

Abstract
Purpose of Review:
Factor XII (FXII), the precursor of the protease FXIIa, contributes to pathologic processes including angioedema and thrombosis. Here we review recent work on structure-function relationships for FXII based on studies using recombinant FXII variants.
Recent Findings:
FXII is a homolog of Pro-hepatocyte growth factor activator (Pro-HGFA). We prepared FXII in which domains are replaced by corresponding parts of Pro-HGA, and tested them in FXII activation and activity assays. In solution FXII and prekallikrein undergo reciprocal activation to FXIIa and kallikrein. The rate of this process is restricted by the FXII fibronectin type-2 and kringle domains. Pro-HGA replacements for these domains accelerate FXII and prekallikrein activation. When FXII and PK bind to negatively charged surfaces, reciprocal activation is enhanced. The FXII EGF1 domain is required for surface binding.
Summary:
We propose a model in which FXII is normally maintained in a closed conformation resistant to activation by intramolecular interactions involving the fibronectin type-2 and kringle domains. These interactions are disrupted when FXII binds to a surface through EGF1, enhancing FXII activation and PK activation by FXIIa. These observations have important implications for understanding the contributions of FXII to disease, and for developing therapies to treat thrombo-inflammatory disorders.
Keywords: Factor XII, Factor XIIa, Prekallikrein, Angioedema, Thrombosis
Introduction
Factor XII (FXII), also called Hageman factor, is the zymogen precursor of the trypsin-like protease FXIIa (FXIIa) [1,2]. FXII was first described in 1955 as a blood constituent missing in some individuals whose plasma clotted slowly when exposed to negatively charged substances such as glass. While this initially suggested a role for the protein in normal blood coagulation (hemostasis), it is now clear that complete absence of FXII is not associated with abnormal bleeding [3,4]. However, despite its limited role in hemostasis, there is considerable interest in FXII as a therapeutic target, centered on its contributions to pathologic coagulation (thrombosis) and inflammation [1,5–7]. Here we discuss recent work on FXII structure and enzymology that provides insight into mechanisms through which this protein contributes to pathologic processes.
FXII, along with the zymogen prekallikrein (PK) and the cofactor high molecular weight kininogen (HK), comprise the plasma kallikrein-kinin system (KKS) [5,7]. In solution, FXII and PK undergo reciprocal proteolytic conversion to the active serine proteases factor XIIa (FXIIa) and plasma kallikrein (PKa), respectively (Figure 1A) [8,9]. PKa, in addition to activating FXII, cleaves HK, releasing the vasoactive nanopeptide bradykinin [5,9]. These reactions are accelerated when the KKS proteins bind to a variety of substances or surfaces by a process called contact activation (Figure 1B) [5,7,10]. Contact activation is initiated by autocatalytic conversion of FXII to FXIIa on a surface (Figure 1C) [1,5,7,8]. Inorganic materials such as silicate-containing compounds (glass, kaolin, celite) [4], and biological polyanionic macromolecules such as polyphosphates [11–13] and nucleic acids [14] are examples of surfaces/substances that support FXII autoactivation and contact activation.
Figure 1. The Kallikrein-Kinin System and Contact Activation.
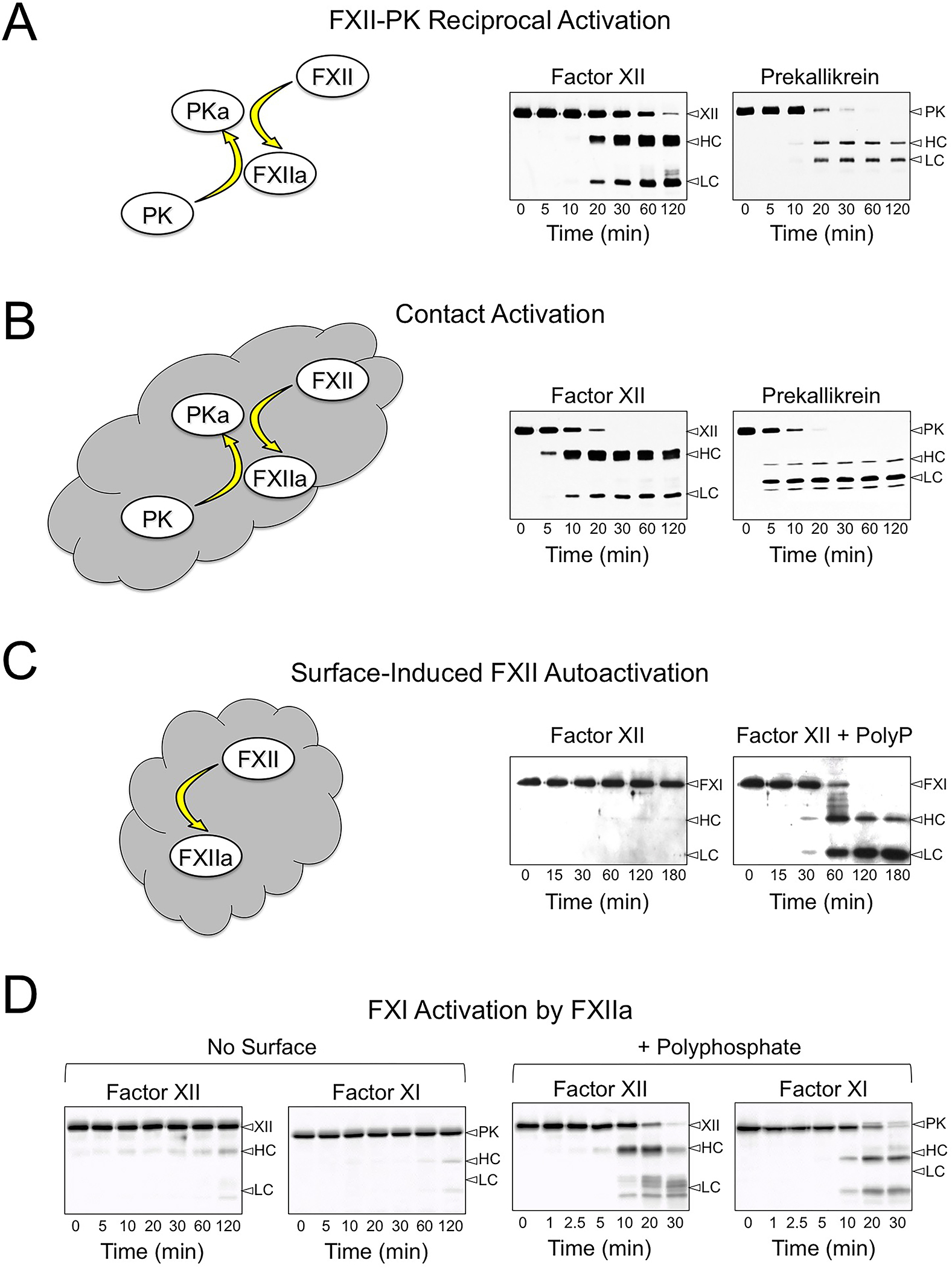
Shown are western blots of time courses for reactions containing FXII (200 nM), PK (300 nM) and/or FXI (30 nM) in 20mM HEPES, pH7.4, 100mM NaCl, 0.1% PEG-8000, 10 μM ZnCl2 incubated at 37 °C. At the indicated times, samples were removed into reducing SDS-sample buffer. Samples were size fractionated on 10% SDS-polyacrylamide gels, transferred to nitrocellulose and developed with goat polyclonal antibodies against human FXII, PK or FXI. (A) Reciprocal activation of FXII and PK in the absence of a surface. (B) Reciprocal activation of FXII and PK in the presence of 100 μg/ml leukocyte DNA. (C) Autoactivation of FXII in the absence (left panel) or presence (right panel) of 70 μM polyphosphate (60–100 unit chain length). (D) Reciprocal activation of FXII and FXI in the absence (no surface) or presence of 70 μM polyphosphate (+ polyphosphate). For all panels, positions of markers for FXII, PK and FXI, and the heavy chains (HC) and light chains (LC) of FXIIa, PKa, and FXIa are shown on the right. Blots in panel A and B are from reference 41, and panel C is from reference 8. Images in Panel D have not been previously published.
Disease Processes Involving Factor XII
In vivo, bradykinin generated during basal FXII-PK reciprocal turnover likely contributes to setting normal vascular tone and permeability by binding to specific bradykinin receptors on blood vessel cells [15,16]. Greater bradykinin production at injury sites increases vascular permeability, tissue swelling, and pain sensation. The term hereditary angioedema (HAE) encompasses several genetic disorders characterized by episodes of painful soft tissue swelling involving the oropharyngeal mucosa, subcutaneous tissues of the hands and face, genitals and gastrointestinal tract [17–19]. Excessive bradykinin production or heightened vascular sensitivity to bradykinin appears to underlie HAE [18,20]. Angioedema in the most common forms of HAE responds to FXIIa-neutralizing antibodies [21], PKa inhibitors [22], and reduction of plasma PK [23], attesting to the importance of the KKS in the disease process.
Exposing plasma to substances that induce contact activation leads to formation of a fibrin clot. This FXIIa-initiated process is the basis of the activated partial thromboplastin time assay used in clinical practice, [3]. Surface-bound FXIIa converts a homolog of PK called factor XI (FXI) to the protease FXIa. FXIa then promotes clot formation by activating factor IX. [24]. While structurally similar to PK, FXI undergoes reciprocal activation with FXII weakly in solution (Figure 1D). Furthermore, FXIIa is a poor FXI activator in the absence of a surface. While FXI activation by FXIIa does not appear to be required for hemostasis [3], it promotes pathologic clot formation (thrombosis) in clinical settings where blood comes into contact with non-biological materials that induce contact activation such as renal dialysis membranes, cardiopulmonary bypass circuits, extracorporeal membrane oxygenators, and central venous catheters [5,25,26].
The Factor XII Molecule
The diagram in Figure 2A shows the amino acid sequence and disulfide bond structure of human FXII [1,2,27,28]. FXII in plasma is synthesized primarily in hepatocytes. The non-catalytic portion of the protein is referred to as the heavy chain (Figure 2B). The FXII heavy chain contains, from the N-terminus, a fibronectin type 2 (FN2), epidermal growth factor-1 (EGF1), fibronectin type 1 (FN1), EGF2, and kringle (KNG) domain and a proline-rich region (PRR). At the FXII C-terminus is a trypsin-like protease domain or light chain (Figure 2B). Conversion of FXII to FXIIa requires a single proteolytic cleavage after Arg353, creating separate heavy and light chain polypeptides that remain connected through the Cys340-Cy467 disulfide bond (Figure 2A). FXII activation can be followed on reducing SDS-polyacrylamide gels, where conversion of 80 kDa FXII to the 50 kDa and 30 kDa heavy and light chains of FXIIa is readily apparent (Figure 1A) [1,2].
Figure 2. Factor XII Structure.
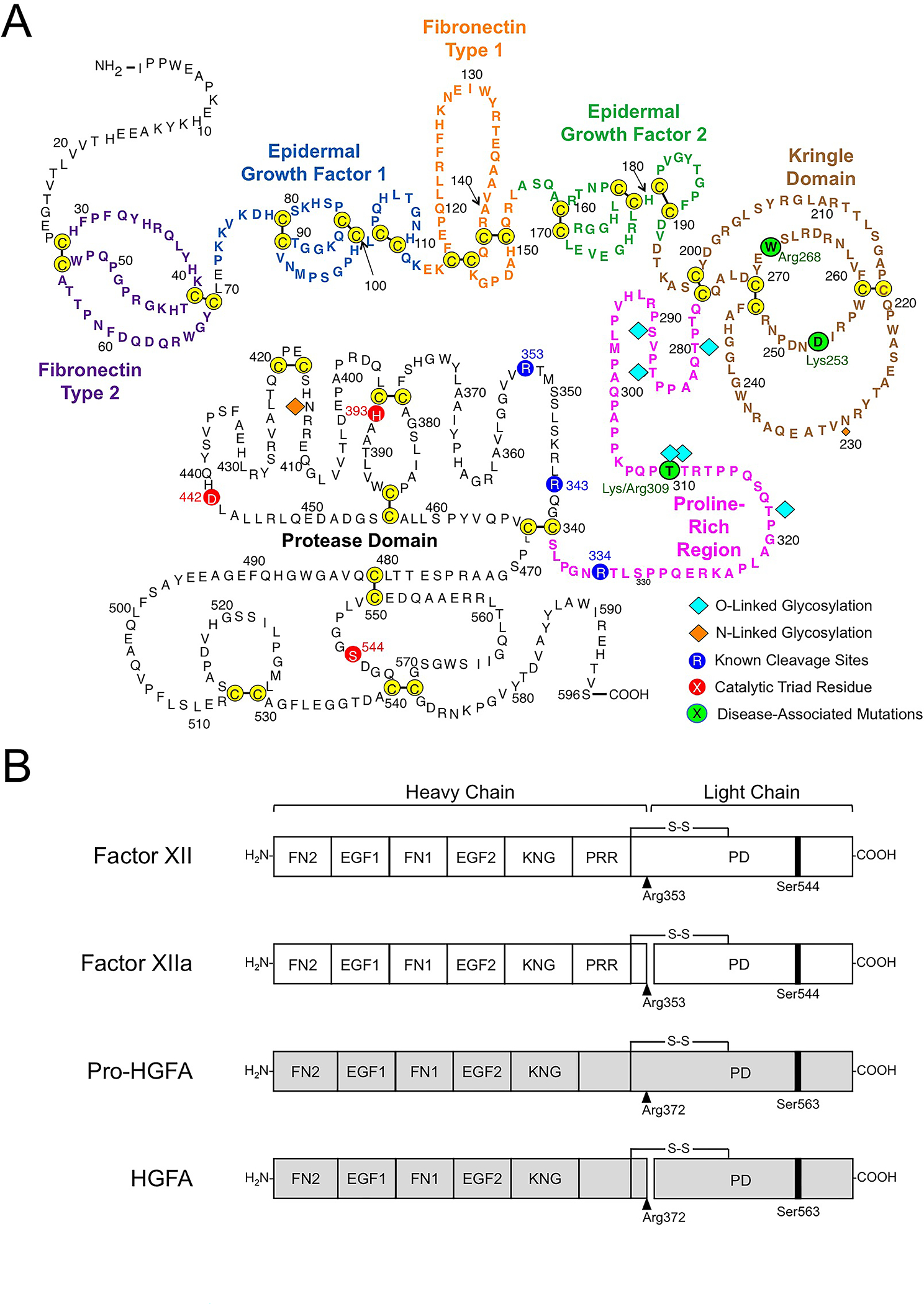
(A) Amino acid sequence and disulfide bonds for human plasma FXII. The heavy chain contains a fibronectin type 2 (purple), epidermal growth factor 1 (blue), fibronectin type 1 (orange), epidermal growth factor 2 (green), and kringle (brown) domain, and a proline-rich regions (magenta). The catalytic triad His393, Asp442 Ser544 in the protease domain (light chain) are indicated in red. Positions of O- and N-linked glycosylation sites are indicated by blue and orange diamonds. Positions of disease-associated mutations are shown in green circles. Image adapted from reference 27. (B) Schematic diagrams of human FXII (white), Pro-HGFA (gray), and their activated forms FXIIa and HGFA. The FXII fibronectin type 2 (FN2), epidermal growth factor 1 (EGF1), fibronectin type 1 (FN1), epidermal growth factor 2 (EGF2), and kringle (KNG) domains, the proline-rich region (PRR), and the protease domain are indicated. Pro-HGFA is organized similarly except that it does not have a PRR, and the corresponding sequence is not assigned a name. Positions of FXII and Pro-HGFA active site serine residues (Ser544 and Ser563, respectively) are indicated by black bars, and sites for proteolytic activation (after Arg353 and Arg372, respectively) are indicated by black arrows. Image adapted from reference 2.
FXII arose from a duplication of the gene for the injury repair protease pro-hepatocyte growth factor activator (Pro-HGFA) [28,29]. Pro-HGFA, like FXII, is converted to its active form (HGFA) by a single cleavage between the heavy and light chains (Figure 2B). While, FXII and Pro-HGFA have similar structures, there are noteworthy differences. The heavy chain of FXII is positively charged, while its protease domain is negatively charged. The situation is reversed in Pro-HGFA [2]. Pro-HGFA also lacks the proline-rich region found in FXII. Despite their structural similarities, Pro-HGFA and FXII are involved in different activities. For the purpose of our discussion, Pro-HGFA cannot substitute for FXII in the KKS in surface-independent reciprocal activation with PK, nor in surface-dependent processes involving contact activation.
Factor XII Activation in the Absence of a Surface
When FXII and PK are mixed in solution (in the absence of a surface) they undergo reciprocal conversion to FXIIa and PKa (Figure 1A) [2,8,9]. The process may be triggered by enzymatic activity inherent in zymogen FXII. FXII in which Arg353 at the activation cleavage site is replaced with alanine (FXII-T) cannot be converted to FXIIa by PKa or by autoactivation on a surface (Figure 3A). However, FXII-T slowly converts PK to PKa indicating it has activity (Figure 3B) [8]. The low level of activity intrinsic to FXII (three orders of magnitude less than FXIIa) likely initiates and maintains reciprocal activation by slowly converting PK to PKa. Amplification of the reaction, however, would require conversion of FXII to FXIIa by PKa. An important implication of intrinsic FXII activity is that FXII-PK reciprocal activation likely occurs in plasma at a basal level, even in the absence of a surface to induce contact activation [8,16,26].
Figure 3. Activity in Factor XII Zymogen and ΔFXII.
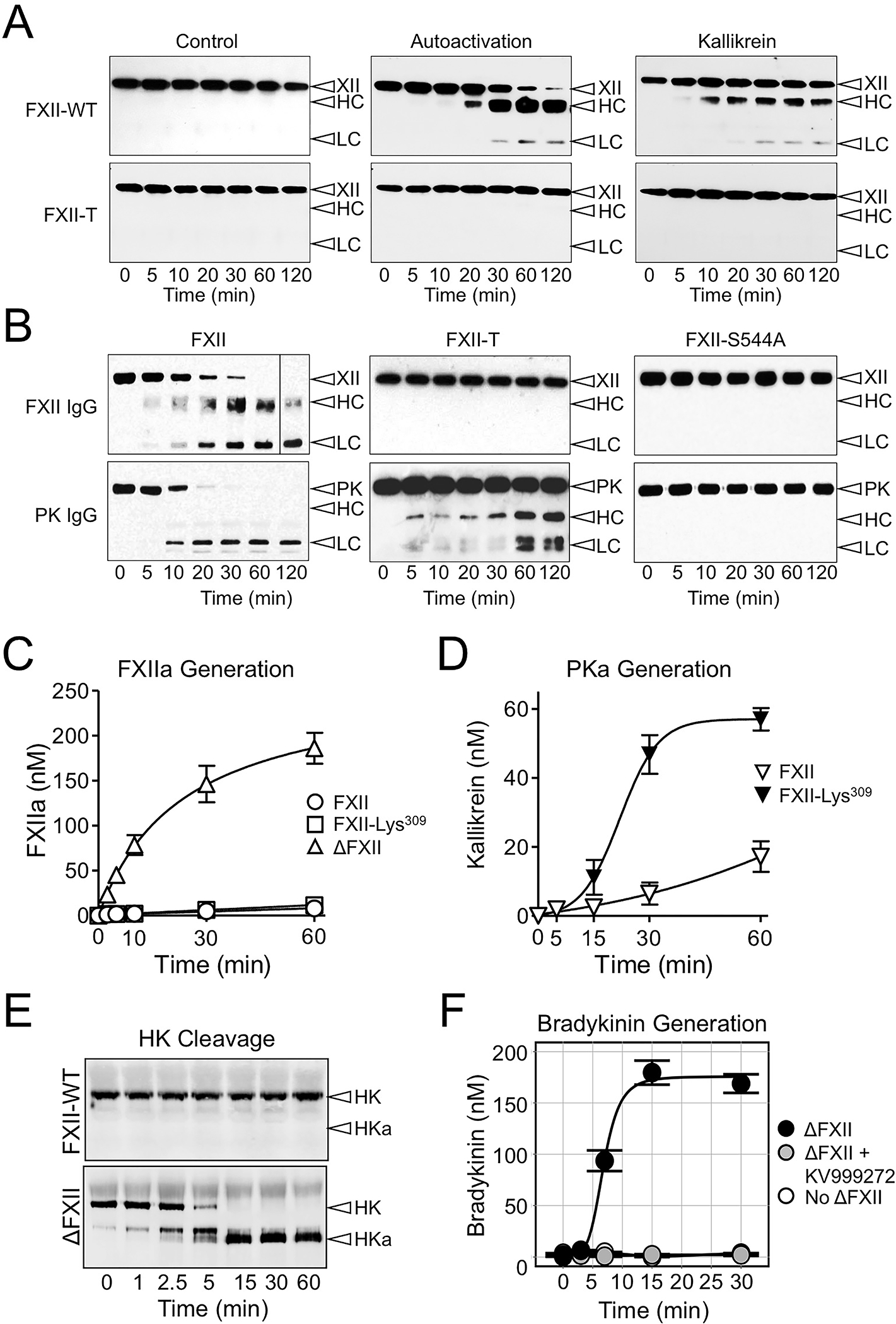
(A & B) FXII activity. (A) FXII and FXII-T (200 nM) were incubated in the absence of an activator (control, left column), with 70 μM Poly-P (center), or with 50 nM kallikrein without a surface (right). (B) FXII, FXII-T or FXII with the active site serine changed to alanine (FXII-S554A), 200 nM, and PK (200 nM) were incubated at 37°C. For panels A and B, at indicated time points, samples were removed into reducing sample buffer. Samples were size fractionated by SDS-PAGE, followed by western blot using a polyclonal anti-human FXII or anti-PK IgG. Positions of standards for FXII (XII) and PK and the heavy chain (HC) and light chain (LC) of FXIIa and PKa are indicated at the right of each image. Data is from reference 8. (C & D) ΔFXII activation and activity. (C) FXII (○), the full-length FXII precursor used to generate ΔFXII (◻), or ΔFXII (△), 200 nM, were incubated at 37°C with PKa (10 nM). FXIIa activity was measured by chromogenic substrate assay. (D) PK (200 nM) was mixed with 12.5 nM FXII (▽) or FXII-Lys309 (▼). The FXII preparations were preincubated with thrombin, which removes the heavy chain from FXII-Lys309 but not from FXII, prior to addition to PK. PKa generation was determined with a chromogenic substrate assay. Data for panels C and D are from reference 8. (E&F) Effects of ΔFXII on high molecular weight kininogen (HK). (E) Western blots of human FXII-deficient plasma supplemented with FXII or ΔFXII (400 nM). At indicated times, samples were removed into non-reducing sample buffer. Western blots were probed with goat anti-human HK IgG (HK). Positions of standards for HK and cleaved HK (HKa) are shown on the right. (F) Bradykinin generation in normal plasma after addition of ΔFXII (160 nM; black), ΔFXII and an inhibitor of kallikrein (KV999272 10 mM; gray), or vehicle (white). Bradykinin was measured by ELISA (Enzo Life Sciences). Data for panels E and F are from reference 42.
In plasma, FXIIa and PKa activities are regulated by the serpin C1-inhibitor (C1-INH) [30]. Inherited C1-INH deficiency is the most common cause of HAE [18]. The episodic nature of angioedema in C1-INH-deficient patients indicates triggering events periodically increase KKS activation that exceeds the regulatory capacity of the reduced C1-INH. It is not known if bradykinin generation in C1-INH-deficient patients requires a surface, but recent work indicates at least one form of HAE is probably not surface-dependent. There are HAE patients with normal C1-INH levels [18,31], and some of these individuals have lysine or arginine substitutions for Thr309 in FXII (FXII-Lys/Arg309, Figure 2A) [18,31]. Several proteases (thrombin, FXIa, plasmin) cleave FXII-Lys/Arg309 after residue 309 resulting in a truncated protein (ΔFXII) lacking most of the heavy chain [9,32]. As the FXII heavy chain is required for surface binding, ΔFXII cannot support contact activation [9]. However, in the absence of a surface, ΔFXII is activated by PKa with a catalytic efficiency 15-fold greater than for FXII (Figure 3C), accelerating reciprocal FXII-PK activation. Adding ΔFXII to normal plasma overwhelms endogenous C1-INH leading to rapid conversion of PK to PKa (Figure 3D), HK cleavage by kallikrein (Figure 3E), and bradykinin release (Figure 3F) [9]. From a structure-function perspective, the properties of ΔFXII imply that, in the absence of a surface, the FXII heavy chain performs an important regulatory function that restricts FXII activation by PKa, and sets a limit on the basal rate of FXII-PK reciprocal activation.
An Inhibitory Role for the FXII Heavy Chain
To investigate FXII heavy chain functions, we took advantage of structural similarities between FXII and Pro-HGFA to create a panel of FXII proteins in which portions of the heavy chain are replaced with corresponding parts from Pro-HGFA (Figure 4A) [2]. Replacing the entire heavy chain results in a protein (HGFAHC/FXIILC) that behaves like ΔFXII in reciprocal activation reactions with PK (Figure 4B, left panel), demonstrating the Pro-HGFA heavy chain cannot replace the normal regulatory function of the FXII heavy chain. For FXII proteins with individual Pro-HGFA domain replacements, rates of reciprocal FXII-PK activation in reactions containing the chimeric proteins FXII-FN2 or FXII-KNG were comparable to those for HGFAHC/FXIILC and ΔFXII (Figure 4C), indicating the FXII FN2 and KNG domains are important for maintaining the protein in a closed conformation that limits activation.
Figure 4. Factor XII/Pro-HGFA Chimeras.
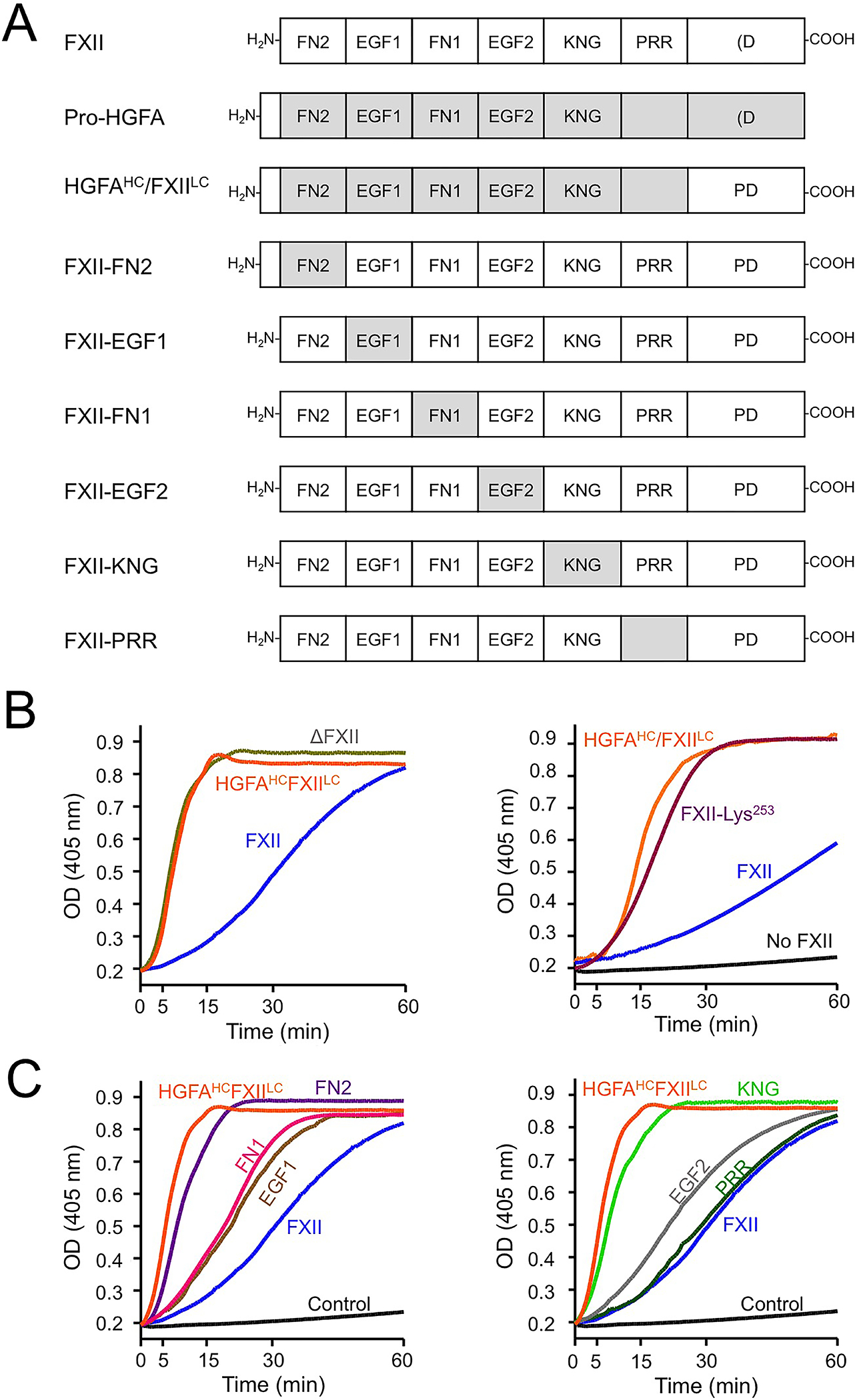
(A) Schematic diagrams for FXII, Pro-HGFA and FXII with domains replaced with corresponding domains from Pro-HGFA. Domain abbreviations are explained in the legend for figure 2. (B&C) Reciprocal activation of FXII and PK. (B) PK (60 nM) was mixed with 12.5 nM FXII, HGFAHC/FXIILC, or ΔFXII (left panel) or FXII-Lys253 (right panel), and 200 mM chromogenic substrate S-2302 at 37°C. Changes in OD of 405 nm were continuously monitored on a spectrophotometer. (C) Reactions set up as in panel B, except that FXII/Pro-HGFA chimeras were used in place of FXII. Data are from reference 2.
KNG domains in the fibrinolytic protease zymogen plasminogen contain Asp-X-Asp/Glu motifs that bind side chains of lysine and arginine [33,34]. These sites mediate intra- and inter-molecular binding interactions that are crucial to the functions of plasminogen and its active form plasmin. In full-length Glu-plasminogen, intramolecular binding interactions involving the KNG domains maintain the protein in a closed form that is relatively resistant to activation. The KNG domain in FXII also contains an Asp-X-Asp/Glu motif (Asp251-Asn-Asp253, Figure 2A). Disrupting the site by replacing Asp253 with lysine (FXII-Lys253) converts FXII from a closed to an open conformation, with activity in reciprocal activation reactions with PK that are comparable to ΔFXII (Figure 4B, right panel) [2]. This suggests that the closed conformation for FXII requires interactions between the KNG domain and lysine/arginine residues elsewhere in the molecule. A Trp286 to arginine substitution in the FXII KNG domain was identified recently in a patient with an autoinflammatory syndrome [35,36]. This mutation also confers an open conformation, suggesting intramolecular interactions involving FXII KNG extend beyond Asp251-Asn-Asp253.
Surface-Dependent Factor XII Activation
FXII bound to a contact surface undergoes autocatalytic conversion to FXIIa that may be initiated by the activity intrinsic to FXII discussed above [1,2,8]. FXII activation by PKa is also enhanced on a surface [2,5]. Surface-binding appears to facilitate FXII activation by two mechanisms. First, binding causes FXII to assume an open conformation that is more susceptible to activation [2,37]. The ability of a relatively short polymer of the polyanion dextran sulfate to enhance FXII activation by PKa demonstrates this effect (Figure 5A). Conformational changes induced by surface binding probably render the activation cleavage site more accessible to an activating protease. Second, substrate (FXII) and activating protease (PKa or FXIIa) can bind in proximity to each other on a surface [2,11]. This “template mechanism” requires a larger binding area than is required for the conformational changes that open the FXII structure, and exhibits a bell-shaped dependency on surface concentration (Figure 5B). The combined enhancing effect of conformational changes and the template mechanism are indicated by the red circles in Figure 5A.
Figure 5. Surface-Dependent FXII activation.
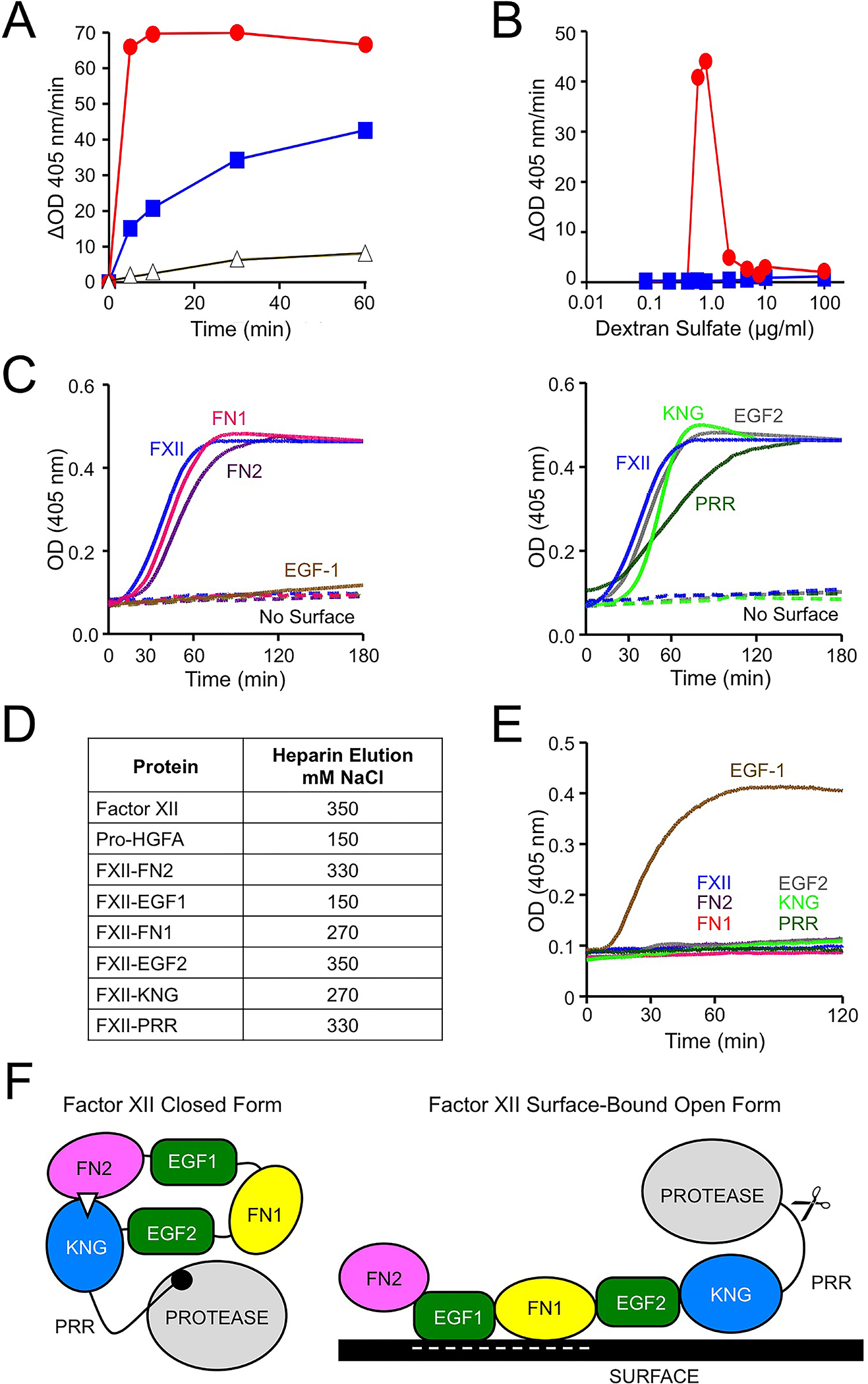
(A) FXII (100 nM) incubated with vehicle (black), 6–10 kDa dextran sulfate (2.5 ug/ml, blue) or 500 kDa dextran sulfate (0.5 ug/ml, red). At indicated times samples were removed and FXIIa activity was detected with a chromogenic substrate. (B) FXII (100 nM) were incubated with varying concentrations of 6–10 kDa dextran sulfate (blue) or 500 kDa dextran sulfate (red). After 30 minutes, FXIIa activity was detected with a chromogenic substrate. (C) FXII autoactivation. FXII or FXII-Pro-HGFA chimeras (figure 4), 200 nM, were incubated with chromogenic substrate S-2302 (200 mM) and polyphosphate (70 μM). Changes in optical density at 405 nm were monitored on a spectrophotometer. Comparable reactions without polyphosphate are indicated by dashed lines. (D) FXII, Pro-HGFA or FXII-Pro-HGFA chimeras (20 μg), were applied to a 1 ml heparin-Sepharose column in buffer containing 100 mM NaCl at pH 7.4. Elution was with a linear NaCl gradient (150–1000 mM). Shown are NaCl concentrations required to elute protein from the column. (E) FXII autoactivation, as in panel C, except that polyphosphate was replaced with 130 nM polyethyleneimine. (F) Model for FXII surface-dependent activation. The image on the left proposes a model for FXII in a closed conformation with the KNG domain binding to lysine/arginine residues elsewhere in the heavy chain through an Asp-X-Asp/Glu lysine-binding motif (represented by the white triangle). In the diagram, the interaction is with residues in the FN2 domain; however, the binding site(s) could reside elsewhere in the molecule. When FXII binds to a negatively charged surface, the internal binding interactions involving KNG are disrupted, exposing the activation cleavage site (indicated by scissors). Surface binding in this model is through the EGF1 domain, although different domains may contribute, depending on the surface. Data in panels, C-F are from reference 2.
The FXII heavy chain is required for surface binding, but there is disagreement as to which domains are most important for the process [2,32,38]. We used the FXII/Pro-HGFA chimera panel to study surface-dependent reactions using polyanionic inducers of contact activation [2]. Results for autoactivation of FXII in the presence of polyphosphate are shown in (Figure 5C), and indicate that the FXII EGF1 domain is required for FXII autoactivation with this polyanion. Similar results were obtained with ellagic acid as the activator. With dextran sulfate, the FXII EGF1 and FN1 were required [2]. PK activation by FXIIa in the presence of a surface also required the EGF1 domain [2]. These data suggest that the FXII EGF1 domain, which carries a net positive charge, contains a binding site for negatively charged surfaces. This notion is supported by studies of FXII binding to the polyanion heparin (Figure 5D). Interestingly, while polyanions do not induce FXII-EGF1 autoactivation, the cationic polymer polyethyleneimine (PEI) does (Figure 5E). This could reflect direct binding of the negatively charged Pro-HGFA EGF1 domain in FXII-EGF1 to PEI, or loss of the positively charged FXII-EGF1 domain allowing interactions between PEI and other parts of the molecule. Regardless of the mechanisms involved, the results with FXII-EGF1 indicate that surface-binding per se, and not a particular surface property such as charge, may be sufficient for FXII autoactivation.
A Model for Surface-Dependent Factor XII Activation
The data presented here, based largely on work with FXII/Pro-HGFA chimeric molecules suggest a model for FXII activation that is summarized in Figure 5F. When not surface bound, FXII is in a closed conformation with restricted access to the activation cleavage site (the Arg353-Val354 peptide bond). The closed conformation requires at least two components of the FXII heavy chain, which may physically mask the activation site, as described for Glu-plasminogen [33,34], and/or directly interact with the protease domain, as observed for plasminogen and prothrombin [33,39,40]. Intramolecular interactions between the KNG domain and other parts of the heavy chain (perhaps FN2) or the light chain may contribute to the closed conformation. Disruption of the closed conformation, either through loss of the heavy chain (ΔFXII) or by changes to its structure (FXII-FN2 and FXII-KNG), enhances FXII activation by PKa. This mechanism suggests a cause for the angioedema and inflammation observed in patients with the Thr309 to Lys/Arg [9,32] or Trp286 to Arg [35,36] substitutions, both of which facilitate removal of the heavy chain.
The FXII heavy chain is required for surface-dependent processes involved in contact activation, including FXII autoactivation, FXII activation by PKa, and FXIIa activation of PK and FXI [1,2,5,10]. Our analysis indicates the EGF1 domain, and perhaps areas adjacent to it on the FN1 domain, mediate binding to polyanions such as polyphosphate, heparin and dextran sulfate. Surface binding appears to neutralize the inhibitory processes mediated through FN2 and KNG, rendering the activation cleavage site more accessible to activating proteases. It is conceivable that FXII binding to some types of surfaces, particularly those that lack a negative charge, involves heavy chain components other than EGF1. Additional work is required to assess this possibility.
Conclusion
Studies over the past thirty years using antibodies, inhibitory peptides, recombinant FXII domains, and FXII deletion variants have provided insights into structure-function relationships for FXII, but have also generated conflicting data regarding the importance of the various heavy chain domains to specific FXII functions. While results with FXII/Pro-HGFA chimeras are consistent with some recently published findings, ultimately, structures for FXII and FXIIa are required to put the data and models into their proper contexts.
The work with recombinant FXII and its variants provides some insight into a clinical conundrum. FXIIa activity is key to tissue edema in most forms of HAE through its capacity to activate PK. FXII also contributes to thrombus formation when blood is exposed to surfaces of medical devices because of its ability to activate FXI. Bradykinin production in HAE is usually thought of in terms of a process that occurs on cell-surfaces or substances that induce contact activation. This begs the question, why aren’t HAE patients prone to thrombosis? This could be explained if bradykinin production in HAE is partly, or even largely, a surface-independent process. The proposed mechanism by which ΔFXII, which lacks a heavy chain, causes HAE in patients with the FXII-Lys/Arg309 substitution is consistent with this hypothesis. FXIIa-induced thrombus formation, on the other hand, may require a surface such as polyphosphate released from platelets because FXIIa is a weak activator of FXI in the absence of a surface. The requirement for a surface may distinguish FXIIa-mediated angioedema from FXIIa-mediated thrombosis.
ACKNOWLEDGMENTS
The authors wish to acknowledge the support of the Ernest W. Goodpasture Chair in Experimental Pathology for Translational Research from Vanderbilt University.
The work discussed in this manuscript was supported by grant HL140025 from the National Heart, Lung and blood Institute.
Footnotes
CONFLICTS OF INTEREST
A.S. and M.L have no conflicts to report. D.G. is a consultant for several pharmaceutical companies that are developing compounds that target factor XI and factor XII for therapeutic purposes.
BIBLIOGRAPHY
(✶ indicates references of special interest)
- 1. Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131(17):1903–1909. ✶An excellent summary on the contributions of factor XII and contact activation to thombo-inflammatory disorders.
- 2. Shamanaev A, Ivanov I, Sun MF, et al. A Model for Surface-Dependent Factor XII Activation: The Roles of Factor XII Heavy Chain Domains. Blood Adv. (in press). ✶This manuscript contains details on most of the structure-function data for FXII covered in this review.
- 3.Gailani D, Wheeler AP, Neff AT. Rare coagulation factor deficiencies. In: Hoffman R, Benz EJ, Silberstein LE, et al. , eds. Hematology: Basic Principles and Practice. 7th ed. Philadelphia, PA: Elsevier; 2018: 2034–2050 [Google Scholar]
- 4.Juang LJ, Mazinani N, Novakowski SK, et al. Coagulation factor XII contributes to hemostasis when activated by soil in wounds. Blood Adv. 2020. Apr 28;4(8):1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14(1):28–39. [DOI] [PubMed] [Google Scholar]
- 6.Weitz JI, Fredenburgh JC. Factors XI and XII as targets for new anticoagulants. Front Med (Lausanne). 2017. Feb 24;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srivastava P, Gailani D. The rebirth of the contact pathway: a new therapeutic target. Curr Opin Hematol. 2020. Sep;27(5):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov I, Matafonov A, Sun MF, et al. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood. 2017;129(11):1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivanov I, Matafonov A, Sun MF, et al. A mechanism for hereditary angioedema with normal C1 inhibitor: an inhibitory regulatory role for the factor XII heavy chain. Blood. 2019;133(10):1152–1163. ✶This manuscript describes experiments explaining the mechanism behind hereditary angioedema caused by the FXII-Lys/Arg309 mutation.
- 10.Long AT, Kenne E, Jung R, et al. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14(3):427–437. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Ivanov I, Smith SA, et al. Polyphosphate, Zn 2+ and high molecular weight kininogen modulate individual reactions of the contact pathway of blood clotting. J Thromb Haemost. 2019. Dec;17(12):2131–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shim YJ, Chatterjee V, Swaidani S, et al. Polyphosphate expression by cancer cell extracellular vesicles mediates binding of factor XII and contact activation. Blood Adv. 2021. Nov 23;5(22):4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malik RA, Zhou J, Fredenburgh JC, et al. Polyphosphate-induced thrombosis in mice is factor XII dependent and is attenuated by histidine-rich glycoprotein. Blood Adv. 2021. Sep 28;5(18):3540–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rangaswamy C, Englert H, Deppermann C, Renné T. Polyanions in Coagulation and Thrombosis: Focus on Polyphosphate and Neutrophils Extracellular Traps. Thromb Haemost. 2021. Aug;121(8):1021–1030. [DOI] [PubMed] [Google Scholar]
- 15.Revenko AS, Gao D, Crosby JR, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011. Nov 10;118(19):5302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwaki T, Castellino FJ. Plasma levels of bradykinin are suppressed in factor XII-deficient mice. Thromb Haemost. 2006. Jun;95(6):1003–10. [DOI] [PubMed] [Google Scholar]
- 17.Lumry WR, Settipane RA. Hereditary angioedema: epidemiology and burden of disease. Allergy Asthma Proc. 2020;41 (6 suppl 1):S08–S13. [DOI] [PubMed] [Google Scholar]
- 18. Wedner HJ. Hereditary angioedema: pathophysiology (HAE type I, HAE type II, and HAE nC1-INH). Allergy Asthma Proc. 2020;41(6 suppl 1):S14–S17. ✶An excellent review of the different types of hereditary angioedema.
- 19.Bork K, Anderson JT, Caballero T, et al. Assessment and management of disease burden and quality of life in patients with hereditary angioedema: a consensus report. Allergy Asthma Clin Immunol. 2021;17(1): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maurer M, Magerl M. Differences and similarities in the mechanisms and clinical expression of bradykinin-mediated vs. mast cell-mediated angioedema. Clin Rev Allergy Immunol. 2021;61(1):40–49. ✶This review offers excellent explanations of the differences behind brady-kinin dependent disorders such as hereditary angioedema, and more common histamine-mediated tissue swelling.
- 21.Craig T, Magerl M, Levy DS, et al. Prophylactic use of an anti-activated factor XII monoclonal antibody, garadacimab, for patients with C1-esterase inhibitor-deficient hereditary angioedema: a randomised, double-blind, placebo-controlled, phase 2 trial Lancet. 2022. Mar 5;399(10328):945–955. [DOI] [PubMed] [Google Scholar]
- 22.Busse P, Kaplan A. Specific Targeting of Plasma Kallikrein for Treatment of Hereditary Angioedema: A Revolutionary Decade. J Allergy Clin Immunol Pract. 2022. Mar;10(3):716–722. [DOI] [PubMed] [Google Scholar]
- 23.Fijen LM, Riedl MA, Bordone L, et al. Inhibition of prekallikrein for hereditary angioedema. N Engl J Med. 2022. Mar 17;386(11):1026–1033. [DOI] [PubMed] [Google Scholar]
- 24.Mohammed BM, Matafonov A, Ivanov I, et al. An update on factor XI structure and function. Thromb Res. 2018. Jan;161:94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tillman B, Gailani D. Inhibition of Factors XI and XII for Prevention of Thrombosis Induced by Artificial Surface. Semin Thromb Hemost. 2018. Feb;44(1):60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mailer RK, Kuta P, Renne T. An update on safe anticoagulation. Haemostaseologie 2022;42(1):65–72. [DOI] [PubMed] [Google Scholar]
- 27.Cool DE, Edgell CJ, Louie GV, et al. Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa. J Biol Chem. 1985. Nov 5;260(25):13666–76. [PubMed] [Google Scholar]
- 28.Ponczek MB, Shamanaev A, LaPlace A, et al. The evolution of factor XI and the kallikrein-kinin system. Blood Adv. 2020. Dec 22;4(24):6135–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyazawa K, Shimomura T, Kitamura A, et al. Molecular cloning and sequence analysis of the cDNA for a human serine protease responsible for activation of hepatocyte growth factor. Structural similarity of the protease precursor to blood coagulation factor XII. J Biol Chem. 1993. May 15;268(14):10024–8. [PubMed] [Google Scholar]
- 30.Karnaukhova E C1-Inhibitor: structure, functional diversity and therapeutic development. Curr Med Chem. 2022;29(3):467–488. [DOI] [PubMed] [Google Scholar]
- 31.Santacroce R, D’Andrea G, Maffione AB, et al. The genetics of hereditary angioedema: a review. J Clin Med. 2021. May 9;10(9):2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Maat S, Clark CC, Boertien M, et al. Factor XII truncation accelerates activation in solution. J Thromb Haemost. 2019. Jan;17(1):183–194. ✶This study investigates the importance of the heavy chain to FXII regulation.
- 33.Law RH, Caradoc-Davies T, Cowieson N, et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012;1(3):185–190. [DOI] [PubMed] [Google Scholar]
- 34.Law RH, Abu-Ssaydeh D, Whisstock JC. New insights into the structure and function of the plasminogen/plasmin system. Curr Opin Struct Biol. 2013;23(6):836–841. [DOI] [PubMed] [Google Scholar]
- 35. Scheffel J, Mahnke NA, Hofman ZLM, et al. Cold-induced urticarial autoinflammatory syndrome related to factor XII activation. Nat Commun. 2020;11(1):179. ✶A very interesting clinical description of a novel autoinflammatory disorder caused by a mutation in FXII.
- 36. Hofman ZLM, Clark CC, Sanrattana W, et al. A mutation in the kringle domain of human factor XII that causes autoinflammation, disturbs zymogen quiescence, and accelerates activation. J Biol Chem. 2020;295(2):363–374. ✶The mechanism behind the disorder described in reference 35 is explored, and lends support to the hypothesis that removal of the FXII heavy chain causes a gain-of-function leading in FXII.
- 37.Rosing J, Tans G, Griffin JH. Surface-dependent activation of human factor XII (Hageman factor) by kallikrein and its light chain. Eur J Biochem. 1985;151(3):531–538. [DOI] [PubMed] [Google Scholar]
- 38.Heestermans M, Naudin C, Mailer RK, et al. Identification of the factor XII contact activation site enables sensitive coagulation diagnostics. Nat Commun. 2021. Sep 22;12(1):5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pozzi N, Chen Z, Zapata F, et al. (2011) Crystal structures of prethrombin-2 reveal alternative conformations under identical solution conditions and the mechanism of zymogen activation, Biochemistry 50, 10195–10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chinnaraj M, Chen Z, Pelc LA, et al. (2018) Structure of prothrombin in the closed form reveals new details on the mechanism of activation, Sci Rep 8, 2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kokoye Y, Ivanov I, Cheng Q, et al. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016. Apr;140:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickeson SK, Kumar S, Sun MF, et al. A Mechanism for Hereditary Angioedema Caused by a Lysine311 to Glutamic Acid Substitution in Plasminogen. Blood (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]