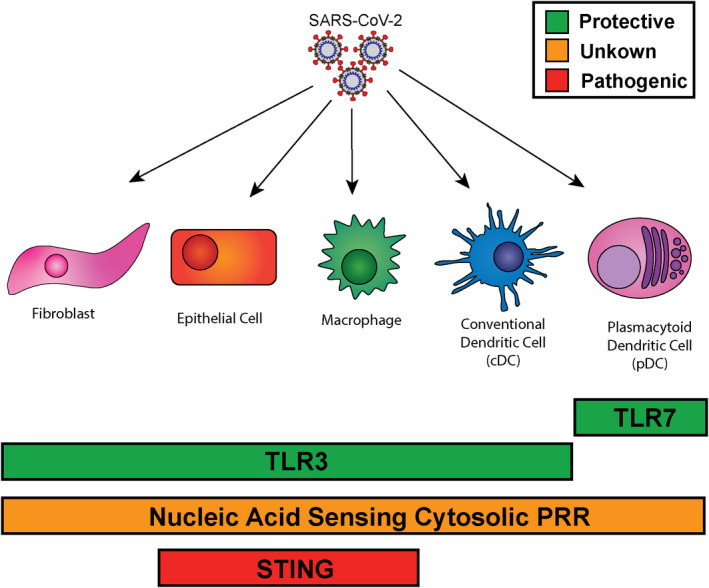
Sensors of SARS‐CoV‐2 that control IFN‐I production. Diagram of pattern recognition receptors shown to recognize SARS‐CoV‐2, their established relationships to protection against COVID‐19, and the breadth of their expression. TLR3 and TLR7 both have genetic associations which show increased COVID severity in patients with genetic deficiencies. TLR7 is primarily expressed in pDC, while TLR3 is expressed in a wider variety of cell types including DC, macrophage, lung epithelial cells, and fibroblasts. Nucleic acid sensing cytosolic receptors are expressed across most cell types, and it is still unclear whether these contribute to COVID severity positively or negatively in human infection. STING has been shown to be activated by SARS‐CoV‐2 infection in lung epithelia and macrophages and its activation is associated with increased pathology. COVID, coronavirus disease; IFN, interferon; pDC, plasmacytoid dendritic cell; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
3.2. Infected Cells, Cytoplasmic Sensors, and TLR3
While the specialized function of pDCs and recognition through TLR7 seems to be essential for the control of SARS‐CoV‐2 infection, smaller quantities of IFN are also produced by other cells that are infected by SARS‐CoV‐2. This is primarily thought to take place through cytosolic nucleic acid sensing PRR. These proteins provide a way for cells to recognize viral material in their cytoplasm. These sensors are broadly expressed and contribute to antiviral cytokine production not only in immune cells, but in non‐immune‐specialized cells as well (reviewed in 65 ). Thus, these PRR may provide a true “first line” of recognition at the frontline of infection. In the case of SARS‐CoV‐2, the cytoplasmic sensors RIG‐I, MDA5, LGP2, and NOD1, which can be activated by viral RNA (reviewed in 66 , 67 ), have all been shown to be involved in the recognition of SARS‐CoV‐2 infection in vitro 68 , 69 , 70 , 71 (Figure 2).
A systematic analysis of cytoplasmic sensors in the human lung epithelial cell line Calu‐3 revealed that MAVS, the common adaptor for RIG‐I and MDA5 66 was essential for the production of IFN‐I in response to SARS‐CoV‐2 infection. 70 Furthermore, knockdown of the sensing proteins MDA5, LGP1, and NOD1 all significantly reduced IFN‐I production after infection with SARS‐CoV‐2. The essential role for MDA5 in IFN‐I production in airway epithelial cell lines has since then been validated by several other groups. 69 , 70 , 71 Notably, several studies have observed distinct contributions of RIG‐I for IFN‐I production in response to SARS‐CoV‐2 infection. While two groups 69 , 70 showed similar IFN‐I levels upon RIG‐I inhibition via siRNA or Cas9 KO, another study showed a significant reduction in IFN‐I production after siRNA knockdown of RIG‐I, 71 suggesting that RIG‐I contribution to IFN‐I production after SARS‐CoV‐2 infection may be context dependent.
Despite its unclear role in IFN‐I production, it has been established that RIG‐I, which induces a transcriptional program beyond IFNs, is an essential restriction factor for viral growth in several lung epithelial cell lines. 68 It is important to note that RIG‐I‐mediated signaling interrupts SARS‐CoV‐2 life‐cycle, and thereby acts as a restriction factor independent of IFN activation. 68
One notable characteristic of IFN induction in Calu‐3 cells is that it occurs after the peak of SARS‐CoV‐2 replication. 71 Potentially as a result of this delay, native IFN‐I production in the airway epithelium may be insufficient to restrict viral spread. Furthermore, in contrast to application of IFN prior to or concurrent with infection, the addition of recombinant IFN as few as 2 hours after infection did not significantly restrict SARS‐CoV‐2 growth in Calu‐3 cells, 71 suggesting that evasion of ISG mediated control is nearly complete once infection is established, and only very early or prophylactic production of IFN‐I may be protective.
3.3. TLR3
TLR3 is an endosomal TLR that senses dsRNA. 72 As with TLR7, TLR3 deficiency is also associated with severe COVID‐19. 30 While TLR3 has not been detected in pDCs, 59 it is expressed in a variety of other cells including immune and non‐immune lineages. 59 , 73 , 74 , 75 , 76 , 77 Indeed, TLR3 controls IFN‐I responses in a variety of non‐hematopoietic cells including fibroblasts 74 , 75 , 76 , 77 and cortical neurons. 74 On the contrary, it has been shown in human peripheral blood mononuclear cells that TLR3 is largely dispensable for the induction of IFN after stimulation with a variety of viruses 76 , 77 and even poly(I:C). 76 TLR3 is also expressed in mixed lung organoids composed of Calu‐3 and MRC‐5, a lung‐fibroblast cell line, and pharmacological inhibition of TLR3 ligation reduces, albeit modestly, their IFN‐I production after SARS‐CoV‐2 infection. 73 Importantly, SV40‐transformed fibroblasts or stem cell derived epithelial cells from patients with inborn errors in TLR3 were compromised in their control of SARS‐CoV‐2 replication. 30 Altogether these studies support a role for TLR3 in the control of SARS‐CoV‐2 infection, and make it tempting to speculate that TLR3‐mediated viral sensing may at least partly take place in the airway epithelium (Figure 2).
3.4. STING
The innate sensing protein stimulator of IFN genes (STING) recognizes cyclic‐di‐nucleotides and is a critical regulator of IFN responses in a variety of conditions. 78 While cyclic‐di‐nucleotides can be produced by bacterial pathogens, 78 they can also be generated by the host enzyme cytoplasmic GMP‐AMP Synthase (cGAS). 78 This enzyme recognizes cytosolic double stranded DNA independent of sequence, and produces cyclic GMP‐AMP which then stimulates STING to drive IFN induction (reviewed in 78 ). Interestingly, it was recently demonstrated that SARS‐CoV‐2 skin lesions have an ISG signature which is abrogated by treatment with the STING inhibitor H‐151. 79 This was also the case when lesions were treated with VBIT‐4, 79 an inhibitor of the mitochondrial voltage dependent anion channel (VDAC1), which has been shown to enable passage of mitochondrial DNA into the cytosol during mitochondrial stress. 80 The authors identified STING activation in both lung macrophages and epithelial cells of COVID‐19 patients, and extended this model into the K18‐hACE2 transgenic mouse model of SARS‐CoV‐2 infection, where they demonstrated that treatment with H‐151 could reduce lung damage. 79 Further studies will be needed to determine how much STING might contribute to the COVID‐19 pathology (Figure 2).
4. MECHANISMS UNDERLYING SARS‐COV‐2 EVASION OF IFNs
Like many other viruses, SARS‐CoV‐2 has developed multiple ways to modulate the IFN‐I response to promote its replication (Figure 3). Given the quantity of redundant systems for IFN production in their hosts, these evasive functions are likely essential for SARS‐CoV‐2 fitness. Additionally, SARS‐CoV‐2 has only been transmitting between human hosts for a relatively short period of time 81 , 82 and so alterations or improvements in these systems may represent potential places where SARS‐CoV‐2 variants may be able to leverage a fitness advantage. 83 There is much information on related coronavirus proteins in humans which modulate the IFN response, however, here, we will focus on those that have been described specifically in SARS‐CoV‐2.
FIGURE 3.
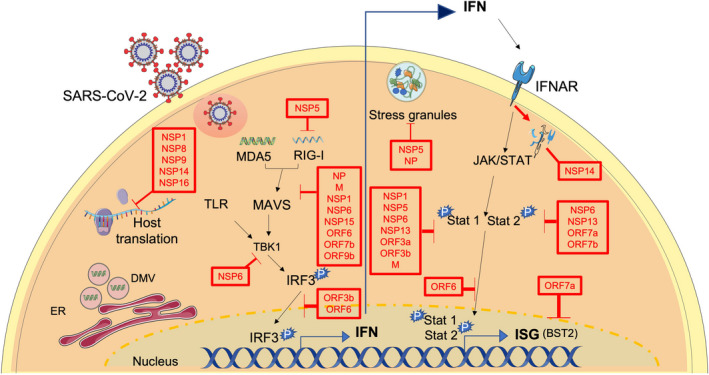
IFN evasion mechanisms of SARS‐CoV‐2. Schematic representation of some of the key pathways that are targeted by SARS‐CoV‐2 proteins to block IFN responses. SARS‐CoV‐2 replicates inside DMVs which helps the virus avoid recognition by the immune system. Additionally, several SARS‐CoV‐2 proteins were found to prevent host translation, while others block stress granule formation. Some viral proteins actively block IFN induction, and/ or signaling at multiple steps upstream and downstream IFN induction. DMVs, double membrane vesicles; IFN, interferon; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
SARS‐CoV‐2 encodes three main groups of proteins: structural proteins, non‐structural proteins (NSP), and accessory proteins. 84 Several of these proteins have been reported to interfere with IFN responses at multiple stages such as sensing of viral RNA, blocking IFN transcription/translation, and targeting ISG effector functions, among others. Multiple proteins disrupt IFN signaling via more than one mechanism, and some share similar targets providing redundancy in their function and highlighting the importance of modulating IFN responses for successful replication of SARS‐CoV‐2.
4.1. Evading immune sensing
Preventing sensing by the immune system is the first line of evasion for SARS‐CoV‐2. In this regard, other Coronaviruses replicate inside double membrane vesicles (DMVs) which helps them avoid recognition by immune receptors (reviewed in 85 ). Similarly, SARS‐CoV‐2 was found to form DMVs containing viral RNA which associated with alterations to the ER and the cytoskeleton apparatus. 86 , 87 Pharmacological disruption of the intermediate filaments network or inhibition of microtubule depolymerization disrupts DMVs and hampers SARS‐CoV‐2 replication. 86 Together, the above observations suggest that DMVs favor SARS‐CoV‐2 growth via segregation of viral RNA and limitation of viral sensing, although more experiments are necessary to fully support this model.
SARS‐CoV‐2 also avoids recognition by targeting stress granule (SG) formation with its protease NSP5 and its nucleoprotein (NP). 88 SGs are membrane‐less organelles that can form after environmental stress, sequestering mRNA and leading to RNA translation inhibition. 89 SGs have also been implicated as contributors of antiviral immunity via recruitment of viral sensors and, as such, many viruses have evolved means to disable SG (reviewed in 90 , 91 ). In particular for SARS‐CoV‐2, NSP5 was shown to attenuate SG formation, independently of its protease activity, while NP interacts with G3BP1, 88 a protein involved in SG assembly. 92
Coronaviruses can also evade recognition by PRRs, specifically RIG‐I, by methylating the 5′‐CAP of viral mRNA and mimicking host mRNA in a manner dependent on NSP13, NSP14, and the NSP16‐NSP10 complex (reviewed in 93 ). While this evasion mechanism has yet to be demonstrated for SARS‐CoV‐2, its NSP16‐NSP10 complex shares high structural similarity with its relatives 94 and is capable of methylating the 5′ CAP of viral RNA. 95
4.2. Inhibition of IFN‐I synthesis and secretion
Another key mechanism by which SARS‐CoV‐2 evades the immune system is by turning off host protein synthesis, which is essential for the production of both IFN and antiviral ISGs. SARS‐CoV‐2 encodes multiple inhibitors of host translation. NSP1, for example, binds the mRNA entry channel of the ribosome to disrupt host, but not viral, protein translation, 96 , 97 , 98 , 99 , 100 , 101 ultimately reducing IFN production and ISG transcription. 100 , 102 NSP14 also inhibits translation and blocks IFN‐I − dependent ISG induction in a manner dependent on its exoribonuclease and N7‐methyltransferase activities. 102 Interestingly, as previously described for SARS‐CoV, NSP14 can form a complex with NSP10 that enhances its capacity to inhibit translation. 102
NSP8, NSP9, and NSP16 were also reported to antagonize IFN via manipulation of translation. 96 , 103 NSP8 and NSP9 do so by binding the host RNA in the signal recognition particle, a complex involved in targeting proteins to their end location, 104 and interfering with protein co‐translational trafficking to the cell membrane. 96 This function has been proposed to prevent the packaging of secreted proteins into vesicles potentially reducing IFN secretion.
Finally, NSP16 suppresses global mRNA splicing by binding to the mRNA recognition domains of the U1 and U2 RNA components of the spliceosome. 96 As a result of this, expression of NSP16 results in alterations in mRNA splicing, and reduced activity from an ISG luciferase reporter. 96
4.3. Suppression of IFN transcriptional induction and IFN receptor signaling
In addition to inhibiting IFN synthesis and secretion, SARS‐CoV‐2 antagonizes IFNs by modulating the induction of IFN mRNA and protein (Figure 3, left center), as well as signaling downstream IFN recognition by interferon alpha receptor (IFNAR) (Figure 3, far right).
Multiple studies have performed systematic analyses of SARS‐CoV‐2 proteins with the ability to suppress IFN transcriptional activation and/or prevent the response to IFN by modulating IFNAR or its downstream signaling through STAT1 and STAT2. 103 , 105 , 106 , 107 Altogether these studies have identified 14 virally encoded proteins (NSP1, NSP3, NSP5, NSP6, NSP10, NSP12, NSP13, NSP14, NSP15, M, ORF3, ORF6, ORF7a, and ORF7b) with the ability to interfere with either the induction of IFN or its downstream signaling through IFNAR. 103 , 105 , 106 , 107 The redundancy of proteins targeting these pathways highlights how critical it is for SARS‐CoV‐2 to block IFN transcriptional induction and prevent the recognition of IFN and the expression of anti‐viral ISGs. It is perhaps due to such abundance of viral proteins blocking IFN production in infected cells, that TLR7 and pDCs, which are not infected by SARS‐CoV‐2, 55 appear more critical to fight SARS‐CoV‐2 than other endemic viruses. Indeed, individuals with inborn errors in TLR7 have not history of serious viral illness despite developing severe COVID‐19. 12 , 23
As described above, the RNA sensing proteins MDA5 and RIG‐I have each been identified as potentially contributing to the production of IFN‐I after SARS‐CoV‐2 infection. These molecules signal through the adapter MAVS, which activates TANK binding kinase 1 (TBK1) ultimately phosphorylating IRF3, which translocates to the nucleus to promote IFN transcription. 66 There are SARS‐CoV‐2 proteins targeted to every step of this process, and many interact with multiple points. NSP6, NSP5, NSP15, ORF6, and ORF7b can block MAVS‐induced IFNβ and/or IFNλ, 105 although the mechanisms by which these proteins interfere with this process have not yet been described. NSP1, discussed above for its function in translational inhibition, also blocks MAVS‐induced IFN promoter activity, 105 , 108 likely through prevention of IRF3 nuclear translocation. 108
NSP3 and NSP5 are the two SARS‐CoV‐2 proteases. 85 NSP3 cleaves ISG15 from host proteins including multiple molecules involved in the activation of IFN transcription, such as IRF3 109 and MDA5. 110 ISGylation of both IRF3 111 and MDA5 110 support the activation of IFN transcription and coordinately the removal of ISG15 by NSP3 leads to attenuation of IFN production. 109 , 110 Additionally, NSP3 can cleave IRF3 itself when the purified proteins are co‐incubated, although the direct relevance of this in infected cells has not yet been investigated. 112 NSP5 may also counteract induction of IFN at multiple steps, as it interferes with MAVS activation and binds RIG‐I and MDA5. 88 NSP5 can remove the 10 most N‐terminal amino acids from RIG‐I, ablating its interaction with MAVS. 113 Moreover, NSP5 promotes the ubiquitination and degradation of MAVS itself, 113 while also blocking RIG‐I ubiquitination. Modulation of ubiquitination is a function shared by ORF9b which interrupts the K63‐linked polyubiquitination of the RIG‐I signaling modulator NEMO. 114
The viral nucleocapsid protein (NP) was also identified as a modulator of IFN responses acting at multiple stages of the IFN induction pathway. The dimerization domain of NP, for example, inhibits Lys63‐linked poly‐ubiquitination and aggregation of MAVS. 115 Intriguingly, when a peptide that interferes with the NP dimerization domain was applied in vivo IFN responses were enhanced in mice infected with SARS‐CoV‐2 while viral loads and organ pathology were attenuated. 115 NP can also interact with RIG‐I and restrict IRF3 phosphorylation and nuclear translocation. 116 On the contrary, the viral membrane glycoprotein M also antagonizes MAVS‐mediated innate immunity by interacting with MAVS and impeding its aggregation and signaling, 117 in addition to interacting with MDA5, TRAF3, IKKϵ, and TBK1, and inducing TBK1 K48‐linked ubiquitination and degradation. 118
Additionally, NSP6 and NSP13 suppress TBK1‐mediated phosphorylation of IRF3, albeit via different mechanisms where only NSP13 blocks TBK1 phosphorylation. 107 ORF6 inhibits nuclear translocation of IRF3 by interacting with the nuclear importin KPNA2. 107 ORF3b was described as a potent IFN antagonist, via suppression of IRF3 nuclear localization for which it seems more efficient than its relative encoded in SARS‐CoV. 119
Fewer SARS‐CoV‐2 proteins have been identified with the capacity to interrupt IFN‐I signaling via IFNAR, though this process may still be quite efficient given that IFN added to SARS‐CoV‐2 infected cells may only restrict viral growth prophylactically. 71 IFN is recognized by IFNAR which signals through STAT1 and STAT2 to induce ISGs 5 that can restrict SARS‐CoV‐2. 53 As with IFN induction, each step of this pathway is targeted by at least one SARS‐CoV‐2 protein, while some steps are targeted redundantly. One protein, NSP14, directly interferes with recognition of IFN through the lysosomal degradation of IFNAR. 106 In contrast, STAT1 and STAT2 are targeted by many viral proteins. NSP1 reduces levels of STAT2 protein. 108 NSP1 also opposes STAT1 phosphorylation (eg, activation), 107 a function that is shared by NSP5, NSP6, NSP13, ORF3a, ORF3b, ORF6, and the viral membrane glycoprotein M. 103 , 107 , 120 While Orf7a, Orf7b, NSP6, and NSP13 have been shown to reduce phosphorylation of STAT2. 107
Finally, SARS‐CoV‐2 proteins can interfere with the function of ISGs. For example, BST2 (tetherin) is an antiviral ISG which inhibits the release of viral particles from HIV and several human coronaviruses 121 , 122 , 123 and has been shown to also inhibit SARS‐CoV‐2 titers in cell culture supernatants. 53 This BST2 activity is antagonized by Orf7a. 53
It is important to note that despite the extremely redundant capacity of SARS‐CoV‐2 to subvert IFN‐I production and downstream signaling, these responses are still evolving, and several studies have identified alterations in the IFN‐I evasive capacity for distinct SARS‐CoV‐2 isolates. Indeed, in the alpha variant of SARS‐CoV‐2, which stimulates less IFN‐I production than earlier variants, there is increased expression of ORF9b, as well as ORF6 and NP. 83 Another study has identified isolates with variations in ORF3b that show increased IFN antagonizing efficiency, 119 and multiple groups have identified truncations of ORF7a, 124 , 125 , 126 some of which impact its ability to disrupt ISG induction after IFN‐I treatment, 124 though it is unclear whether they alter the ability of ORF7a to antagonize BST2. 53 These mutations may represent random events tolerated in the SARS‐CoV‐2 population due to redundancy in the systems that target IFN‐I induction and IFNAR signaling. Alternatively, they could also represent adaptations to improve replicative capacity within human hosts. Further work will be needed to disentangle the epidemiological consequences of these variations.
5. IFNS AS THERAPEUTICS IN COVID‐19
Trials aimed at using IFN as a therapeutic for COVID‐19 can be broken down roughly by the type of IFN used: IFN‐β, IFN‐α, or IFN‐λ. For many of these trials, however, it should be noted that the multidrug regimens used may have confounded the interpretation of the IFN effects.
The most studied of the three aforementioned IFNs in clinical trials is IFN‐β, and this has shown promise in some cases, 127 , 128 , 129 , 130 but not others. 131 , 132 , 133 The most recent and robust trial investigated the efficacy of IFNβ‐1a in combination with the antiviral drug remdesevir. 133 In this trial, they found no significant benefit of IFNβ‐1a over treatment with remdesivir. The authors provide possible reasons on why this study does not show efficacy. This includes the potential for anti‐synergy between two drugs targeted at limiting viral replication. Additionally, they note that they included patients with more advanced disease than some previous studies, 127 and that this may have limited their efficacy. It is unlikely, however, that the this study showed reduced efficacy as the result of low dosing as another study has compared the efficacy of IFNβ‐1a in two doses, one similar to Kalil et al 133 and one 10× higher, and found no benefit of higher vs lower IFNβ‐1a dose. 134 Of note, the last‐mentioned study does not compare with a placebo treated group, and so, the overall efficacy of IFNβ‐1a cannot be assessed. 134
Regarding IFN‐α trials, it is worth highlighting several smaller scale studies analyzing the effect of nebulized IFN‐α2b. An insightful retrospective study of IFN‐α2b treatment showed that early administration of IFN‐α2b in COVID‐19 patients decreased mortality while late treatment resulted in the opposite outcome. 135 In the same line, another study looking at the effect of IFN‐α2b in combination with the antiviral arbidol found that the group receiving IFN‐α2b exhibited decreased time to viral clearance from the upper airways and reduced severity of lung abnormalities compared to the individuals who received arbidol alone. 136 , 137 Notably, in this study, there was a slight skew toward average earlier intervention in the IFN‐α2b group (8 days) as opposed to those getting arbidol alone (17 days), 137 which in the context of the above mentioned observation that early administration of IFN‐α2b is more beneficial, 135 may meaningfully complicate the interpretation of these data. Finally, a phase II trial using IFN‐III (specifically IFNλ‐1), has shown promising results including earlier time to viral clearance. 138
Overall, the results of the aforementioned IFN‐I & IFN‐III trials are mixed. One explanation for this may lie in patient heterogeneity. Indeed, IFN treatment may be especially effective in patients with inborn deficiencies in their capacity to produce IFN‐I. For example, treatment with pegylated‐IFN‐α in two individuals with inborn errors in TLR3 and IRF3, respectively, resulted in quick resolution of infection. 139 While promising, a much larger study will be needed to definitively determine if patients with deficiencies underlying reduced IFN‐induction benefit more from IFN‐I therapy than the general population. Conversely, IFN‐I treatment may be particularly ineffective in patients with IFN neutralizing antibodies if the patients have antibodies against the subtype of IFN that is provided. Since patients with anti‐IFN‐I autoantibodies make up as much as 10% of cases of severe COVID‐19, 11 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 24 , 25 , 26 , 27 , 28 , 29 this may significantly alter the results of a trial which does not account for this heterogeneity, and so, future studies should take into account the IFN‐I autoantibody status of their cohort. This may also allow for matching patients to treatment with subtypes of IFN‐I for which they do not have autoantibodies. This has been suggested and attempted in a study of a single patient with IFN‐α neutralizing autoantibodies who showed attenuation of symptoms upon treatment with IFN‐β. 14
6. DISCUSSION
The SARS‐CoV‐2 pandemic has been met with an abundance of research into this novel pathogen and the immune responses that attempts to control it. Across this literature, it has become apparent that Type I and III IFNs are crucial for the control of SARS‐CoV‐2 and are produced in response to engagement of a number of PRR (Figure 2). It is clear that SARS‐CoV‐2 can be sensed via TLR7 in pDCs, 9 , 10 , 12 , 23 as well as TLR3 in other cells, potentially lung epithelium or fibroblasts. 29 , 73 Importantly, loss of either of these systems is associated with the development of severe COVID‐19. 9 , 10 , 12 , 23 , 30 On the contrary, a significant body of work indicates that cytosolic RNA‐sensing PRR 69 , 70 , 71 and cGAS‐STING pathway 79 can promote IFN‐I production in response to SARS‐CoV‐2 infection. The non‐redundant impact of cytosolic PRR signaling on the course of SARS‐CoV‐2 infection in humans is not yet clear as, at the writing of this review, deficiencies in these PRRs nor their adaptors have been identified as risk factors for severe COVID‐19.
Deficiencies in IFN‐I or IFN‐III production or signaling, 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 19 , 20 , 21 , 22 , 26 , 28 , 29 , 30 or the antiviral proteins they induce 37 , 38 , 39 , 40 are highly associated with severe disease. This is at first blush seemingly at odds with studies that identify a positive correlation between IFN signatures and disease severity. 31 , 32 , 33 , 34 , 35 However, when incorporating timing, and location these observations can be reconciled. A commonly proposed model is that severe COVID‐19 is the result of initial IFN deficiency or delayed IFN responses 44 , 140 , 141 (Figure 1). This compromises early viral control, which leads to increases in viral titers that drive up late inflammatory and IFN responses that could cause pathology (Figure 1). Alternatively, there may also be patients predisposed to secrete excessive IFN that could lead to exaggerated inflammation and more severe COVID‐19 (Figure 1).
Like other viruses, SARS‐CoV‐2 has evolved mechanisms which block IFN responses (Figure 3). Interestingly, as SARS‐CoV‐2 has adapted within its new human host population variants with alterations in IFN‐evasive mechanisms have emerged. For example, alpha variant isolates have been reported to have increased expression levels of the immunoevasin ORF9b, and coordinately reduced IFN induction as compared to early wave isolates. 83 Coincidentally, similar genetic alterations are observed in the Delta and Omicron variants. 83 In the same line, identification of SARS‐CoV‐2 isolates with increased ORF3b capacity to inhibit IFN‐I further suggests that this virus may have space to improve on its IFN antagonization activity in humans. 119 While it is tempting to speculate that increased capacity for IFN subversion could confer benefit to these later variants it is important to acknowledge the complication of causally linking specific mutations in a particular variant with disease phenotypes.
Altogether it is critical to recognize IFN as a central protective force against SARS‐CoV‐2 and COVID‐19, with potential for increasing pathology under specific circumstances. There is heterogeneity in both the human response (eg, genetic variants and autoantibodies), as well as the strain of virus (eg, Orf9b expression levels) which hold the potential to inform us as to the efficacy of applied IFN therapies in the prevention of COVID‐19. Moving forward, leveraging our understanding of these intricacies will allow us to personalize IFN based interventions, and to even develop new methods of treatment centered around increasing IFN responses where they are beneficial and decreasing them where they are detrimental. Furthermore, because of its ancient and central role in antiviral responses, it is likely that the lessons learned from SARS‐CoV‐2 may someday find themselves useful to inform currently understudied and emerging diseases, and perhaps guide the design of common IFN‐based therapies to treat multiple human illnesses.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
We thank members of the Zuniga lab for fruitful discussions. Research in the Zuniga lab is supported by N.I.H. grants AI145314, AI113923 and AI132122. Figure 3 was created using icons modified from Servier Medical Art, https://smart.servier.com/, a publicly available service provided by Les Laboratoires Servier. Images have been adapted for the purposes of the Figure and their use does not imply endorsement by Les Laboratoires Servier.
Chiale C, Greene TT, Zuniga EI. Interferon induction, evasion, and paradoxical roles during SARS‐CoV‐2 infection. Immunol Rev. 2022;309:12‐24. doi: 10.1111/imr.13113
*This article is part of a series of reviews covering SARS‐CoV‐2 Immunity appearing in Volume 309 of Immunological Reviews.
Carolina Chiale and Trever T. Greene contributed equally to the manuscript.
DATA AVAILABILITY STATEMENT
Data sharing not applicable ‐ no new data generated.
REFERENCES
- 1. Woolhouse ME, Gowtage‐Sequeria S. Host range and emerging and reemerging pathogens. Emerg Infect Dis. 2005;11:1842‐1847. doi: 10.3201/eid1112.050997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16‐34. doi: 10.3109/08830185.2010.529976 [DOI] [PubMed] [Google Scholar]
- 3. Venkatesh B, Lee AP, Ravi V, et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature. 2014;505:174‐179. doi: 10.1038/nature12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Secombes CJ, Zou J. Evolution of interferons and interferon receptors. Front Immunol. 2017;8:209. doi: 10.3389/fimmu.2017.00209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneider WM, Chevillotte MD, Rice CM. Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513‐545. doi: 10.1146/annurev-immunol-032713-120231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:2061. doi: 10.3389/fimmu.2018.02061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dowling JW, Forero A. Beyond good and evil: molecular mechanisms of type I and III IFN functions. J Immunol. 2022;208:247‐256. doi: 10.4049/jimmunol.2100707 [DOI] [PubMed] [Google Scholar]
- 8. Gonzalez‐Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125‐135. doi: 10.1038/nri3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Made CI, Simons A, Schuurs‐Hoeijmakers J, et al. Presence of genetic variants among young men with severe COVID‐19. JAMA. 2020;324:663‐673. doi: 10.1001/jama.2020.13719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fallerini C, Daga S, Mantovani S, et al. Association of Toll‐like receptor 7 variants with life‐threatening COVID‐19 disease in males: findings from a nested case‐control study. Elife. 2021;10. doi: 10.7554/eLife.67569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abers MS, Rosen LB, Delmonte OM, et al. Neutralizing type‐I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID‐19. Immunol Cell Biol. 2021;99:917‐921. doi: 10.1111/imcb.12495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asano T, Boisson B, Onodi F, et al. X‐linked recessive TLR7 deficiency in ~1% of men under 60 years old with life‐threatening COVID‐19. Sci Immunol. 2021;6. doi: 10.1126/sciimmunol.abl4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bastard P, Gervais A, Le Voyer T, et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID‐19 deaths. Sci Immunol. 2021;6. doi: 10.1126/sciimmunol.abl4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bastard P, Levy R, Henriquez S, Bodemer C, Szwebel TA, Casanova JL. Interferon‐beta therapy in a patient with incontinentia pigmenti and autoantibodies against type I IFNs infected with SARS‐CoV‐2. J Clin Immunol. 2021;41:931‐933. doi: 10.1007/s10875-021-01023-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science. 2020;370. doi: 10.1126/science.abd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chauvineau‐Grenier A, Bastard P, Servajean A, et al. Autoantibodies neutralizing type I interferons in 20% of COVID‐19 deaths in a French hospital. J Clin Immunol. 2022. doi: 10.1007/s10875-021-01203-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goncalves D, Mezidi, M , Bastard, P , et al. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID‐19 patients. Clin Transl Immunology. 2021;10:e1327. doi: 10.1002/cti2.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koning R, Bastard P, Casanova JL, Brouwer MC, van de Beek D, with the Amsterdam UMCC‐BI . Autoantibodies against type I interferons are associated with multi‐organ failure in COVID‐19 patients. Intensive Care Med. 2021;47:704‐706. doi: 10.1007/s00134-021-06392-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Solanich X, Rigo‐Bonnin R, Gumucio VD, et al. Pre‐existing autoantibodies neutralizing high concentrations of type I interferons in almost 10% of COVID‐19 patients admitted to intensive Care in Barcelona. J Clin Immunol. 2021;41:1733‐1744. doi: 10.1007/s10875-021-01136-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Troya J, Bastard P, Planas‐Serra L, et al. Neutralizing autoantibodies to type I IFNs in >10% of patients with severe COVID‐19 pneumonia hospitalized in Madrid, Spain. J Clin Immunol. 2021;41:914‐922. doi: 10.1007/s10875-021-01036-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Wijst MGP, Vazquez SE, Hartoularos GC, et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID‐19. Sci Transl Med. 2021;13:eabh2624. doi: 10.1126/scitranslmed.abh2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vazquez SE, Bastard P, Kelly K, et al. Neutralizing autoantibodies to type I interferons in COVID‐19 convalescent donor plasma. J Clin Immunol. 2021;41:1169‐1171. doi: 10.1007/s10875-021-01060-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Solanich X, Vargas‐Parra G, van der Made CI, et al. Genetic screening for TLR7 variants in young and previously healthy men with severe COVID‐19. Front Immunol. 2021;12:719115. doi: 10.3389/fimmu.2021.719115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Acosta‐Ampudia Y, Monsalve DM, Rojas M, et al. COVID‐19 convalescent plasma composition and immunological effects in severe patients. J Autoimmun. 2021;118:102598. doi: 10.1016/j.jaut.2021.102598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang SE, Feng A, Meng W, et al. New‐onset IgG autoantibodies in hospitalized patients with COVID‐19. Nat Commun. 2021;12:5417. doi: 10.1038/s41467-021-25509-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raadsen MP, Gharbharan A, Jordans CCE, et al. Interferon‐alpha2 auto‐antibodies in convalescent plasma therapy for COVID‐19. J Clin Immunol. 2021. doi: 10.1007/s10875-021-01168-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang EY, Mao T, Klein J, et al. Diverse functional autoantibodies in patients with COVID‐19. Nature. 2021;595:283‐288. doi: 10.1038/s41586-021-03631-y [DOI] [PubMed] [Google Scholar]
- 28. Ziegler CGK, Miao VN, Owings AH, et al. Impaired local intrinsic immunity to SARS‐CoV‐2 infection in severe COVID‐19. Cell. 2021;184:4713‐4733.e4722. doi: 10.1016/j.cell.2021.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shaw ER, Rosen LB, Cheng A, et al. Temporal dynamics of anti‐type 1 interferon autoantibodies in COVID‐19 patients. Clin Infect Dis. 2021. doi: 10.1093/cid/ciab1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science. 2020;370. doi: 10.1126/science.abd4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584:463‐469. doi: 10.1038/s41586-020-2588-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galani IE, Rovina N, Lampropoulou V, et al. Untuned antiviral immunity in COVID‐19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol. 2021;22:32‐40. doi: 10.1038/s41590-020-00840-x [DOI] [PubMed] [Google Scholar]
- 33. Sposito B, Broggi A, Pandolfi L, et al. The interferon landscape along the respiratory tract impacts the severity of COVID‐19. Cell. 2021;184:4953‐4968.e4916. doi: 10.1016/j.cell.2021.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee JS, Park S, Jeong HW, et al. Immunophenotyping of COVID‐19 and influenza highlights the role of type I interferons in development of severe COVID‐19. Sci Immunol. 2020;5. doi: 10.1126/sciimmunol.abd1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dorgham K, Quentric P, Gokkaya M, et al. Distinct cytokine profiles associated with COVID‐19 severity and mortality. J Allergy Clin Immunol. 2021;147:2098‐2107. doi: 10.1016/j.jaci.2021.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang S, Wang L, Cheng G. The battle between host and SARS‐CoV‐2: innate immunity and viral evasion strategies. Mol Ther. 2022;30:1869‐1884. doi: 10.1016/j.ymthe.2022.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wickenhagen A, Sugrue E, Lytras S, et al. A prenylated dsRNA sensor protects against severe COVID‐19. Science. 2021;374:eabj3624. doi: 10.1126/science.abj3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soveg FW, Schwerk J, Gokhale NS, et al. Endomembrane targeting of human OAS1 p46 augments antiviral activity. Elife. 2021;10. doi: 10.7554/eLife.71047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huffman JE, Butler‐Laporte G, Khan A, et al. Multi‐ancestry fine mapping implicates OAS1 splicing in risk of severe COVID‐19. Nat Genet. 2022. doi: 10.1038/s41588-021-00996-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou S, Butler‐Laporte G, Nakanishi T, et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID‐19 susceptibility and severity. Nat Med. 2021;27:659‐667. doi: 10.1038/s41591-021-01281-1 [DOI] [PubMed] [Google Scholar]
- 41. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369:718‐724. doi: 10.1126/science.abc6027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trouillet‐Assant S, Viel S, Gaymard A, et al. Type I IFN immunoprofiling in COVID‐19 patients. J Allergy Clin Immunol. 2020;146:206‐208.e202. doi: 10.1016/j.jaci.2020.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu C, Martins AJ, Lau WW, et al. Time‐resolved systems immunology reveals a late juncture linked to fatal COVID‐19. Cell. 2021;184:1836‐1857.e1822. doi: 10.1016/j.cell.2021.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greene TT, Zuniga EI. Type I interferon induction and exhaustion during viral infection: plasmacytoid dendritic cells and emerging COVID‐19 findings. Viruses. 2021;13. doi: 10.3390/v13091839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med. 2011;208:2367‐2374. doi: 10.1084/jem.20110654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Channappanavar R, Fehr AR, Zheng J, et al. IFN‐I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129:3625‐3639. doi: 10.1172/JCI126363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4:374‐386. doi: 10.1016/j.chom.2008.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stacey AR, Norris PJ, Qin L, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719‐3733. doi: 10.1128/JVI.01844-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang X, Rayner S, Luo MH. Does SARS‐CoV‐2 has a longer incubation period than SARS and MERS? J Med Virol. 2020;92:476‐478. doi: 10.1002/jmv.25708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lauer SA, Grantz KH, Bi Q, et al. The incubation period of coronavirus disease 2019 (COVID‐19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. 2020;172:577‐582. doi: 10.7326/M20-0504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Y, Yan LM, Wan L, et al. Viral dynamics in mild and severe cases of COVID‐19. Lancet Infect Dis. 2020;20:656‐657. doi: 10.1016/S1473-3099(20)30232-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bermejo‐Martin JF, Gonzalez‐Rivera M, Almansa R, et al. Viral RNA load in plasma is associated with critical illness and a dysregulated host response in COVID‐19. Crit Care. 2020;24:691. doi: 10.1186/s13054-020-03398-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin‐Sancho L, Lewinski MK, Pache L, et al. Functional landscape of SARS‐CoV‐2 cellular restriction. Mol Cell. 2021;81:2656‐2668.e2658. doi: 10.1016/j.molcel.2021.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15:471‐485. doi: 10.1038/nri3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Onodi F, Bonnet‐Madin L, Meertens L, et al. SARS‐CoV‐2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;218. doi: 10.1084/jem.20201387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Severa M, Diotti RA, Etna MP, et al. Differential plasmacytoid dendritic cell phenotype and type I interferon response in asymptomatic and severe COVID‐19 infection. PLoS Pathog. 2021;17:e1009878. doi: 10.1371/journal.ppat.1009878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cervantes‐Barragan L, Zust R, Weber F, et al. Control of coronavirus infection through plasmacytoid dendritic‐cell‐derived type I interferon. Blood. 2007;109:1131‐1137. doi: 10.1182/blood-2006-05-023770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moreno‐Eutimio MA, Lopez‐Macias C, Pastelin‐Palacios R. Bioinformatic analysis and identification of single‐stranded RNA sequences recognized by TLR7/8 in the SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV genomes. Microbes Infect. 2020;22:226‐229. doi: 10.1016/j.micinf.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kadowaki N, Ho S, Antonenko S, et al. Subsets of human dendritic cell precursors express different toll‐like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863‐869. doi: 10.1084/jem.194.6.863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bender AT, Tzvetkov E, Pereira A, et al. TLR7 and TLR8 differentially activate the IRF and NF‐kappaB pathways in specific cell types to promote inflammation. Immunohorizons. 2020;4:93‐107. doi: 10.4049/immunohorizons.2000002 [DOI] [PubMed] [Google Scholar]
- 61. Greene TT, Jo YR, Zuniga EI. Infection and cancer suppress pDC derived IFN‐I. Curr Opin Immunol. 2020;66:114‐122. doi: 10.1016/j.coi.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369:1210‐1220. doi: 10.1126/science.abc6261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Laing AG, Lorenc A, Del Molino Del Barrio I, et al. A dynamic COVID‐19 immune signature includes associations with poor prognosis. Nat Med. 2020;26:1623‐1635. doi: 10.1038/s41591-020-1038-6 [DOI] [PubMed] [Google Scholar]
- 64. Peruzzi B, Bencini S, Capone M, et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS‐CoV‐2 infection. Immunology. 2020;161:345‐353. doi: 10.1111/imm.13254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461‐488. doi: 10.1146/annurev-immunol-032713-120156 [DOI] [PubMed] [Google Scholar]
- 66. Rehwinkel J, Gack MU. RIG‐I‐like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20:537‐551. doi: 10.1038/s41577-020-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Trindade BC, Chen GY. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol Rev. 2020;297:139‐161. doi: 10.1111/imr.12902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamada T, Sato S, Sotoyama Y, et al. RIG‐I triggers a signaling‐abortive anti‐SARS‐CoV‐2 defense in human lung cells. Nat Immunol. 2021;22:820‐828. doi: 10.1038/s41590-021-00942-0 [DOI] [PubMed] [Google Scholar]
- 69. Rebendenne A, Valadao ALC, Tauziet M, et al. SARS‐CoV‐2 triggers an MDA‐5‐dependent interferon response which is unable to control replication in lung epithelial cells. J Virol. 2021. doi: 10.1128/JVI.02415-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yin X, Riva L, Pu Y, et al. MDA5 governs the innate immune response to SARS‐CoV‐2 in lung epithelial cells. Cell Rep. 2021;34:108628. doi: 10.1016/j.celrep.2020.108628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thorne LG, Reuschl AK, Zuliani‐Alvarez L, et al. SARS‐CoV‐2 sensing by RIG‐I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021;40:e107826. doi: 10.15252/embj.2021107826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double‐stranded RNA and activation of NF‐kappaB by toll‐like receptor 3. Nature. 2001;413:732‐738. doi: 10.1038/35099560 [DOI] [PubMed] [Google Scholar]
- 73. Bortolotti D, Gentili V, Rizzo S, et al. TLR3 and TLR7 RNA sensor activation during SARS‐CoV‐2 infection. Microorganisms. 2021;9. doi: 10.3390/microorganisms9091820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gao D, Ciancanelli MJ, Zhang P, et al. TLR3 controls constitutive IFN‐beta antiviral immunity in human fibroblasts and cortical neurons. J Clin Invest. 2021;131. doi: 10.1172/JCI134529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lim HK, Huang SXL, Chen J, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. 2019;216:2038‐2056. doi: 10.1084/jem.20181621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guo Y, Audry M, Ciancanelli M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208:2083‐2098. doi: 10.1084/jem.20101568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522‐1527. doi: 10.1126/science.1139522 [DOI] [PubMed] [Google Scholar]
- 78. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS‐STING signalling. Nat Rev Mol Cell Biol. 2020;21:501‐521. doi: 10.1038/s41580-020-0244-x [DOI] [PubMed] [Google Scholar]
- 79. Di Domizio J, Gulen MF, Saidoune F, et al. The cGAS‐STING pathway drives type I IFN immunopathology in COVID‐19. Nature. 2022. doi: 10.1038/s41586-022-04421-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim J, Gupta R, Blanco LP, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus‐like disease. Science. 2019;366:1531‐1536. doi: 10.1126/science.aav4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boni MF, Lemey P, Jiang X, et al. Evolutionary origins of the SARS‐CoV‐2 sarbecovirus lineage responsible for the COVID‐19 pandemic. Nat Microbiol. 2020;5:1408‐1417. doi: 10.1038/s41564-020-0771-4 [DOI] [PubMed] [Google Scholar]
- 82. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thorne LG, Bouhaddou M, Reuschl AK, et al. Evolution of enhanced innate immune evasion by SARS‐CoV‐2. Nature. 2021. doi: 10.1038/s41586-021-04352-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. doi: 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nat Rev Microbiol. 2021;19:155‐170. doi: 10.1038/s41579-020-00468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cortese M, Lee JY, Cerikan B, et al. Integrative imaging reveals SARS‐CoV‐2‐induced reshaping of subcellular morphologies. Cell Host Microbe. 2020;28:853‐866.e855. doi: 10.1016/j.chom.2020.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Klein S, Cortese M, Winter SL, et al. SARS‐CoV‐2 structure and replication characterized by in situ cryo‐electron tomography. Nat Commun. 2020;11:5885. doi: 10.1038/s41467-020-19619-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zheng Y, Deng J, Han L, et al. SARS‐CoV‐2 NSP5 and N protein counteract the RIG‐I signaling pathway by suppressing the formation of stress granules. Signal Transduct Target Ther. 2022;7:22. doi: 10.1038/s41392-022-00878-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Anderson P, Kedersha N. Stress granules. Curr Biol. 2009;19:R397‐R398. doi: 10.1016/j.cub.2009.03.013 [DOI] [PubMed] [Google Scholar]
- 90. Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014;35:420‐428. doi: 10.1016/j.it.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McCormick C, Khaperskyy DA. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol. 2017;17:647‐660. doi: 10.1038/nri.2017.63 [DOI] [PubMed] [Google Scholar]
- 92. Matsuki H, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells. 2013;18:135‐146. doi: 10.1111/gtc.12023 [DOI] [PubMed] [Google Scholar]
- 93. Nencka R, Silhan J, Klima M, et al. Coronaviral RNA‐methyltransferases: function, structure and inhibition. Nucleic Acids Res. 2022;50:635‐650. doi: 10.1093/nar/gkab1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rosas‐Lemus M, Minasov G, Shuvalova L, et al. High‐resolution structures of the SARS‐CoV‐2 2'‐O‐methyltransferase reveal strategies for structure‐based inhibitor design. Sci Signal. 2020;13. doi: 10.1126/scisignal.abe1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Viswanathan T, Arya S, Chan SH, et al. Structural basis of RNA cap modification by SARS‐CoV‐2. Nat Commun. 2020;11:3718. doi: 10.1038/s41467-020-17496-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Banerjee AK, Blanco MR, Bruce EA, et al. SARS‐CoV‐2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. 2020;183:1325‐1339.e1321. doi: 10.1016/j.cell.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lapointe CP, Grosely R, Johnson AG, Wang J, Fernandez IS, Puglisi JD. Dynamic competition between SARS‐CoV‐2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc Natl Acad Sci USA. 2021;118. doi: 10.1073/pnas.2017715118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mendez AS, Ly M, Gonzalez‐Sanchez AM, et al. The N‐terminal domain of SARS‐CoV‐2 nsp1 plays key roles in suppression of cellular gene expression and preservation of viral gene expression. Cell Rep. 2021;37:109841. doi: 10.1016/j.celrep.2021.109841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Schubert K, Karousis ED, Jomaa A, et al. SARS‐CoV‐2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. 2020;27:959‐966. doi: 10.1038/s41594-020-0511-8 [DOI] [PubMed] [Google Scholar]
- 100. Thoms M, Buschauer R, Ameismeier M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science. 2020;369:1249‐1255. doi: 10.1126/science.abc8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yuan S, Peng L, Park JJ, et al. Nonstructural protein 1 of SARS‐CoV‐2 is a potent pathogenicity factor redirecting host protein synthesis machinery toward viral RNA. Mol Cell. 2020;80:1055‐1066.e1056. doi: 10.1016/j.molcel.2020.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hsu JC, Laurent‐Rolle M, Pawlak JB, Wilen CB, Cresswell P. Translational shutdown and evasion of the innate immune response by SARS‐CoV‐2 NSP14 protein. Proc Natl Acad Sci USA. 2021;118. doi: 10.1073/pnas.2101161118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Akopian D, Shen K, Zhang X, Shan SO. Signal recognition particle: an essential protein‐targeting machine. Annu Rev Biochem. 2013;82:693‐721. doi: 10.1146/annurev-biochem-072711-164732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shemesh M, Aktepe TE, Deerain JM, et al. SARS‐CoV‐2 suppresses IFNbeta production mediated by NSP1, 5, 6, 15, ORF6 and ORF7b but does not suppress the effects of added interferon. PLoS Pathog. 2021;17:e1009800. doi: 10.1371/journal.ppat.1009800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hayn M, Hirschenberger M, Koepke L, et al. Systematic functional analysis of SARS‐CoV‐2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021;35:109126. doi: 10.1016/j.celrep.2021.109126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xia H, Cao Z, Xie X, et al. Evasion of type I interferon by SARS‐CoV‐2. Cell Rep. 2020;33:108234. doi: 10.1016/j.celrep.2020.108234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kumar A, Ishida R, Strilets T, et al. SARS‐CoV‐2 nonstructural protein 1 inhibits the interferon response by causing depletion of key host signaling factors. J Virol. 2021;95:e0026621. doi: 10.1128/JVI.00266-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shin D, Mukherjee R, Grewe D, et al. Papain‐like protease regulates SARS‐CoV‐2 viral spread and innate immunity. Nature. 2020;587:657‐662. doi: 10.1038/s41586-020-2601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu G, Lee JH, Parker ZM, et al. ISG15‐dependent activation of the sensor MDA5 is antagonized by the SARS‐CoV‐2 papain‐like protease to evade host innate immunity. Nat Microbiol. 2021;6:467‐478. doi: 10.1038/s41564-021-00884-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang D, Zhang DE. Interferon‐stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res. 2011;31:119‐130. doi: 10.1089/jir.2010.0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Moustaqil M, Ollivier E, Chiu HP, et al. SARS‐CoV‐2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect. 2021;10:178‐195. doi: 10.1080/22221751.2020.1870414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liu Y, Qin C, Rao Y, et al. SARS‐CoV‐2 Nsp5 demonstrates two distinct mechanisms targeting RIG‐I and MAVS to evade the innate immune response. mBio. 2021;12:e0233521. doi: 10.1128/mBio.02335-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wu J, Shi Y, Pan X, et al. SARS‐CoV‐2 ORF9b inhibits RIG‐I‐MAVS antiviral signaling by interrupting K63‐linked ubiquitination of NEMO. Cell Rep. 2021;34:108761. doi: 10.1016/j.celrep.2021.108761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang S, Dai T, Qin Z, et al. Targeting liquid‐liquid phase separation of SARS‐CoV‐2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat Cell Biol. 2021;23:718‐732. doi: 10.1038/s41556-021-00710-0 [DOI] [PubMed] [Google Scholar]
- 116. Chen K, Xiao F, Hu D, et al. SARS‐CoV‐2 nucleocapsid protein interacts with RIG‐I and represses RIG‐mediated IFN‐beta production. Viruses. 2020;13. doi: 10.3390/v13010047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Fu YZ, Wang SY, Zheng ZQ, et al. SARS‐CoV‐2 membrane glycoprotein M antagonizes the MAVS‐mediated innate antiviral response. Cell Mol Immunol. 2021;18:613‐620. doi: 10.1038/s41423-020-00571-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sui L, Zhao Y, Wang W, et al. SARS‐CoV‐2 membrane protein inhibits type I interferon production through ubiquitin‐mediated degradation of TBK1. Front Immunol. 2021;12:662989. doi: 10.3389/fimmu.2021.662989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Konno Y, Kimura I, Uriu K, et al. SARS‐CoV‐2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185. doi: 10.1016/j.celrep.2020.108185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wu Y, Ma L, Zhuang Z, et al. Main protease of SARS‐CoV‐2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct Target Ther. 2020;5:221. doi: 10.1038/s41392-020-00332-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV‐1 Vpu. Nature. 2008;451:425‐430. doi: 10.1038/nature06553 [DOI] [PubMed] [Google Scholar]
- 122. Wang SM, Huang KJ, Wang CT. BST2/CD317 counteracts human coronavirus 229E productive infection by tethering virions at the cell surface. Virology. 2014;449:287‐296. doi: 10.1016/j.virol.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Taylor JK, Coleman CM, Postel S, et al. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol. 2015;89:11820‐11833. doi: 10.1128/JVI.02274-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nemudryi A, Nemudraia A, Wiegand T, et al. SARS‐CoV‐2 genomic surveillance identifies naturally occurring truncation of ORF7a that limits immune suppression. Cell Rep. 2021;35:109197. doi: 10.1016/j.celrep.2021.109197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Addetia A, Xie H, Roychoudhury P, et al. Identification of multiple large deletions in ORF7a resulting in in‐frame gene fusions in clinical SARS‐CoV‐2 isolates. J Clin Virol. 2020;129:104523. doi: 10.1016/j.jcv.2020.104523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Holland LA, Kaelin EA, Maqsood R, et al. An 81‐nucleotide deletion in SARS‐CoV‐2 ORF7a identified from sentinel surveillance in Arizona (January to March 2020). J Virol. 2020;94. doi: 10.1128/JVI.00711-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hung IF, Lung KC, Tso EY, et al. Triple combination of interferon beta‐1b, lopinavir‐ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID‐19: an open‐label, randomised, phase 2 trial. Lancet. 2020;395:1695‐1704. doi: 10.1016/S0140-6736(20)31042-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Davoudi‐Monfared E, Rahmani H, Khalili H, et al. A randomized clinical trial of the efficacy and safety of interferon beta‐1a in treatment of severe COVID‐19. Antimicrob Agents Chemother. 2020;64. doi: 10.1128/AAC.01061-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Monk PD, Marsden RJ, Tear VJ, et al. Safety and efficacy of inhaled nebulised interferon beta‐1a (SNG001) for treatment of SARS‐CoV‐2 infection: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Respir Med. 2021;9:196‐206. doi: 10.1016/S2213-2600(20)30511-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rahmani H, Davoudi‐Monfared E, Nourian A, et al. Interferon beta‐1b in treatment of severe COVID‐19: a randomized clinical trial. Int Immunopharmacol. 2020;88:106903. doi: 10.1016/j.intimp.2020.106903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ader F, Peiffer‐Smadja N, Poissy J, et al. An open‐label randomized controlled trial of the effect of lopinavir/ritonavir, lopinavir/ritonavir plus IFN‐beta‐1a and hydroxychloroquine in hospitalized patients with COVID‐19. Clin Microbiol Infect. 2021;27:1826‐1837. doi: 10.1016/j.cmi.2021.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Consortium, W. H. O. S. T , Pan H, Peto R, et al. Repurposed antiviral drugs for Covid‐19 ‐ interim WHO solidarity trial results. N Engl J Med. 2021;384:497‐511. doi: 10.1056/NEJMoa2023184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kalil AC, Mehta AK, Patterson TF, et al. Efficacy of interferon beta‐1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID‐19: a double‐bind, randomised, placebo‐controlled, phase 3 trial. Lancet Respir Med. 2021;9:1365‐1376. doi: 10.1016/S2213-2600(21)00384-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Alavi Darazam I, Hatami F, Mahdi Rabiei M, et al. An investigation into the beneficial effects of high‐dose interferon beta 1‐a, compared to low‐dose interferon beta 1‐a in severe COVID‐19: the COVIFERON II randomized controlled trial. Int Immunopharmacol. 2021;99:107916. doi: 10.1016/j.intimp.2021.107916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wang N, Zhan Y, Zhu L, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID‐19 patients. Cell Host Microbe. 2020;28:455‐464.e452. doi: 10.1016/j.chom.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhou Q, MacArthur MR, He X, et al. Interferon‐alpha2b treatment for COVID‐19 is associated with improvements in lung abnormalities. Viruses. 2020;13. doi: 10.3390/v13010044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhou Q, Chen V, Shannon CP, et al. Interferon‐alpha2b treatment for COVID‐19. Front Immunol. 2020;11:1061. doi: 10.3389/fimmu.2020.01061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Feld JJ, Kandel C, Biondi MJ, et al. Peginterferon lambda for the treatment of outpatients with COVID‐19: a phase 2, placebo‐controlled randomised trial. Lancet Respir Med. 2021;9:498‐510. doi: 10.1016/S2213-2600(20)30566-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Levy R, Bastard P, Lanternier F, Lecuit M, Zhang SY, Casanova JL. IFN‐alpha2a therapy in two patients with inborn errors of TLR3 and IRF3 infected with SARS‐CoV‐2. J Clin Immunol. 2021;41:26‐27. doi: 10.1007/s10875-020-00933-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID‐19: a timeline of immunological insights. Nat Rev Immunol. 2021;21:245‐256. doi: 10.1038/s41577-021-00522-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zhang Q, Bastard P, Bolze A, et al. Life‐threatening COVID‐19: defective interferons unleash excessive inflammation. Med (N Y). 2020;1:14‐20. doi: 10.1016/j.medj.2020.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable ‐ no new data generated.