

Abstract
Protein phosphatase 2A (PP2A) is a major cellular phosphatase with many protein substrates. As expected for a signaling molecule with many targets, inhibition of PP2A disrupts fundamental aspects of cellular physiology including cell division and survival. In post-mitotic neurons, the microtubule associated protein Tau is a particularly well-studied PP2A substrate as hyperphosphorylation of Tau is a hallmark of Alzheimer’s disease. Although many cellular targets are likely altered by loss of PP2A, here we find that activation of a single pathway can explain important aspects of the PP2A loss-of-function phenotype in neurons. We demonstrate that PP2A inhibits activation of the neuronal stress kinase DLK and its Drosophila ortholog Wallenda. In the fly, PP2A inhibition activates a DLK/Wallenda-regulated transcriptional program that induces synaptic terminal overgrowth at the neuromuscular junction. In cultured mammalian neurons, PP2A inhibition activates a DLK-dependent apoptotic program that induces cell death. Since hyperphosphorylated Tau is toxic, we wished to test the hypothesis that dephosphorylation of Tau by PP2A is required for neuronal survival. Contrary to expectations, in the absence of Tau PP2A inhibition still activates DLK and induces neuronal cell death, demonstrating that hyperphosphorylated Tau is not required for cell death in this model. Moreover, hyperphosphorylation of Tau following PP2A inhibition does not require DLK. Hence, loss of PP2A function in cortical neurons triggers two independent neuropathologies: 1) Tau hyperphosphorylation and 2) DLK activation and subsequent neuronal cell death. These findings demonstrate that inhibition of the DLK pathway is an essential function of PP2A required for normal Drosophila synaptic terminal development and mammalian cortical neuron survival.
Keywords: PP2A, DLK, Tau, neurodegeneration, synapse development
Introduction
Protein phosphatase 2A (PP2A) is a major cellular serine/threonine phosphatase of the phosphoprotein phosphatase (PPP) family, with the catalytic subunit of this multimeric protein making up roughly 0.1% of total protein in some cells types (Ruediger et al., 1991). Dysregulation of PP2A function leads to an increase in phosphorylation of its substrates, and a variety of cell-type specific cellular dysfunctions including an increase in cell death, defects in mitotic processes, and cytoskeletal dysregulation (Janssens and Goris, 2001). In post-mitotic neurons PP2A functions to coordinate proper development and maintain cellular survival (Rui et al., 2020; Tanaka et al., 1995; Van Kanegan and Strack, 2009; Viquez et al., 2009, 2006; Zhu et al., 2010). PP2A likely affects these disparate processes through multiple key substrates. One substrate of particular interest in post-mitotic neurons is the microtubule associated protein Tau (Janssens and Goris, 2001; Taleski and Sontag, 2018). Loss of PP2A leads to hyperphosphorylation of Tau, a hallmark of Alzheimer’s disease (AD) and other neurodegenerative tauopathies (Gong et al., 1993; Merrick et al., 1997). Hence, the link between PP2A and the hyperphosphorylation of Tau has been well-studied and hypothesized to contribute to the increase in neuronal cell death following PP2A inhibition. However, Tau is not the only substrate disrupted following PP2A inhibition in neurons and many cellular processes are dysregulated (Rui et al., 2020; Van Kanegan and Strack, 2009; Zhu et al., 2010). Despite the many protein substrates of PP2A, we have defined a single kinase cascade downstream of PP2A inhibition that is required for both proper synaptic terminal development at the Drosophila neuromuscular junction and for apoptotic cell death in mammalian cortical neurons. Here, we demonstrate the MAP3K dual leucine zipper (DLK, also called MAP3K12) stress kinase signaling cascade is activated following PP2A inhibition to alter synaptic development and to promote cell death in a Tau-independent manner.
We previously identified a role for PP2A in Drosophila synaptic development, demonstrating that PP2A acts antagonistically to glycogen synthases kinase 3β (GSK-3β, Shaggy in flies) to modulate arborization of the neuromuscular junction (NMJ). At the time, we hypothesized that the phosphatase/kinase pair affected synaptic development through dysregulation of the cytoskeleton (Viquez et al., 2009). Later, we discovered that cytoskeletal dysregulation activates DLK in mammals and its ortholog Wallenda (Wnd) in Drosophila (Valakh et al., 2015, 2013). In flies, elevated Wnd signaling stimulates a transcriptional program that induces increased synaptic arborization at the Drosophila NMJ, a similar phenotype to that observed in flies with decreased PP2A activity (Collins et al., 2006). In mammals, increased DLK signaling triggers a transcriptional program that promotes axon regeneration in the peripheral nervous system but can induce apoptosis in CNS neurons (Asghari Adib et al., 2018; Xiong et al., 2010). Importantly, DLK signaling is increased in the brains of AD model mice and likely in AD patients, and inhibition of DLK extends lifespan and improves behavioral outcomes in mouse models of familial AD (Le Pichon et al., 2017). This confluence of findings led us to revisit our prior studies of PP2A, and to explore the hypothesis that PP2A inhibition may function through the DLK/Wnd pathway in both Drosophila and mammalian neurons.
Here we demonstrate that loss of neuronal PP2A activity activates Wnd/DLK signaling in both Drosophila motor neurons and cultured mammalian cortical neurons. In the fly, increased Wnd signaling leads to activation of a transcriptional stress response program and resulting changes in synaptic terminal morphology. Pharmacological inhibition of PP2A in mouse cortical neurons leads to DLK-dependent apoptosis. Surprisingly, loss of PP2A activity in Tau knockout neurons still activates DLK and induces apoptosis, demonstrating that the accumulation of hyperphosphorylated Tau and the activation of DLK apoptotic signaling are parallel pathways downstream of PP2A dysfunction. Hence, this study identifies PP2A as an evolutionarily conserved inhibitor of DLK stress kinase signaling, and demonstrates that activated DLK is a central effector of PP2A inhibition in post-mitotic neurons with potential roles in both development and disease.
Methods
Fly Stocks and Husbandry.
Drosophila were maintained using standard techniques and raised at 25°C and 60% relative humidity. The following stocks were used in this study: DVGlut-gal4 (Daniels et al., 2008), UAS-PP2A-DN, a dominant negative form of PP2A catalytic subunit microtubule stars obtained from Suzanne Eaton (Hannus et al., 2002), Wallenda RNAi (Vienna stock center, #103410), PucLacZE69(Martín-Blanco et al., 1998), elav-gal4 (Yao and White, 1994). Fly stocks obtained from the Bloomington Stock Center included: UAS-PP2A-C RNAi, an RNAi targeting the catalytic subunit of PP2A microtubule stars (#27723), UAS-LexA RNAi (#67947), UAS-PP5 RNAi (#42794 and #57307), UAS-Fos-DN (#7214). Gal4 drivers were crossed to UAS-LexA RNAi as a control for expression of experimental constructs. Roughly equal numbers of male and female flies were tested, with the exception of crosses using the DVGlut-gal4 driver, in which cases only males were examined. Flies raised on DLK inhibitor were crossed in vials with 35 μM GNE-3511 (MedChem Express HY-12947) or an equivalent volume of DMSO in Instant Drosophila Blue Food Medium (Carolina, #173210) (Russo and DiAntonio, 2019).
Immunohistochemistry.
Wandering third instar larvae were dissected in cold PBS and fixed in either bouin’s fixative for 10 minutes (DVGlut, HRP) or 4% PFA in PBS for 20 minutes (Elav, beta-galactosidase). After fixing, larvae were washed in PBS + 0.1% Triton X-100 (PBS-T) followed by blocking in 5% normal goat serum in PBS-T for at least 30 minutes. All antibodies were incubated in 5% normal goat serum blocking solution. The following antibodies were used: rabbit anti-DVGlut (1:5,000 (Daniels et al., 2004)), rat anti-Elav (1:50, Developmental Studies Hybridoma Bank AB_528218), mouse anti-beta-galactosidase (1:100, Developmental Studies Hybridoma Bank AB_2314509). The following secondary antibodies were used at 1:1000 for 90 minutes: cy3-conjugated goat anti-HRP (Jackson ImmunoResearch, Cat# 123-165-021; AB_2338959), AlexaFluor 488 goat anti-rabbit (ThermoScientific – Invitrogen, Cat# A-11034; AB_2576217), cy3 goat anti-mouse (Jackson ImmunoResearch, Cat# 115-165-146; AB_2338680), and AlexaFluor 488 chicken anti-rat (ThermoScientific – Invitrogen Cat# A-21470; AB_2535873). After secondary staining, larval preps were stored in 70% glycerol in PBS until being mounted in vectashield (Vector Laboratories).
Imaging and Image Analysis in Drosophila.
Synaptic growth was assessed using larval preps stained for DVGlut and HRP which were imaged using the 40X objective on a Leica TCS SPE confocal microscope. Images shown were all taken at the same gain, adjusted to be as bright as possible without oversaturation, and with identical z-stack step sizes. Bouton number was quantified as previously described, in which n represents the number of neurons analyzed, as each NMJ represents the arborization of a single motor neuron on to a single target muscle (Collins et al., 2006). DVGlut-positive boutons were manually counted on muscle 4 of larval segments A2-A4. PucLacZ measurements were taken by staining larval ventral nerve cords and taking images with a Leica TCS SPE confocal microscope using the 20X objective. Average PucLacZ intensity was quantified in the neuronal nuclei along the dorsal midline of the ventral nerve cord as described previously (Xiong et al., 2010). Briefly, neurons in the z-stack images are identified using an Elav stain and the average intensity of beta-galactosidase is measured in the defined area. All images were taken at the same gain and with the same z-step size for all conditions in each experiment.
Primary Cortical Neuron Culture.
Primary neurons were cultured from E16.5-E18.5 mouse embryos. For Figure 4 CD1 mice were used, and for Figure 5 C57BL/6 mice (Charles River Laboratories) were used as wildtype controls for Tau KO mice obtained from John Cirrito (Washington University in St. Louis). Cortices were dissected in ice cold Hank’s Buffered Salt Solution and digested in Accumax (StemCell Technologies, cat# 07921) for 25 minutes. Digested cells were then rinsed once in neurobasal media with 10% fetal bovine serum, followed by trituration of the cell pellet in this media and plating at 250,000 cells per mL. Four hours after plating, media was changed to maintenance media of neurobasal media with 1:50 B-27 supplement (Thermo Fischer Scientific, cat# 17504044). Half of media was changed every 3 days and included a 1:200 addition of FDU to prevent growth of non-neuronal cells. On DIV 3 lentivirus was added, either a GFP control or a BCL-XL construct. Cells were treated with 1 μM LB-100 on DIV 9 or an equivalent volume of DMSO then treated 24 hours later for cell survival studies or collected 4 hours later for Western blotting assays. Cells treated with DLK inhibitor were treated 30 minutes prior to LB-100 treatment with 500 nM GNE-3511 (MedChem Express HY-12947) or an equivalent volume of DMSO.
Figure 4. PP2A inhibition triggers DLK-dependent neuronal cell death independent of Tau.
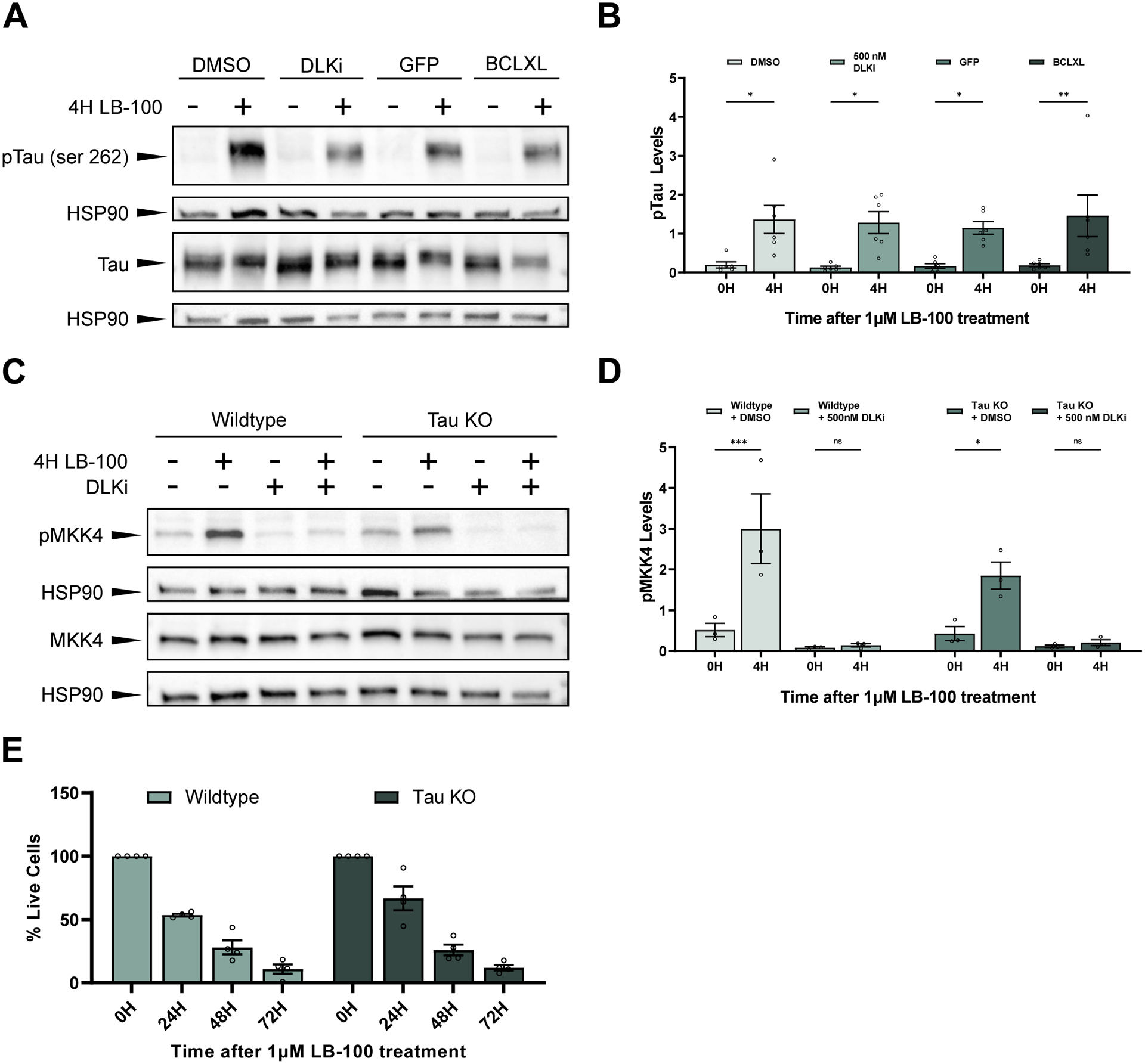
(A) Representative western blots stained with pTau (serine 262), total Tau (Tau-5), and HSP90. Primary mouse cortical neurons were treated with 1 μM LB-100 for 4 hours. Cells were expressing either a GFP control or BCLXL or were treated 30 minutes before LB-100 treatment with either DMSO or 500 nM DLKi.
(B) Quantification of the mean (± SEM) relative levels of pTau (serine 262) at 0 or 4 hours following exposure to 1 μM LB-100 in primary mouse cortical neurons. Cells were expressing a GFP control or the anti-apoptotic protein BCLXL or were pre-treated 30 minutes before the addition of LB-100 with DMSO or 500 nM DLKi. Data points represent biological replicates of separate neuronal preps. There was a significant increase in pTau levels in cells 4 hours after LB-100 treatment as measured by two-way ANOVA with Šídák’s multiple comparisons test (p = 0.043503 for GFP-expressing cells, p = 0.004952 for BCLXL-expressing cells, p = 0.011190 for DMSO treated cells, and p = 0.012500 for DLKi treated cells).
(C) Representative western blots showing pMKK4 (serine 257/threonine 261), MKK4, and HSP90 levels in primary cortical neurons from wildtype (C57BL/6) or Tau KO mice treated with 1 μM LB-100 for 4 hours. Cells were pre-treated 30 minutes before the addition of LB-100 with either DMSO or 500 nM DLKi.
(D) Quantification of the mean (± SEM) relative levels of pMKK4 following LB-100 treatment in primary cortical neurons from wildtype (C57BL/6) and Tau KO mice pre-treated with DMSO or 500 nM DLKi. Both wildtype and Tau KO mice show a significant increase in pMKK4 following LB-100 treatment (p = 0.000342 and p = 0.034282 respectively), while neurons pre-treated with DLKi showed no difference in pMKK4 levels following LB-100 treatment (p = 0.999909 for wildtype neurons and p = 0.999612 for Tau KO neurons). Data points represent biological replicates of separate neuronal preps. Statistical significance was measured by two-way ANOVA with Šídák’s multiple comparisons test.
(E) Quantification of mean (± SEM) relative amount of living cells as measured by an MTT assay in primary cortical neurons from wildtype and Tau KO mice following treatment with 1 μM LB-100 for 0, 24, 48, and 72 hours. Each data point represents a biological replicate which is the average of at least 4 technical replicates. There is no significant difference in the percentage of living cells at 24, 48, or 72 hours after LB-100 treatment between wildtype and Tau KO neurons (p = 0.169797, p = 0.996235, and p = 0.999647 respectively). Statistical significance was measured using a two-way ANOVA with Šídák’s multiple comparisons test.
Western Blots.
To analyze phosphorylation evens on Tau and MKK4 following LB-100 treatment protein was collected and analyzed by Western blot from DIV 10 primary cortical neurons. Protein was collected in RIPA buffer with protease inhibitor (Millipore Sigma, cat# 11873580001) and phosphatase inhibitor (Sigma-Aldrich, cat# P0044) after washing cells in ice cold PBS. Cells were spun down for 4 minutes at 4,000 RCF, then protein was mixed with Laemmli buffer and 2-mercaptoethanol (Sigma-Aldrich, cat# M3148) and boiled at 100°C for 10 minutes. Samples were run on 4–20% Mini-PROTEAN® TGX Precast Protein Gels (Bio-Rad Laboratories, cat# 4561093) in duplicate, with one gel being reserved for the phosphorylated form of the protein and the other for the non-phosphorylated control. Gels were transferred to nitrocellulose membranes using Trans-Blot® Turbo Midi Nitrocellulose Transfer Packs (Bio-Rad Laboratories, cat# 1704159). Membranes were blocked in 5% bovine serum albumin (Sigma-Aldrich, cat# A3912-50G) in TBS+0.1% tween for 60 minutes. Membranes were then incubated in one of the following antibodies overnight: 1:1,000 rabbit anti-MKK4 (Cell Signaling Technologies, cat# 9152), 1:1,000 rabbit anti-phospho-MKK4 (ser257/thr261) (Cell Signaling Technologies, cat# 9156), 1:500 mouse anti-Tau-5 (Thermo Fischer Scientific, cat# MA512808), 1:200 rabbit anti-phospho-Tau (ser262) (Thermo Fischer Scientific, cat# 44750G), 1:1,000 rabbit anti-phosphorylated cJun (ser73) (Cell Signaling Technologies, cat# 3270S), 1:1000 rabbit anti-cJun (Cell Signaling Technologies, cat# 9165P), 1:5,000 rabbit anti-HSP90 (Cell Signaling Technologies, cat# 4877S). Membranes were then incubated in the following secondary antibodies in PBST + 5% BSA for 60 minutes: Peroxidase AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch Laboratories cat# 111-035-045) at 1:5,000 for MKK4, pMKK4, and Tau-5 blots and 1:10,000 for HSP90 blots, Peroxidase AffiniPure Goat Anti-Mouse IgG (Jackson ImmunoResearch Laboratories, cat# 115-035-003) at 1:5,000. Western blots for phosphorylated cJun were stripped with Restore™ PLUS Western Blot Stripping Buffer (Thermo Scientific cat# 46430) for 15 minutes at 37°C before being re-probed for total cJun. Western blots were developed with Immobilon Western Chemiluminescent Substrate (EMD Millipore) and imaged on a G:Box Chemi-XX6 (Syngene). Blots were quantified using ImageJ (NIH, v1.53c), Phosphorylated and non-phosphorylated proteins were first normalized to their respective loading controls and then phosphorylated protein was divided by total protein such that the final result was (phosphorylated protein of interest/loading control)/(total protein of interest/loading control) to determine the relative level of phosphorylated protein in each treatment group.
MTT Assay.
Cortical neurons were cultured as described above in 96 well plates. On DIV 9, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Thermo Fischer Scientific, cat# M6494) was dissolved in sterile PBS at 5mg/mL. An appropriate volume was added to media such that changing half of the media in the 96 well plate resulted in a final concentration of 0.5mg/mL. Cells were incubated with MTT for 4 hours then lysed by replacing media with DMSO (50 μL per well). The active cells converted MTT into formazan, resulting in a purple solution the intensity of which was measured using the absorbance setting on a POLARstar OPTIMA plate reader (BMG LabTech). Background was subtracted from all cells based on the average reading of 8 wells without cells. Percent of cell death was calculated by comparing individual well readings to the average intensity of untreated wells on the same plate.
Experimental Design and Statistical Analyses.
For quantification of synaptic boutons at the neuromuscular junction at least 3 larvae were quantified for each genotype with each NMJ being an N of 1, as each motor neuron targets a single muscle 4 hemi-segment. For the quantification of PucLacZ intensity in the ventral nerve cord at least 3 larvae were used per genotype, with each larva representing an N of 1. Experimental genotypes were compared to control genotypes on the same slide and from the same dissection, with all measurements being performed blinded to the genotype. Western blot experiments were run at least 3 times with each experiment being a separate biological replicate, ie: collected neurons were from different mice. Protein collected was pooled from 2–3 technical replicates (wells on the same plate). MTT assays were repeated with at least 3 biological replicates (neuronal preparations from separate embryos), which are each an average of at least 4 technical replicates (wells on the same plate). Graphed data points represent biological replicates.
All data is represented as mean ± SEM with exact values represented as individual data points where possible. Statistical tests are described below each Figure, including p-value. GraphPad Prism 9 was used to perform all statistical tests.
Results
PP2A inhibits DLK signaling to restrain synaptic terminal growth at the Drosophila NMJ
Neuronal PP2A function is required for restraining growth of the synaptic terminal at the Drosophila neuromuscular junction (NMJ) during development (Viquez et al., 2009, 2006), however the molecular mechanisms linking PP2A activity to proper synaptic development are unknown. We previously inhibited PP2A via neuronal expression of a dominant negative transgene (Viquez et al., 2009, 2006). We confirmed this result here, through expression of the dominant negative (PP2A-DN) and independently verified PP2A’s role at the NMJ via expression of an RNAi transgene targeting the PP2A catalytic subunit, Microtubule Stars, (PP2A-C RNAi). Third instar larvae expressing either of these two transgenes under the control of motoneuron-specific DVGlut-Gal4 were dissected and stained for the pre-synaptic marker Drosophila Vesicular Glutamate Transporter (DVGlut) to identify synaptic boutons and with anti-horse radish peroxidase (HRP), which stains the nerve membrane (Jan and Jan, 1982). The UAS-dicer2 (UAS-dcr) transgene was expressed with RNAi constructs to improve knockdown, as Dcr2 is required for translational repression by small interfering RNAs (Lee et al., 2004). Each of these manipulations leads to synaptic overgrowth, as quantified by an increase in the number of synaptic boutons on the type 1b muscle 4 NMJ compared to animals expressing a control transgene. There is also a decrease in the levels of DVGlut, which is commonly seen in synaptic overgrowth mutants (Collins et al., 2006) (Fig. 1 A and B). Neuronal expression of either of two separate RNAi’s against the related protein phosphatase PP5 induced no change in the number of synaptic boutons compared to expression of a control RNAi, demonstrating selectivity of the PP2A phenotype (Fig. 1 A and B). Having confirmed and extended our prior studies demonstrating that PP2A inhibition induces synaptic terminal overgrowth, we next tested the hypothesis that Wnd/DLK signaling functions downstream of PP2A to drive the increase in synaptic bouton number.
Figure 1. PP2A inhibits DLK signaling to restrain synaptic terminal growth at the Drosophila NMJ.
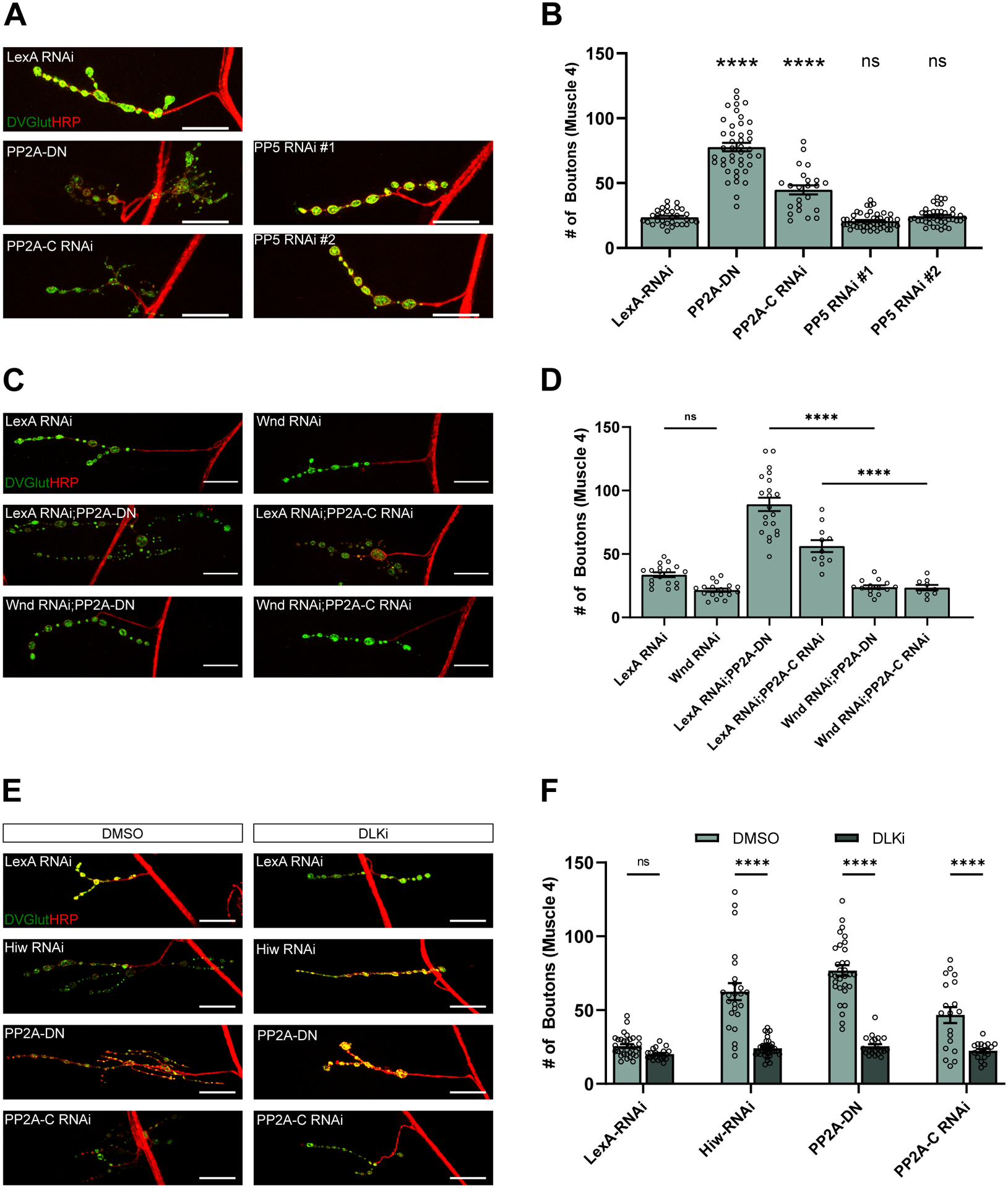
(A) Representative images of synaptic terminal overgrowth at the NMJ of third instar larvae. Transgenes are being driven by DVGlutgal4 in the presence of UAS-Dcr to improve RNAi knockdown. Tissue is stained for the presynaptic marker DVGlut (green) and the nerve membrane with anti-HRP (red). Scale bar is 25 μM.
(B) Quantification of the mean (± SEM) number of boutons on the type 1b synapse on muscle 4. Expression of PP2A-DN or PP2A-C RNAi significantly increases the number of boutons compared to an animal expressing a control RNAi (p < 0.0001) as measured by a one-way ANOVA with Tukey’s multiple comparison test. Expression of either of two RNAis against PP5 is not significantly different from animals expressing a control RNAi (p = 0.8450 and p = 0.9991 respectively). Data points represent biological replicates of individual neurons analyzed at the level of the NMJ.
(C) Representative images of the suppression of synaptic terminal overgrowth from PP2A inhibition by Wnd RNAi in third instar larvae. Transgenes are being driven by DVGlutgal4 in the presence of UAS-Dcr to improve RNAi knockdown. Tissue is stained for the presynaptic marker DVGlut (green) and the nerve membrane with anti-HRP (red). Scale bar is 25 μM.
(D) Quantification of the mean (± SEM) number of boutons in larvae driving constructs using DVGlutgal4 and UAS-Dcr. Co-expression of Wnd RNAi compared to co-expression a LexA RNAi control with PP2A inhibition significantly suppresses an increase in synaptic bouton number (p < 0.0001 for PP2A-DN and p = 0.0004 for PP2A-C RNAi). There is no significant difference between the number of boutons in flies expressing LexA RNAi and Wnd RNAi (p = 0.1029). Data points represent biological replicates of individual neurons analyzed at the level of the NMJ. Statistical significance was measured by a one-way ANOVA with Tukey’s multiple comparison test.
(E) Representative images of the suppression of synaptic terminal overgrowth in third instar larvae from PP2A inhibition by DLK inhibitor (GNE-3511, 35 μM). Transgenes are being driven by DVGlutgal4 in the presence of UAS-Dcr to improve RNAi knockdown. Tissue is stained for the presynaptic marker DVGlut (green) and the nerve membrane with anti-HRP (red). Scale bar is 25 μM.
(F) Quantification of the mean (± SEM) number of boutons in larvae fed either DMSO or DLK inhibitor (GNE-3511, 35 μM). Being raised on DLKi as compared to DMSO significantly suppressed synaptic overgrowth in larvae expressing Hiw RNAi (p < 0.0001), PP2A-DN (p <0.0001), or PP2A-C RNAi (p <0.0001) under a DVGlutgal4 with UAS-Dcr. Data points represent biological replicates of individual neurons analyzed at the level of the NMJ. Statistical significance was determined using a two-way ANOVA with Šídák’s multiple comparisons test.
To investigate the role of Wnd/DLK signaling in the PP2A-dependent synaptic terminal overgrowth phenotype, we co-expressed a validated Wnd RNAi (Valakh et al., 2013) along with the dominant negative or an RNAi targeting the catalytic subunit of PP2A in motoneurons. Inhibition of Wnd/DLK leads to a significant reduction in the number of synaptic boutons at the NMJ, suppressing the synaptic bouton number to wildtype levels (Fig. 1 C and D). This knockdown also appears to restore DVglut levels, the reduction of which is a well described effect of increased Wnd signaling (Collins et al., 2006). As previously demonstrated, expression of Wnd RNAi in a wildtype background does not affect bouton number (Fig. 1 C and D), consistent with the role of Wnd/DLK as a stress signal (Brace et al., 2014; Collins et al., 2006; Valakh et al., 2013; Xiong et al., 2010). As an independent method of suppressing Wnd, we used an inhibitor of the mammalian ortholog DLK, GNE-3511, that we previously validated inhibits Drosophila Wnd (Russo and DiAntonio, 2019). Pharmacological inhibition of Wnd significantly reduced the number of boutons in animals expressing either a dominant negative PP2A or an RNAi against the catalytic subunit (Fig. 1 E and F). Treatment with GNE-3511 was also sufficient to suppress the DLK-dependent synaptic overgrowth in larvae mutant for the E3 ubiquitin ligase Highwire (Hiw), which targets Wnd for degradation (Collins et al., 2006). Hence, genetic and pharmacological studies demonstrate that Wnd signaling is required for the synaptic terminal overgrowth induced by impaired neuronal PP2A activity.
A DLK-regulated transcriptional program is activated by loss of PP2A function
In our original study demonstrating that decreased neuronal PP2A activity induces synaptic terminal overgrowth we hypothesized that this was due to local signaling converging on the cytoskeleton (Viquez et al., 2009). However, the known role of Wnd/DLK in regulating a transcriptional program to control synaptic terminal growth (Collins et al., 2006) inspired us to test whether this transcriptional program contributes to the phenotypes induced by reduced PP2A activity. We first tested whether inhibition of PP2A causes transcriptional changes in a well-known DLK-dependent cellular stress response program. To do this we employed a Puckered LacZ enhancer trap (PucLacZ). Expression of Puckered, a phosphatase which targets JNK, is positively regulated by JNK activity. Thus, increased Wnd signaling results in elevated levels of Puckered transcription and higher intensity of LacZ staining (Martín-Blanco et al., 1998; Xiong et al., 2010). Expressing either dominant negative PP2A or RNAi targeting PP2A-C in neurons triggers a substantial increase in Puckered expression. Expression of RNAi lines targeting the related protein phosphatase PP5 did not significantly alter levels of Puckered expression (Fig. 2 A and B). These data demonstrate that loss of PP2A activity alters transcription in neurons and with our previous findings lead us to hypothesize that this transcriptional change was Wnd-dependent. Co-expression of Wnd RNAi with either dominant negative PP2A or PP2A-C RNAi lowered PucLacZ expression back to wildtype levels (Fig. 2 C and D). Therefore, we conclude that loss of neuronal PP2A activity results in the induction of a Wnd-dependent transcriptional response.
Figure 2. A DLK-regulated transcriptional program is activated by loss of PP2A function.
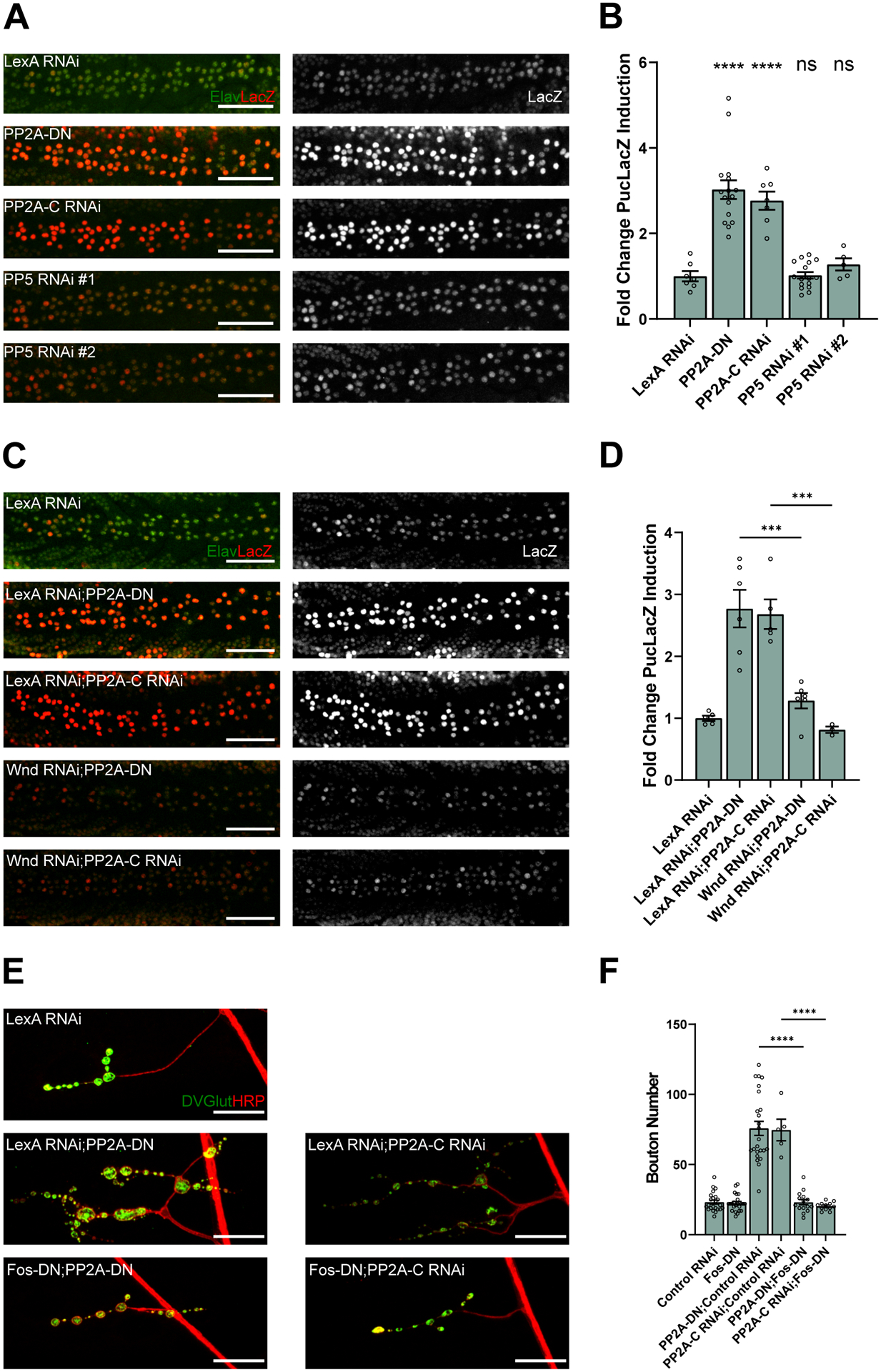
(A) Representative images of ventral nerve cords in larvae expressing neuronal transgenes to inhibit neuronal PP2A or PP5. The neuronal protein Elav is stained in green and LacZ from the PucLacZ enhancer trap is stained in red. Transgenes are expressed using the pan-neuronal driver ElavGal4 along with UAS-Dcr to improve RNAi knock down. Scale bars are 50 μM.
(B) Quantification of the mean (± SEM) PucLacZ fold induction in neurons of the ventral nerve cord. Expression of PP2A-DN or PP2A-C RNAi (p <0.0001 for both) significantly increases PucLacZ induction compared to expression of a control LexA RNAi, but PP5 RNAi expression has no significant effect (p >0.9999 for PP5 RNAi #1 and p = 0.9296 for PP5 RNAi #2) from a one-way ANOVA analysis with Tukey’s multiple comparison test. Data points represent biological replicates of ventral nerve cords from separate animals.
(C) Representative images of the ventral nerve cords in larvae expressing transgenes under the neuronal driver ElavGal4. The neuronal protein Elav is stained in green and LacZ from the PucLacZ enhancer trap is stained in red. Transgenes are expressed using the pan-neuronal driver ElavGal4 along with UAS-Dcr to improve RNAi knock down. Scale bars are 50 μM.
(D) Quantification of the mean (± SEM) PucLacZ fold induction in neurons of the ventral nerve cord. Expression of Wnd RNAi with PP2A-DN (p =0.0002) or PP2A-C RNAi (p =0.0002) significantly suppresses PucLacZ induction compared to co-expression with a control LexA RNAi. Data points represent biological replicates of ventral nerve cords from separate animals. Statistical significance was measured by one-way ANOVA with Tukey’s multiple comparison test.
(E) Representative images of neuromuscular junctions on muscle 4 of third instar larvae. Tissue is stained for presynaptic marker DVGlut (green) and neuronal membrane marker HRP (red). Expression of transgenes is driven by the glutamatergic neuron driver DVGlutgal4 in the presence of UAS-Dcr to improve RNAi processing. Scale bars are 25 μM.
(F) Quantification of the mean (± SEM) number of boutons on muscle 4. Driving Fos-DN suppresses an increase in synaptic bouton number when co-expressed with PP2A-DN or PP2A-C RNAi compared to co-expression of a control LexA RNAi (p <0.0001 for both forms of PP2A inhibition). Data points represent biological replicates of individual neurons analyzed at the level of the NMJ. Statistical significance was measured by one-way ANOVA with Tukey’s multiple comparison test.
We hypothesized that this Wnd-dependent transcriptional program is required for the synaptic overgrowth induced by loss of PP2A activity. Previously, we demonstrated that the transcription factor Fos is required for Wnd-dependent transcriptional changes and synaptic terminal overgrowth (Collins et al., 2006). Hence, we expressed a dominant negative Fos transgene in neurons and found it blocks the increase in number of synaptic boutons induced by PP2A inhibition (Fig. 2 E and F). Therefore, we conclude that loss of neuronal PP2A activity induces synaptic terminal overgrowth by activating a Wnd- and Fos-dependent transcriptional program.
PP2A inhibition in mammalian cortical neurons induces DLK-dependent cell death
Loss of PP2A activity is observed in Alzheimer’s disease patients and is thought to contribute to disease progression via an undefined mechanism (Shentu et al., 2018; Sontag et al., 2004; Vogelsberg-Ragaglia et al., 2001). While PP2A inhibition likely impacts multiple substrates, one potential mechanism that has gotten a great deal of attention is the formation of toxic species of hyperphosphorylated Tau, a well-studied substrate of PP2A (Gong et al., 1993; Liu et al., 2005; Sontag et al., 1996). Inhibition of PP2A activity, either through genetic manipulations or pharmaceutical methods, in mammalian cortical neurons in vitro leads to neuronal cell death (Kim et al., 1999a; Kins et al., 2001). Wnd and DLK function are conserved between flies and mice in the axon injury response pathway (Miller et al., 2009; Shin et al., 2012b; Xiong et al., 2010). Here we investigate the hypothesis that PP2A-dependent inhibition of DLK signaling is also evolutionarily conserved, and that DLK may trigger neuronal cell death upon loss of PP2A function in mammalian cortical neurons.
We first asked whether PP2A inhibition in cultured mouse cortical neurons activates DLK signaling. To assess DLK signaling, we assayed phosphorylation of the direct DLK substrate mitogen-activated protein kinase kinase 4 (MKK4). Primary mouse cortical neurons were cultured from E16–18 embryos for 12 days before treatment for 4 hours with either vehicle or the PP2A inhibitor, LB-100. LB-100 is a potent second-generation inhibitor of PP2A activity that is more selective than previously characterized PP2A inhibitors (Hong et al., 2015). A pharmacological inhibitor was used as shRNA expression in cortical neurons often leads to increased rates of cell death that is target independent and because there are multiple PP2A catalytic subunits expressed in mouse (Khew-Goodall and Hemmings, 1988). LB-100 treatment led to significantly higher levels of phosphorylated MKK4 compared to those treated with a DMSO control. Heat shock protein 90 (HSP90) was used as a loading control in these Western blot assays as it is an abundant, ubiquitously expressed protein. While HSP90 acts a chaperone for DLK, DLK signaling does not affect HSP90 levels (Karney-Grobe et al., 2018). To test whether phosphorylation of MKK4 is DLK-dependent, neurons were pre-treated with either DLK inhibitor GNE-3511 or DMSO 30 minutes before LB-100 application. Pre-treatment with DLK inhibitor blocks the increase in pMKK4 (Fig. 3 A and B). We further probed whether the activation of a transcriptional response downstream of DLK activation was conserved in mammalian cortical neurons treated with PP2A inhibitor. To assay this, we measured levels of the transcription factor cJun phosphorylated at serine 73 (p-cJun) via Western blot from cortical neurons treated with LB-100 for four hours. PP2A inhibition leads to a significant increase in levels of p-cJun, which is blocked by a 30 minute pre-treatment with DLK inhibitor (Fig.3 C and D). Hence, PP2A inhibition in mammalian cortical neurons activates DLK signaling.
Figure 3. PP2A inhibition in mammalian cortical neurons induces DLK-dependent cell death.
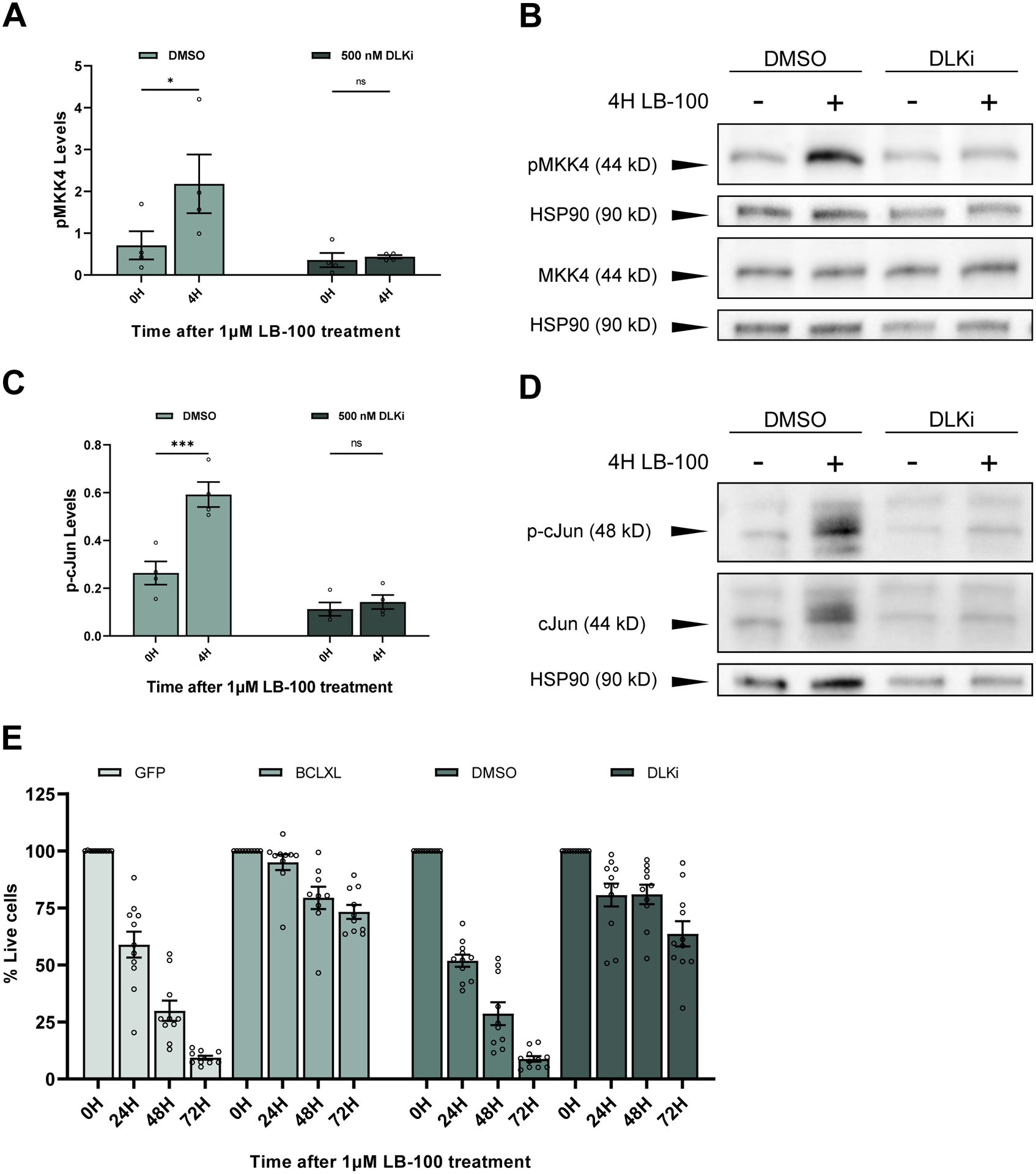
(A) Quantification of the mean (± SEM) levels of pMKK4 in mouse cortical neurons after 4 hours of LB-100 treatment. pMKK4 levels significantly increase following LB-100 treatment in cells pre-treated with DMSO (p = 0.045616) while cells treated 30 minutes prior with 500 nM DLKi do not show an increase in pMKK4 levels (p = 0.988571). Data points represent biological replicates of separate neuronal preps. Statistical analysis was performed using a two-way ANOVA with Šídák’s multiple comparisons test.
(B) Representative Western blots stained for pMKK4 (serine 257/threonine 261) and HSP90 on one blot and MKK4 and HSP90 on a second blot. Primary mouse cortical neurons were treated with 1 μM LB-100 on DIV 12 after being pre-treated by 30 minutes with either DMSO or 500 nM DLKi.
(C) Quantification of the mean (± SEM) levels of phosphorylated cJun in mouse cortical neurons after 4 hours of LB-100 treatment. Phosphorylated cJun levels significantly increase following LB-100 treatment in cell pre-treated with DMSO (p = 0.012677) while cells pre-treated with 500 nM DLKi do not show an increase (p = 0.997312). Data points represent biological replicates of separate neuronal preps. Statistical significance was determined by two-way ANOVA with Šídák’s multiple comparisons test.
(D) Representative Western blots stained for phosphorylated cJun (serine 73), total cJun, and HSP90. Western blot was first probed for phosphorylated cJun, stripped, and re-probed for total cJun. Primary mouse cortical neurons were treated with 1 μM LB-100 on DIV 12 after being pre-treated for 30 minutes with either DMSO or 500 nM DLKi.
(E) Quantification of the mean (± SEM) relative amount of living cells as measured by MTT assay. Biological replicates were normalized to the level of living cells after 0 hours of 1 μM LB-100 treatment for 24 hours, 48 hours, and 72 hours following treatment. Cells were expressing a GFP control or the anti-apoptotic protein BCLXL or were pre-treated for 30 minutes before LB-100 treatment with either DMSO or 500 nM DLKi. Each data point represents a biological replicate which is the average of at least 4 technical replicates. There are significantly more living cells in the BCLXL group compared to the GFP-expressing control group at 24, 48, and 72 hours after LB-100 treatment (p <0.000001, p <0.000001, p <0.000001 respectively). There are significantly more living cells in the DLKi-treated group compared to the DMSO-treated group at 24, 48, and 72 hours after LB-100 treatment (p <0.000001, p <0.000001, p <0.000001 respectively). Each data point represents a biological replicate which is the average of at least 4 technical replicates. Statistical significance was determined by ordinary two-way ANOVA with Šídák’s multiple comparisons test.
We next investigated whether DLK signaling promotes neuronal cell death upon PP2A inhibition. DLK signaling is pro-apoptotic in other contexts (Chen et al., 2008; Fernandes et al., 2014; Ghosh et al., 2011; Pozniak et al., 2013; Watkins et al., 2013; Welsbie et al., 2013), so we first tested whether the cell death caused by PP2A inhibition is apoptotic. Primary cortical neurons were treated with LB-100 and cell death/viability was assessed every 24 hours for three days. To block apoptosis, we infected cortical neurons with lentivirus expressing the anti-apoptotic protein BCL-XL or a control GFP construct at DIV 3. Cortical neurons were treated on DIV 9 with either LB-100 or DMSO and cell viability was measured using an MTT survival assay. BCL-XL-expressing cells were significantly protected at all timepoints compared to GFP-expressing controls. Next, we tested whether the cell death caused by PP2A inhibition was DLK-dependent. Cells were treated with DLK inhibitor or DMSO 30 minutes prior to LB-100 treatment and the relative cell death was measured as described above. DLK inhibitor pre-treatment phenocopied BCL-XL expression, potently protecting neurons treated with LB-100 out to 72 hours after treatment (Fig. 3E). This cell death can also be observed by fragmentation of neurites and loss of cell bodies 48 hours after LB-100 treatment, which is inhibited by both DLK inhibition and BCL-XL expression (Fig. S1). These findings demonstrate that PP2A inhibition in cultured cortical neurons leads to a cell death that is both apoptotic and DLK-dependent.
PP2A inhibition Triggers DLK-dependent Neuronal Cell Death Independent of Tau
The microtubule-associated protein Tau is a well-defined target of PP2A. Loss of PP2A activity leads to an alteration in the phosphorylation pattern of Tau, which can form a toxic protein species that is a hallmark of Alzheimer’s disease and tauopathies. We investigated whether hyperphosphorylated Tau participates in DLK-dependent cell death following PP2A inhibition. We first asked whether DLK signaling influences Tau phosphorylation at serine 262, a known target of PP2A and a site of increased phosphorylation in neurodegenerative diseases (Keramidis et al., 2020; Liu et al., 2005; Qian et al., 2010). Primary mouse cortical neurons expressed either BCL-XL or a GFP control or were treated with either DMSO or DLK inhibitor 30 minutes prior to treatment with LB-100. Following 4 hours of LB-100 treatment, protein was collected and levels of Tau phosphorylated at serine 262 (pTau) was measured by Western blot. PP2A inhibition led to the expected dramatic increase in pTau, and neither BCL-XL expression nor DLK inhibition blocked this effect (Fig. 4 A and B). Hence, levels of Tau phosphorylated at serine 262 are not changed by conditions that block neuronal cell death following PP2A inhibition.
Having demonstrated that DLK signaling is not required for increased phosphorylation of Tau at serine 262, we next asked the converse question—is hyperphosphorylated Tau necessary to activate DLK signaling following PP2A inhibition? This is an attractive hypothesis because we previously demonstrated that disrupting the mammalian cytoskeleton can activate neuronal DLK signaling (Valakh et al., 2015, 2013), and a dysregulated cytoskeleton is observed in cases of increased Tau phosphorylation (Merrick et al., 1997; Sontag et al., 1999). We cultured neurons from Tau knock out mice, treated them with LB-100, and assessed DLK activation by measuring levels of phosphorylated MKK4. Results from Tau knockout and wildtype neurons were very similar, showing an increase in pMKK4 four hours after treatment with LB-100 (Fig. 4 C and D). We confirmed Tau knockout via Western blot and demonstrated that the levels of pTau increased in wildtype cells following 4 hours of 1 μM LB-100 treatment (Fig. S2). These results demonstrate that hyperphosphorylated Tau is not required for the activation of DLK signaling following PP2A inhibition.
The findings above lead us to hypothesize that cell death triggered by PP2A inhibition is DLK-dependent and Tau-independent. To test this, we treated neurons cultured from Tau KO mice with LB-100 and measured cell death, predicting that the neurons would die even in the absence of hyperphosphorylated Tau. Tau KO neurons showed significant cell death 72 hours after LB-100 treatment, phenocopying results observed in wildtype neurons and demonstrating that Tau is not necessary for PP2A inhibition to trigger cell death (Fig. 4 E and F). Hence, PP2A inhibition activates two independent pro-degenerative pathways, DLK activation and Tau hyperphosphorylation. Targeting both DLK and Tau may offer benefits for the treatment of Alzheimer’s disease and other disorders with prominent PP2A dysfunction.
Discussion
Here we demonstrate that PP2A acts to inhibit DLK signaling in neurons. Loss of PP2A activity affects many cellular processes, including promoting mammalian neuronal cell death and Drosophila NMJ overgrowth. Remarkably, inhibition of DLK suppresses both of these phenotypes. Moreover, we show that PP2A-dependent inhibition of DLK-induced neuronal cell death and the hyperphosphorylation of the PP2A substrate Tau are independent. These findings identify a mechanistic link between PP2A inhibition and DLK activation with implications for the pathogenesis of neurodegenerative diseases.
PP2A and DLK/Wallenda signaling
Our work demonstrates that PP2A inhibition activates DLK signaling and subsequently a downstream transcriptional program in neurons. We have two hypotheses for the mechanism of DLK activation following PP2A inhibition. The first is that the disruption of cytoskeletal structure, which has previously been observed following PP2A inhibition, triggers DLK activation (Merrick et al., 1997). PP2A regulates the status of a family of substrates including many microtubule-associated proteins, ultimately affecting the organization of the cytoskeleton (Janssens and Goris, 2001). DLK is a major neuronal stress kinase that is activated by cytoskeletal disruption in both Drosophila and mammals (Asghari Adib et al., 2018; Valakh et al., 2015, 2013), suggesting that DLK acts as a sensor and/or effector downstream of cytoskeletal perturbations in neurites. We hypothesize that that PP2A acts to maintain cytoskeletal homeostasis that in turn keeps DLK signaling at a low basal state. When PP2A activity is disrupted, the cytoskeleton is perturbed and DLK is activated, triggering the stress response appropriate for that cell, including death of cortical neurons (Chen et al., 2008; Shin et al., 2012a; Watkins et al., 2013; Welsbie et al., 2013; Xiong et al., 2010; Yan et al., 2009). The second hypothesis, which is not mutually exclusive with the first, is that DLK itself is a direct target of PP2A. Previous work has demonstrated that treating cultured cells with okadaic acid, a pharmaceutical inhibitor of serine/threonine phosphatases including PP2A, leads to an increase in the molecular weight of DLK consistent with phosphorylation (Mata et al., 1996). Because DLK becomes phosphorylated when activated through indirect mechanisms such as trophic factor withdrawal (Huntwork-Rodriguez et al., 2013), these experiments did not definitively demonstrate whether or not DLK is a direct PP2A target. Hence, the restraint of DLK signaling by PP2A could be either direct or indirect. Regardless of the exact mechanism of action, we show here that major deleterious outcomes attributed to inhibition of neuronal PP2A are due to the secondary activation of DLK. This is an exciting finding because while many PP2A targets are undoubtedly disrupted in both our fly and cortical neuron paradigms, DLK inhibition is sufficient to suppress both developmental and degenerative phenotypes. Hence, we propose that DLK activation is a particularly significant consequence of PP2A dysfunction in post-mitotic neurons.
PP2A signaling, Tau, and the mechanism of neuronal cell death
PP2A inhibition causes cell death both in vitro and in vivo indicating that PP2A function is essential for neuron survival in the central nervous system (Kamat et al., 2013; Kim et al., 1999b; Kins et al., 2001). Loss of PP2A function is observed in Alzheimer’s disease patient brains, due to lowered mRNA levels of the PP2A catalytic subunit, increased expression of the inhibitor protein cancerous inhibitor of PP2A (CIP2A), and changes in expression of methyltransferases that regulate PP2A activity (Gong et al., 1995, 1993; Liu et al., 2005; Park et al., 2018; Shentu et al., 2018; Sontag et al., 2004; Vogelsberg-Ragaglia et al., 2001). Taken together, the strong evidence that PP2A activity is lowered in patient brains has motivated mechanistic studies of the role of PP2A in neuronal survival. One of the best studied targets of PP2A in the nervous system is Tau, which forms toxic species when hyperphosphorylated that are present not only in Alzheimer’s disease patient brains but also in tauopathies (Gong et al., 1995; Liu et al., 2005; Merrick et al., 1997; Sontag et al., 1999, 1996; Sontag and Sontag, 2014). The dual role of PP2A in suppressing Tau hyperphosphorylation and neuronal cell death has led to the hypothesis that this neuronal cell death is due to the effects of hyperphosphorylated Tau (Iqbal et al., 2009; Taleski and Sontag, 2018). Here we test this model directly by assessing the impact of PP2A inhibition in the absence of Tau and explore the alternative hypothesis that cell death following PP2A inhibition in post-mitotic neurons requires DLK signaling.
We find that PP2A inhibition activates a DLK signaling cascade that promotes cortical neuron cell death. The MAP triple kinase DLK is a major neuronal stress kinase, sensing cellular stress and launching a transcriptional program in response (Asghari Adib et al., 2018; Valakh et al., 2015; Xiong et al., 2010). Increased DLK signaling is implicated in neurodegenerative diseases—signaling is elevated in murine models of Alzheimer’s disease and amyotrophic lateral sclerosis (ALS) and DLK inhibition improved neuronal survival and behavioral phenotypes in these models. Moreover, increased levels of a key target of DLK signaling, the phosphorylated form of the transcription factor Jun, is observed in post-mortem nervous tissue of Alzheimer’s and ALS disease patients (Le Pichon et al., 2017). Together, our findings and published work support the hypothesis that inappropriate activation of DLK is common to multiple neurodegenerative diseases, and that dampening this stress pathway has potential as a treatment.
Finally, we explored potential mechanistic links between the Tau hyperphosphorylation and increased DLK signaling that occur as a consequence of PP2A inhibition. Surprisingly, we find that when PP2A is inhibited, hyperphosphorylation of Tau is not required for either DLK activation or neuronal death. This exciting result extends our understanding of neurodegenerative mechanisms downstream of PP2A inhibition. In patients, we expect that both hyperphosphorylated Tau and activated DLK will contribute to neuropathology, suggesting that therapies targeting both Tau and DLK may provide benefits for the treatment of these devastating neurodegenerative diseases.
Supplementary Material
Supplemental Figure S2. (A) Representative western blots stained with pTau (serine 262), total Tau (Tau-5), and HSP90. Primary mouse cortical neurons were cultured from either wildtype (C57/B6) or Tau KO mice, then treated with 1 μM LB-100 for 4 hours. Cells were pre-treated for 30 minutes with either DMSO control or 500 nM DLK inhibitor (GNE-3511). There is no Tau or pTau observed in the Tau KO animals, but protein is present as confirmed by HSP90 loading control.
(B) Quantification of the mean (± SEM) relative levels of pTau (serine 262) at 0 or 4 hours following exposure to 1 μM LB-100 in primary cortical neurons from wildtype (C57/B6) mice. Cells were pre-treated 30 minutes before the addition of LB-100 with DMSO or 500 nM DLKi. Data points represent biological replicates of separate neuronal preps.
Supplemental Figure S1. PP2A inhibition increases cell death in mammalian cortical neurons Embryonic mouse cortical neurons were treated with DMSO control or LB-100. An increase in fragmentation of neurites and a loss of cell bodies were observed 48 hours after treatment in those with PP2A inhibition compared to controls. Pre-treatment with DLK inhibitor or expression of BCL-XL blocked these observed phenotypes.
Acknowledgements
We thank John Cirrito for the Tau knockout mice used in these experiments. We are also grateful to all members of the DiAntonio lab for rigorous discussion and kind support.
Funding
This work was supported by NIH R37 NS065053 awarded to A.D.
Footnotes
Competing Interest Statement: None
Citations
- Asghari Adib E, Smithson LJ, Collins CA, 2018. An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr. Opin. Neurobiol 10.1016/j.conb.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brace EJ, Wu C, Valakh V, DiAntonio A, 2014. SkpA Restrains Synaptic Terminal Growth during Development and Promotes Axonal Degeneration following Injury. J. Neurosci 34, 8398–8410. 10.1523/JNEUROSCI.4715-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rzhetskaya M, Kareva T, Bland R, During MJ, Tank AW, Kholodilov N, Burke RE, 2008. Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J. Neurosci 28, 672–680. 10.1523/JNEUROSCI.2132-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A, 2006. Highwire Restrains Synaptic Growth by Attenuating a MAP Kinase Signal. Neuron 51, 57–69. 10.1016/j.neuron.2006.05.026 [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A, 2004. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J. Neurosci 24, 10466–10474. 10.1523/JNEUROSCI.3001-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Gelfand MV, Collins CA, DiAntonio A, 2008. Visualizing glutamatergic cell bodies and synapses in Drosophila larval and adult CNS. J. Comp. Neurol 508, 131–52. 10.1002/cne.21670 [DOI] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, John SW, Shrager P, Libby RT, 2014. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis 69, 108–116. 10.1016/j.nbd.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW, 2011. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol 194, 751–764. 10.1083/jcb.201103153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C‐X, Shaikh S, Wang J‐Z, Zaidi T, Grundke‐Iqbal I, Iqbal K, 1995. Phosphatase Activity Toward Abnormally Phosphorylated τ: Decrease in Alzheimer Disease Brain. J. Neurochem 65, 732–738. 10.1046/j.1471-4159.1995.65020732.x [DOI] [PubMed] [Google Scholar]
- Gong C‐X, Singh TJ, Grundke‐Iqbal I, Iqbal K, 1993. Phosphoprotein Phosphatase Activities in Alzheimer Disease Brain. J. Neurochem 61, 921–927. 10.1111/j.1471-4159.1993.tb03603.x [DOI] [PubMed] [Google Scholar]
- Hannus M, Feiguin F, Heisenberg CP, Eaton S, 2002. Planar cell polarization requires widerborst, a B’ regulatory subunit of protein phosphatase 2A. Development 129, 3493–3503. 10.1242/dev.129.14.3493 [DOI] [PubMed] [Google Scholar]
- Hong CS, Ho W, Zhang C, Yang C, Elder JB, Zhuang Z, 2015. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther 16, 821. 10.1080/15384047.2015.1040961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW, 2013. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J. Cell Biol 202, 747–763. 10.1083/jcb.201303066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, del Alonso AC, Grundke-Iqbal I, 2009. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 10.1007/s00401-009-0486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY, Jan YN, 1982. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci 79, 2700–2704. 10.1073/PNAS.79.8.2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J, 2001. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J 10.1042/0264-6021:3530417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat PK, Rai S, Nath C, 2013. Okadaic acid induced neurotoxicity: An emerging tool to study Alzheimer’s disease pathology. Neurotoxicology. 10.1016/j.neuro.2013.05.002 [DOI] [PubMed] [Google Scholar]
- Karney-Grobe S, Russo A, Frey E, Milbrandt J, Diantonio XA, 2018. HSP90 is a chaperone for DLK and is required for axon injury signaling. Proc. Natl. Acad. Sci. U. S. A 115, E9899–E9908. 10.1073/PNAS.1805351115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramidis I, Vourkou E, Papanikolopoulou K, Skoulakis EMC, 2020. Functional Interactions of Tau Phosphorylation Sites That Mediate Toxicity and Deficient Learning in Drosophila melanogaster. Front. Mol. Neurosci 13, 191. 10.3389/fnmol.2020.569520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khew-Goodall Y, Hemmings BA, 1988. Tissue-specific expression of mRNAs encoding alpha- and beta-catalytic subunits of protein phosphatase 2A. FEBS Lett. 238, 265–268. 10.1016/0014-5793(88)80493-9 [DOI] [PubMed] [Google Scholar]
- Kim D, Su J, Cotman CW, 1999a. Sequence of neurodegeneration and accumulation of phosphorylated tau in cultured neurons after okadaic acid treatment. Brain Res. 839, 253–262. 10.1016/S0006-8993(99)01724-2 [DOI] [PubMed] [Google Scholar]
- Kim D, Su J, Cotman CW, 1999b. Sequence of neurodegeneration and accumulation of phosphorylated tau in cultured neurons after okadaic acid treatment. Brain Res. 839, 253–262. 10.1016/S0006-8993(99)01724-2 [DOI] [PubMed] [Google Scholar]
- Kins S, Crameri A, Evans DRH, Hemmings BA, Nitsch RM, Götz J, 2001. Reduced Protein Phosphatase 2A Activity Induces Hyperphosphorylation and Altered Compartmentalization of Tau in Transgenic Mice. J. Biol. Chem 276, 38193–38200. 10.1074/jbc.m102621200 [DOI] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Sengupta Ghosh A, Jiang Z, Lee S-H, Maloney J, Gandham VD, Pozniak CD, Wang B, Lee S, Siu M, Patel S, Modrusan Z, Liu X, Rudhard Y, Baca M, Gustafson A, Kaminker J, Carano RAD, Huang EJ, Foreman O, Weimer R, Scearce-Levie K, Lewcock JW, 2017. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med 9, eaag0394. 10.1126/scitranslmed.aag0394 [DOI] [PubMed] [Google Scholar]
- Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, Carthew RW, 2004. Distinct Roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA Silencing Pathways. Cell 117, 69–81. 10.1016/S0092-8674(04)00261-2 [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX, 2005. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci 22, 1942–1950. 10.1111/j.1460-9568.2005.04391.x [DOI] [PubMed] [Google Scholar]
- Martín-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A, 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Merritt SE, Fan G, Geng Yu G, Holzman LB, 1996. Characterization of Dual Leucine Zipper-bearing Kinase, a Mixed Lineage Kinase Present in Synaptic Terminals Whose Phosphorylation State Is Regulated by Membrane Depolarization via Calcineurin*. 10.1074/jbc.271.28.16888 [DOI] [PubMed] [Google Scholar]
- Merrick SE, Trojanowski JQ, Lee VMY, 1997. Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons (NT2N cells). J. Neurosci 17, 5726–5737. 10.1523/jneurosci.17-15-05726.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, Diantonio A, 2009. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci 12, 387–389. 10.1038/nn.2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Lee KW, Oh S, Yan R, Zhang J, Beach TG, Adler CH, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM, 2018. Protein phosphatase 2A and its methylation modulating enzymes LCMT-1 and PME-1 are dysregulated in tauopathies of progressive supranuclear palsy and Alzheimer disease. J. Neuropathol. Exp. Neurol 77, 139–148. 10.1093/jnen/nlx110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Ghosh AS, Gogineni A, Hanson JE, Lee SH, Larson JL, Solanoy H, Bustos D, Li H, Ngu H, Jubb AM, Ayalon G, Wu J, Scearce-Levie K, Zhou Q, Weimer RM, Kirkpatrick DS, Lewcock JW, 2013. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J. Exp. Med 210, 2553–2567. 10.1084/jem.20122832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Shi J, Yin X, Iqbal K, Grundke-Iqbal I, Gong C-X, Liu F, 2010. PP2A Regulates Tau Phosphorylation Directly and also Indirectly via Activating GSK-3β. J. Alzheimer’s Dis 19, 1221–1229. 10.3233/JAD-2009-1317 [DOI] [PubMed] [Google Scholar]
- Ruediger R, Van Wart Hood JE, Mumby M, Walter’ G, 1991. Constant Expression and Activity of Protein Phosphatase 2A in Synchronized Cells. Mol. Cell. Biol 4282–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui M, Ng KS, Tang Q, Bu S, Yu F, 2020. Protein phosphatase PP2A regulates microtubule orientation and dendrite pruning in Drosophila. EMBO Rep. 21. 10.15252/EMBR.201948843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, DiAntonio A, 2019. Wnd/DLK Is a Critical Target of FMRP Responsible for Neurodevelopmental and Behavior Defects in the Drosophila Model of Fragile X Syndrome. Cell Rep. 28, 2581–2593.e5. 10.1016/j.celrep.2019.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shentu YP, Huo Y, Feng XL, Gilbert J, Zhang Q, Liuyang ZY, Wang XL, Wang G, Zhou H, Wang XC, Wang JZ, Lu YM, Westermarck J, Man HY, Liu R, 2018. CIP2A Causes Tau/APP Phosphorylation, Synaptopathy, and Memory Deficits in Alzheimer’s Disease. Cell Rep. 24, 713–723. 10.1016/j.celrep.2018.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A, 2012a. Dual Leucine Zipper Kinase Is Required for Retrograde Injury Signaling and Axonal Regeneration. Neuron 74, 1015–1022. 10.1016/j.neuron.2012.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A, 2012b. SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U. S. A 109, E3696–705. 10.1073/pnas.1216204109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL, 2004. Altered Expression Levels of the Protein Phosphatase 2A ABαC Enzyme Are Associated with Alzheimer Disease Pathology. J. Neuropathol. Exp. Neurol 63, 287–301. 10.1093/jnen/63.4.287 [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC, 1996. Regulation of the phosphorylation state and microtubule-binding activity of tau by protein phosphatase 2A. Neuron 17, 1201–1207. 10.1016/S0896-6273(00)80250-0 [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, Mumby MC, Bloom GS, 1999. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem 274, 25490–25498. 10.1074/jbc.274.36.25490 [DOI] [PubMed] [Google Scholar]
- Sontag JM, Sontag E, 2014. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci 7, 16. 10.3389/fnmol.2014.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taleski G, Sontag E, 2018. Protein phosphatase 2A and tau: an orchestrated ‘Pas de Deux.’ FEBS Lett. 10.1002/1873-3468.12907 [DOI] [PubMed] [Google Scholar]
- Tanaka H, Katagiri M, Arima S, Matsuzaki K, Inokoshi J, Omura S, 1995. Neuronal Differentiation of Neuro 2a Cells by Lactacystin and Its Partial Inhibition by the Protein Phosphatase Inhibitors Calyculin A and Okadaic Acid. Biochem. Biophys. Res. Commun 216, 291–297. 10.1006/BBRC.1995.2623 [DOI] [PubMed] [Google Scholar]
- Valakh V, Frey E, Babetto E, Walker LJ, Diantonio A, 2015. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury HHS Public Access. Neurobiol Dis 77, 13–25. 10.1016/j.nbd.2015.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valakh V, Walker LJ, Skeath JB, DiAntonio A, 2013. Loss of the Spectraplakin Short Stop Activates the DLK Injury Response Pathway in Drosophila. J. Neurosci 33, 17863. 10.1523/JNEUROSCI.2196-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kanegan MJ, Strack S, 2009. The Protein Phosphatase 2A Regulatory Subunits B′β and B′δ Mediate Sustained TrkA Neurotrophin Receptor Autophosphorylation and Neuronal Differentiation. Mol. Cell. Biol 29, 662–674. 10.1128/MCB.01242-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Füger P, Valakh V, Daniels RW, Rasse TM, DiAntonio A, 2009. PP2A and GSK-3β act antagonistically to regulate active zone development. J. Neurosci 29, 11484–11494. 10.1523/JNEUROSCI.5584-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viquez NM, Li CR, Wairkar YP, DiAntonio A, 2006. The B′ protein phosphatase 2A regulatory subunit well-rounded regulates synaptic growth and cytoskeletal stability at the Drosophila neuromuscular junction. J. Neurosci 26, 9293–9303. 10.1523/JNEUROSCI.1740-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VMY, 2001. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol 168, 402–412. 10.1006/exnr.2001.7630 [DOI] [PubMed] [Google Scholar]
- Watkins Trent A, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW, 2013. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U. S. A 110, 4039–44. 10.1073/pnas.1211074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins Trent A., Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW, 2013. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci 110, 4039–4044. 10.1073/PNAS.1211074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Fuller J, Fu J, Cao LH, Han B, Auld D, Xue T, Hirai SI, Germain L, Simard-Bisson C, Blouin R, Nguyen JV, Davis CHO, Enke RA, Boye SL, Merbs SL, Marsh-Armstrong N, Hauswirth WW, Diantonio A, Nickells RW, Inglese J, Hanes J, Yau KW, Quigley HA, Zack DJ, 2013. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. U. S. A 110, 4045–4050. 10.1073/pnas.1211284110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, DiAntonio A, Collins CA, 2010. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J. Cell Biol 191, 211–223. 10.1083/jcb.201006039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y, 2009. The DLK-1 Kinase Promotes mRNA Stability and Local Translation in C. elegans Synapses and Axon Regeneration. Cell 138, 1005–1018. 10.1016/j.cell.2009.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao KM, White K, 1994. Neural specificity of elav expression: Defining a Drosophila promoter for directing expression to the nervous system. J. Neurochem 63, 41–51. 10.1046/j.1471-4159.1994.63010041.x [DOI] [PubMed] [Google Scholar]
- Zhu L-Q, Zheng H-Y, Peng C-X, Liu D, Li H-L, Wang Q, Wang J-Z, 2010. Protein Phosphatase 2A Facilitates Axonogenesis by Dephosphorylating CRMP2. J. Neurosci 30, 3839–3848. 10.1523/JNEUROSCI.5174-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S2. (A) Representative western blots stained with pTau (serine 262), total Tau (Tau-5), and HSP90. Primary mouse cortical neurons were cultured from either wildtype (C57/B6) or Tau KO mice, then treated with 1 μM LB-100 for 4 hours. Cells were pre-treated for 30 minutes with either DMSO control or 500 nM DLK inhibitor (GNE-3511). There is no Tau or pTau observed in the Tau KO animals, but protein is present as confirmed by HSP90 loading control.
(B) Quantification of the mean (± SEM) relative levels of pTau (serine 262) at 0 or 4 hours following exposure to 1 μM LB-100 in primary cortical neurons from wildtype (C57/B6) mice. Cells were pre-treated 30 minutes before the addition of LB-100 with DMSO or 500 nM DLKi. Data points represent biological replicates of separate neuronal preps.
Supplemental Figure S1. PP2A inhibition increases cell death in mammalian cortical neurons Embryonic mouse cortical neurons were treated with DMSO control or LB-100. An increase in fragmentation of neurites and a loss of cell bodies were observed 48 hours after treatment in those with PP2A inhibition compared to controls. Pre-treatment with DLK inhibitor or expression of BCL-XL blocked these observed phenotypes.