

Abstract
Approximately 25% of prostate cancer (PCa) cases experience biochemical recurrence (BCR) following radical prostatectomy (RP). The patients with BCR, especially with BCR ≤2 year after RP (early BCR), are more likely to develop clinical metastasis and castration resistance. Now decision-making regarding BCR after RP relies solely on clinical parameters. We thus attempted to establish an early BCR-risk prediction model by combining a molecular signature with clinicopathological features for guiding clinical decision-making. In this study, an 8-gene signature was derived, and these eight genes were SPTBN2, LGI3, TGM3, LENG9, HAS3, SLC25A27, PCDHGA1, and ADPRHL1. The Kaplan-Meier analysis revealed a significantly prolonged BCR-free survival in the patients with low-risk scores compared to those with high-risk scores in both training and validation datasets. Harrell’s concordance index and time-dependent receiver operating characteristic analysis demonstrated that this gene signature tended to outperform three commercial panels at early BCR prediction. Moreover, this signature was also proven as an independent predictor of BCR-free survival. A nomogram, incorporating the gene signature and clinicopathologic features, was constructed and excellently predicted 1-, 2- and 3-year BCR-free survival of localized PCa patients after RP. Gene set enrichment analysis, tumor immunity, and mRNA expression profiling analysis showed that the high-risk group was more prone to the immunosuppressive microenvironment and impaired DNA damage response than the low-risk group. Collectively, we successfully developed a novel 8-gene signature as a powerful predictor for early BCR after RP and created a prognostic nomogram, which may help inform the clinical management of PCa.
Keywords: Prostate cancer, biochemical recurrence, gene signature, risk stratification, biochemical recurrence-free survival
Introduction
Prostate cancer (PCa), the most common site of cancer in aging males, is one of the leading causes of cancer-associated death across the globe. Long-term declines in mortality for PCa have halted, with an estimated 34,130 deaths and 248,530 new cases in 2021 cancer statistics [1]. Biochemical recurrence (BCR) defined by a rise in the blood level of prostate-specific antigen (PSA) is widely used for reporting the outcome of radical prostatectomy (RP). Although PCa has a better overall survival (OS) than other malignancies, its recurrence rate is significantly high with BCR in approximately 20-30% of patients after standard treatment such as RP and radiation therapy [2]. BCR within the first two years after RP is usually considered to be an early BCR. The patients with BCR, especially with early BCR, are more likely to end up developing clinical recurrence and metastasis, while the median survival for the patients presenting with metastatic PCa is only 30 months [3]. It is thus vital to recognize early BCR in PCa. The serum PSA (sPSA) test has become a routine clinical test for early detection, risk stratification, and monitoring of PCa. But in recent years, sPSA screening test has garnered much criticism for its poor specificity eliciting the potential for overdetection and overtreatment of PCa. In addition, the Gleason score is a decisive prognostic factor recommended by the National Comprehensive Cancer Network (NCCN) Guidelines for PCa, but not perfect due to the limitation of interobserver variability, subjective assessment, and sampling error [4].
With the development of tumor molecular biology, new prediction models based on prognosis-associated genes are emerging and could reflect tumor progression at the molecular level, which might be advantageous to achieve a more accurate prognosis prediction and personalized management of PCa. In fact, with the emergence of gene chips and high-throughput sequencing, it has become a reality that gene signatures derived from abnormal transcriptional profiles can predict prognosis for PCa. In the last decade, multiple gene signatures for PCa prognosis prediction have been proposed. For example, given PCa with heterogeneous hypoxia, Yang et al. proposed a 28-gene hypoxia-related signature as a powerful predictor for 5-year BCR-free survival (BFS) in PCa patients after RP or receiving post-prostatectomy radiotherapy independent of clinicopathological factors and commercially available prognostic signatures [5]. Klein et al. developed a 17-gene signature, effectively predicting clinical recurrence, PCa-specific death, and adverse pathology [6]. Several other studies also utilized similar methods to construct mRNA expression signatures consisting of various numbers of genes [6-12]. These reports primarily focused on long-term BFS and OS.
Even more successful, three commercialized prognostic gene signatures, Decipher with 22 genes, OncotypeDX Prostate with 17 genes, and Prolaris with 46 genes, are on the market and recommended by the NCCN Guidelines for PCa risk-stratification [13]. Specifically, these three tests can help clinical decision-making for patients with a positive biopsy. Additionally, Decipher and Prolaris are also available for PCa patients undergoing RP to predict tumor progression and thus are helpful in decision-making on secondary treatment. Likewise, only long-term metastasis-free survival data was released for these tests, including 5-year and 10-year metastatic PCa risk. Few studies investigate the molecular signatures for early BCR. In-depth analysis of the public datasets identify prognostic-associated genes and develop a robust gene signature for predicting early BCR, which will help inform clinical management of PCa.
Herein, we constructed an 8-gene signature using the sample data from The Cancer Genome Atlas Project (TCGA) database, following validation in multiple independent cohorts. The predictive performance of our signature for early BCR was compared with that of three commercial panels (Decipher, Oncotype DX, and Prolaris). A nomogram was developed combining this gene signature with independent clinicopathological factors and might provide clinicians with a preferable tool for risk stratification, prognosis prediction, and thus clinical decision-making.
Materials and methods
Data collection
The mRNA expression and clinical data of PCa patients were collected from publicly accessible databases, including TCGA (https://portal.gdc.cancer.gov/), Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), and German Cancer Research Centre (DKFZ) (https://www.cbioportal.org/study/clinicalData?id=prostate_dkfz_2018). The exploratory TCGA dataset contained 422 PCa samples and 59 normal tissue samples. Two independent GEO datasets with the accessing number of GSE107299 and GSE70769 consisted of 76 and 94 PCa specimens, respectively, and the DKFZ2018 included 292 PCa cases [14].
Differential gene expression analysis
In the TCGA cohort, the raw RNA-Seq count data of the 422 PCa samples and 59 normal tissue samples were normalized, and log2-scale transformed. The differentially expressed genes (DEGs) between PCa and normal samples were identified using the limma R package [15]. The gene expression data were filtered by count per million >1 in ≥50% of all samples to avoid random correlations between low-expressing genes. The gene was deemed a candidate DEG if a false discovery rate (FDR) of <0.05 and |Log2 fold change| >1 were detected and used for subsequent analysis. A volcano plot of DEGs was drawn using R package ggplot2 (version 3.3.5) and ggrepel (version 0.9.1).
Establishing the 8-gene signature
As previously described [16], a gene signature for BFS prediction was developed. Briefly, we combined the expression level of the screened 1,184 DEGs with the BFS information to establish a multi-gene signature that could predict BFS in PCa. Of note, 60% of the 422-patient TCGA cohort were randomly selected as the training set while the rest 40% as the inner-validation set by adopting the createDataPartition function included in the caret package. DEGs significantly related to BFS were screened out using univariate Cox regression analysis, followed by continuously narrowed-down by LASSO Cox regression analysis designed using the glmnet R package [17]. Then 100 iterations of 10-fold cross-validation with binomial deviance minimization criteria were performed to identify the optimal tuning parameter lambda and corresponding LASSO regression coefficients. The optimal gene set was constructed based lambda according to 1-SE (standard error) rule, and the best combination of genes was determined by best subsets regression using the regsubsets function of the leaps R package [18]. Finally, an 8-gene signature was constructed in the TCGA discovery cohort and presented as a risk score which can be computed for each patient by the following formula:
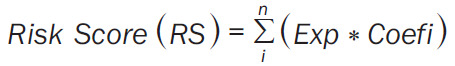 |
Among them, n is the number of BFS-associated genes, Exp indicates the expression value, and Coefi represents the genetic regression coefficient.
Validating the 8-gene signature
The BCR prediction efficiency of the developed 8-gene panel was first confirmed in the inner-validation dataset with 168 patients, which was randomly split from the 422-patient TCGA cohort. The GSE107299, GSE70769, and DKFZ2018 study were served as the separate external validation cohorts. The risk score was computed per the specific risk score formula. The median risk score as a threshold value separated the patients into the high- and low-risk groups. The prognostic differences between the high- and low-risk groups were compared via Kaplan-Meier survival curves combined with a log-rank test. The predictive capability of this signature for 1-, 2-, and 3-year BFS after surgery was assessed by time-dependent receiver operating characteristic (ROC) and concordance index (C-index) analysis. The performance of this gene signature was also detected in the subgroup of the discovery cohort and compared with that of three commercial prognostic gene panels.
Identifying the independent predictors for BCR
To identify independent markers of BCR in PCa, the 8-gene signature and other clinical parameters, including age, Gleason score, clinical TNM stage, pathologic TNM stage, sPSA, and lymph node involvement (LNI), were submitted to Cox regression analyses.
Developing a predictive nomogram
All significant prognostic markers selected by Cox regression analysis were combined to construct a nomogram using cph function from the rms package, allowing for predicting 1-, 2-, and 3-year BFS of PCa patients in the TCGA dataset. The nomogram’s discrimination, calibration, and reliability were assessed, wherein the discrimination was evaluated by C-index, the calibration by comparing predicted and observed OS, and the reliability via decision curve analysis (DCA).
Restricted mean survival time (RMST) analysis
In longitudinal clinical studies to compare two groups, RMST is useful in quantifying underlying differences concerning a time-to-event endpoint. RMST analysis was used in the discovery cohort to examine BFS rates in the high- and low-risk groups at different time points (5 and 10 years of follow-up) via the ‘survRM2’ package.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was conducted in the TCGA cohort to identify gene sets whose expression was enriched or depleted in the high- and low-risk patients using the GSEA function of clusterProfiler R package. The enriched or depleted gene sets correlated with survival were identified via Molecular Signatures Database v.7.4 [19].
Tumor immunity and mRNA expression profiling analysis
xCell, a novel method integrating the advantages of gene set enrichment with deconvolution algorithm, can estimate the scores of 64 infiltrating cell subtypes using the normalized RNA-seq data [20]. Here, the xCell algorithm was adopted to characterize the tumor microenvironment (TME) of PCa tissues in the high- and low-risk groups and further investigated the association between the risk score and the immune infiltrate in the TCGA discovery cohort.
The mRNA expression of genes associated with TME and DNA damage response in PCa was also analyzed in the two risk groups. The expression of TME and DNA damage response-related genes was visualized using ggboxplot function in R package ggpubr (version 0.4.0), and was compared by Student t test between the high- and low-risk groups. Heat maps were plotted using R package pheatmap (version 1.0.12). P-values for high-risk versus low-risk groups were added using the function stat_compare_means in the boxplots.
Statistical analyses
All analytical methods above and R packages were conducted by R software v 4.0 (R Foundation for Statistical Computing, Vienna, Austria). Cox and LASSO regression were utilized to screen the BFS-related variables. The log-rank test was adopted to compare Kaplan-Meier curves. Unless otherwise stipulated, a P-value below 0.05 was considered statistically significant.
Results
Identifying DEGs
A flowchart was graphed to describe this study visually (Figure 1A). Through analysis of gene expression data of 422 primary PCa samples and 59 normal tissue samples in the TCGA discovery cohort, a total of 1,184 DEGs were identified, consisting of 333 up-regulated and 851 down-regulated genes in tumor tissues (Figure 1B). DLX1 showed the most significant differential expression, up-regulated approximately 55-fold in tumor tissue (Figure 1B).
Figure 1.
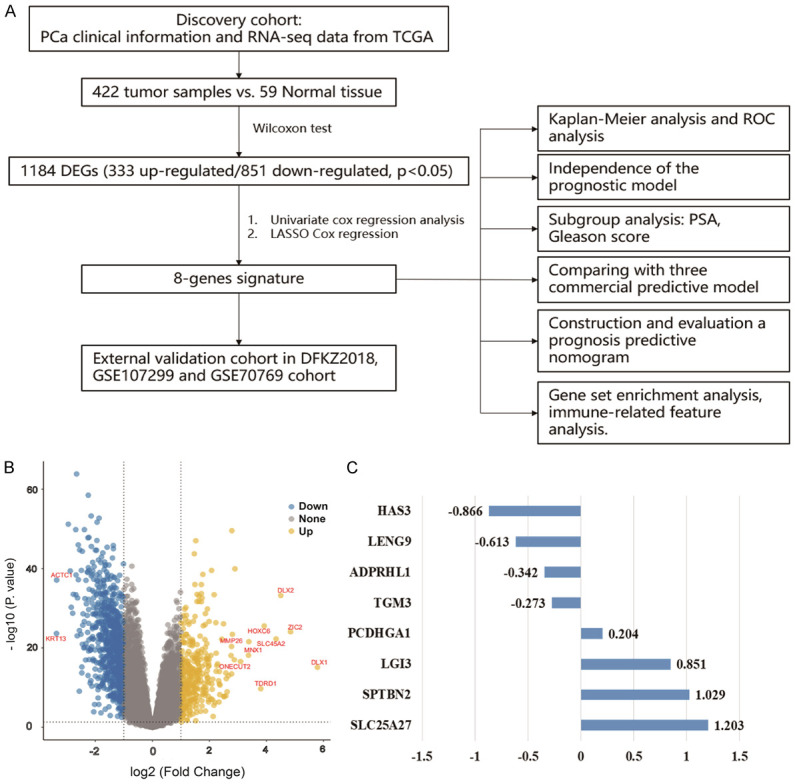
Screening BFS-correlated DEGs and establishing the 8-gene prognostic signature. A. The flow chart presents the process of establishing the gene signature and prognostic nomogram for PCa. B. The volcano plot displays the DEGs, which meet the criteria of a false discovery rate of <0.05, and |log2 fold change| >1. C. Eight BFS-correlated DEGs are shown with corresponding estimated regression coefficients. BFS: biochemical recurrence-free survival; DEGs: differentially expressed genes; PCa: prostate cancer.
Screening BFS-correlated DEGs and establishing the 8-gene prognostic signature
The 422-patient TCGA cohort was randomly assigned in a 6:4 ratio into a training dataset (N=254) and an inner-validation dataset (N=168). The DEGs with significant prognostic values were first screened out in the training dataset via univariate Cox proportional hazards regression analysis and then were narrowed down by a LASSO Cox regression model and an all subset regression model. Finally, eight genes associated with PCa prognosis, including spectrin beta non-erythrocytic 2 gene (SPTBN2), leucine-rich glioma inactivated 3 (LGI3), Transglutaminase 3 (TGM3), leukocyte receptor cluster member 9 (LENG9), hyaluronan synthase 3 (HAS3), uncoupling protein-4 (UCP4/SLC25A27), protocadherin gamma-A1 (PCDHGA1), and ADP-Ribosylhydrolase Like 1 (ADPRHL1) were identified using the regsubsets function of the leaps R package and then used to establish the prognostic gene signature. The down-regulated HAS3, LENG9, ADPRHL1, and TGM3 were deemed tumor suppressors, and the up-regulated PCDHGA1, LGI3, SPTBN2, and SLC25A27 were considered as oncogenes (Figure 1C). Then the risk score for prognosis prediction was computed according to following formula: risk score = (1.029×Exp SPTBN2) + (0.851×Exp LGI3) + (-0.273×Exp TGM3) + (-0.613×Exp LENG9) + (-0.866×Exp HAS3) + (1.203×Exp SLC25A27) + (0.204×Exp PCDHGA1) + (-0.342×Exp ADPRHL1).
Validating the 8-gene signature
The median risk score of 0.79 was determined as the optimal threshold for risk stratification of PCa patients in the training dataset, while this cut-off value of 0.79 also served as a risk indicator for both the inner-validation dataset and the whole TCGA dataset. After stratification, the high-risk group separately comprised 127 patients in the training dataset, 92 in the inner-validation dataset, and 219 in the entire TCGA dataset. The clinicopathological features in the high- and low-risk patients of the TCGA dataset were compared using the twogrps function in R package CBCgrps (version 2.8.2), wherein no significant differences were seen among all the clinical characteristics between the two groups (Table 1). Time-dependent ROC and Kaplan-Meier curves were utilized to assess the performance of this 8-gene signature for the prediction of 1-, 2-, and 3-year BFS. As shown in the top panels of Figure 2A-C, the area under the ROC curve (AUC) values for 1-, 2-, and 3-year BFS predictions were 0.705, 0.732, and 0.751 for the training dataset, 0.822, 0.791, and 0.707 for the inner-validation dataset, and 0.737, 0.760, and 0.737 for the entire TCGA cohort, respectively. Kaplan-Meier plots indicated that in all three datasets, a significantly poor BFS existed in the patients with high-risk scores compared to the patients with low-risk scores (P=0.003 for the training dataset, P=0.0069 for the inner-validation dataset, and P<0.0001 for the entire TCGA discovery cohort) (Figure 2A-C, bottom panels). The RMST for the whole TCGA cohort was also evaluated. The 5- and 10-year RMST were 4.16 (95% CI, 3.92-4.40) and 6.99 (95% CI, 6.07-7.91) years for the high-risk group, and 4.70 (95% CI, 4.55-4.84) and 8.98 (95% CI, 8.50-9.50) years for the low-risk group with significant difference between the high- and low-risk groups in 5- (P=0.0003) and 10-year (P=0.0005) (Figure 2D). Subgroup analysis showed that when patients were stratified by PSA level, the patients with a PSA level of ≤10 or 20 ng/ml had a significantly favorable prognosis versus the patients with a PSA level of >10 or 20 ng/ml, while 8-gene signature-based risk group stratification still performed well in predicting BFS in the subgroup with a PSA level of ≤10 or 20 ng/ml (Figure 2E and 2F). Similarly, 8-gene signature-based risk group stratification also performed well in the subgroup with clinically staged T1-2, M0, or pathologically staged N0 PCa (Supplementary Figure 1). These results indicated that the 8-gene signature was an effective predictor of BFS for PCa patients even across subgroups stratified according to PSA level or TNM status.
Table 1.
Comparison of clinical baseline characteristics between the high- and low-risk patients in the TCGA cohort
| Variables | Total (n=422) | High-risk (n=219) | Low-risk (n=203) | p value |
|---|---|---|---|---|
| Age, Median (Q1, Q3) | 61 (56, 66) | 61 (56, 65.5) | 62 (57, 66) | 0.12 |
| gleason_score, n (%) | 0.804 | |||
| ≤6 | 39 (9) | 19 (9) | 20 (10) | |
| >6 | 383 (91) | 200 (91) | 183 (90) | |
| clinical_T, n (%) | 0.661 | |||
| T1/2 | 304 (86) | 157 (85) | 147 (88) | |
| T3/4 | 48 (14) | 27 (15) | 21 (12) | |
| clinical_M, n (%) | 1 | |||
| M0 | 397 (99) | 202 (100) | 195 (99) | |
| M1 | 2 (1) | 1 (0) | 1 (1) | |
| pathologic_T, n (%) | 0.402 | |||
| pT1/2 | 153 (37) | 75 (35) | 78 (39) | |
| pT3/4 | 264 (63) | 142 (65) | 122 (61) | |
| pathologic_N, n (%) | 0.154 | |||
| N0 | 302 (82) | 151 (79) | 151 (85) | |
| N1 | 66 (18) | 40 (21) | 26 (15) | |
| psa_value, n (%) | 0.642 | |||
| <10 | 396 (97) | 202 (96) | 194 (97) | |
| ≥10 | 13 (3) | 8 (4) | 5 (3) | |
| LNI, n (%) | 1 | |||
| No | 60 (14) | 31 (14) | 29 (14) | |
| Yes | 362 (86) | 188 (86) | 174 (86) | |
| radiation_therapy, n (%) | 31 (7) | 14 (6) | 17 (8) | 0.586 |
| No | 337 (80) | 179 (82) | 158 (78) | |
| Yes | 54 (13) | 26 (12) | 28 (14) | |
| residual_tumor, n (%) | 10 (2) | 4 (2) | 6 (3) | 0.437 |
| R0 | 269 (64) | 139 (63) | 130 (64) | |
| R1 | 128 (30) | 69 (32) | 59 (29) | |
| R2 | 3 (1) | 0 (0) | 3 (1) | |
| RX | 12 (3) | 7 (3) | 5 (2) |
Figure 2.
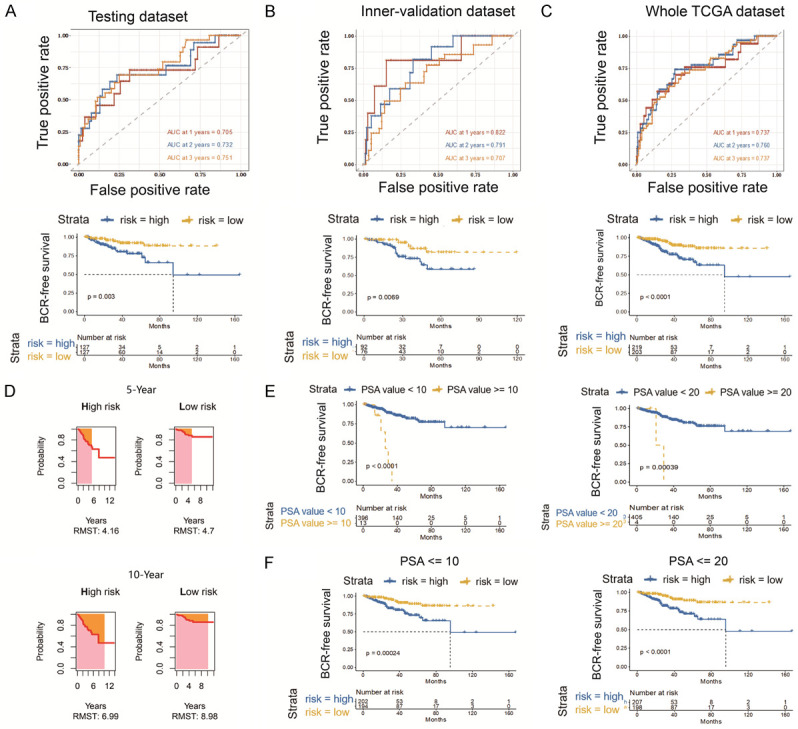
Prognostic analysis and performance evaluation of the 8-gene signature in the discovery set. The time-dependent ROC analyses and Kaplan-Meier analysis of the 8-gene signature for predicting 1-, 2-, and 3-year BFS in the training dataset (A), inner-validation dataset (B), and whole TCGA dataset (C). Patients are stratified into high- and low-risk groups with a cut-off of the median value in the training set. (D) The 5- and 10-year RMST of the high- and low-risk groups in the TCGA discovery cohort. (E) Kaplan-Meier curves for BFS in the patients with low and high PSA levels (cut-off value 10 or 20 ng/ml). (F) Kaplan-Meier BFS analyses of the 8-gene predicative model in the subgroup with a PSA level of ≤10 or 20 ng/ml. ROC: receiver operating characteristic; BFS: biochemical recurrence-free survival; RMST: restricted mean survival time; TCGA: The Cancer Genome Atlas Project; PSA: prostate-specific antigen.
To confirm the predictive value of this signature, three external sets, DKFZ2018, GSE107299, and GSE70769, were utilized to assess the findings from the discovery cohort. The AUC values for predicting 1-, 2-, and 3-year BFS were 0.803, 0.793, and 0.834 for the DKFZ2018 dataset, 0.759, 0.743, and 0.727 for the GSE107299 dataset, and 0.646, 0.770, and 0.733 for the GSE70769 dataset, respectively (Figure 3A-C, top panels). Patients were classified into the high- and low-risk groups using the median risk score derived from each dataset. As expected, the high-risk group had a significantly inferior BFS as compared to the low-risk group (P=0.019 for the DKFZ2018 dataset, P<0.0001 for the GSE107299 dataset, and P=0.0015 for the GSE70769 dataset) (Figure 3A-C, bottom panels).
Figure 3.

Validation of the 8-gene signature. The time-dependent ROC and Kaplan-Meier curves for evaluating the performance of the 8-gene signature in the validation datasets, DFKZ 2018 (A), GSE107299 (B), and GSE70769 (C). (D) Comparison of the 8-gene signature with three commercial panels using ROC analyses for predicting 1-, 2- and 3-year BFS. (E) The nomogram plot for predicting 1-, 2- and 3-year BFS in PCa patients. (F) The calibration curves of the nomogram showing the probability of BFS at 1-, 2- and 3-year after radical prostatectomy. (G) The DCA curves comparing the net clinical benefit among all significant clinical factors, including the nomogram. ROC: receiver operating characteristic; BFS: biochemical recurrence-free survival; DCA: decision curve analysis.
Comparing the 8-gene signature with commercial prognostic gene panels
In addition, in view that the marketed Decipher, Oncotype DX Prostate, or Prolaris tumor-based molecular panel has been recommended by the NCCN Guidelines for initial risk stratification in the appropriate PCa patients, the performance of this 8-gene signature was also evaluated via comparison with that of these three commercial prognostic gene products. The genes of these three commercial panels were listed in Supplementary Table 1. As demonstrated by time-dependent ROC curves in Figure 3D and Supplementary Table 2, the AUC values of the 8-gene signature were numerically higher than those of three commercial panels for predicting 1- and 2-year BFS (1-year AUC: 0.737 vs. 0.612, 0.712, and 0.718; and 2-year AUC: 0.760 vs. 0.619, 0.698, and 0.726), though the differences were not significant. For 3-year BFS prediction, the AUC values of the 8-gene signature and three commercial panels were similar (3-year AUC: 0.737 vs. 0.702, 0.761, and 0.784). Additionally, the C-index of this signature was also comparable to that of three commercial panels (0.646 vs. 0.639, 0.669, and 0.650). These results revealed the 8-gene signature’s good performance for early BCR prediction in PCa.
Identifying the independent prognostic markers and building a predictive nomogram
The independent prognostic factors for PCa were determined by Cox regression analyses of data from the TCGA cohort. The unadjusted univariate Cox regression analysis revealed that clinical T stage (P<0.001), pathologic N (P=0.047), and T stage (P<0.001), PSA (P<0.001), and 8-gene signature-based risk group classifier (P<0.001) were significantly correlated with BFS in PCa patients. In addition, multivariate Cox regression analysis displayed that risk score remained a strong predictor for BFS alongside clinical T stage, pathologic T stage, and PSA (Table 2).
Table 2.
Univariate and multivariate Cox regression analyses of risk score and clinical pathological variables in the TCGA cohort
| Characteristics | No Of Pts | Univariate Cox | Multivariate Cox | ||
|---|---|---|---|---|---|
|
|
|
||||
| HR (95% CI) | P. value | HR (95% CI) | P. value | ||
| Age (≥60 vs. <60) | 422 | 1.52 (0.88-2.65) | 0.1300 | ||
| Gleason score (>6 vs. ≤6) | 422 | 2.71 (0.66-11.12) | 0.1500 | ||
| Clinical M (M1 vs. M0) | 399 | 0 (0-Inf) | 0.8200 | ||
| Clinical T (T3/4 vs. T1/2) | 352 | 3.9 (2.16-7.04) | 1.00E-06 | 2.57 (1.33-4.94) | 0.0047 |
| Pathologic N (N1 vs. N0) | 368 | 1.84 (1-3.4) | 0.0470 | 1.1 (0.55-2.17) | 0.7900 |
| Pathologic T (pT3/4 vs. pT1/2) | 417 | 5.4 (2.31-12.6) | 1.20E-05 | 3.31 (1.24-8.84) | 0.0170 |
| PSA (≥10 vs. <10) | 409 | 9.14 (3.57-23.43) | 2.10E-08 | 4 (1.37-11.68) | 0.0110 |
| LNI (Yes vs. No) | 417 | 2.04 (0.74-5.63) | 0.1600 | ||
| LNI_N (≥ median vs. < median) | 366 | 0.6 (0.34-1.06) | 0.0740 | ||
| Risk (High vs. Low) | 422 | 0.32 (0.18-0.57) | 5.30E-05 | 0.31 (0.15-0.62) | 0.00094 |
For the development of a more reliable prediction tool for clinical practice, a nomogram was established via integrating all significant clinical factors, including the risk score, clinical T stage, pathologic N and T stage, and PSA, to predict 1-, 2- and 3-year BFS in PCa (Figure 3E). Calibration plots showed the nomogram with a strong predictive ability for 1-, 2-, and 3-year BFS, as demonstrated by the consistency between predicted and actual survival time (Figure 3F). DCA curves showed that the net clinical benefit of the nomogram was higher than that of any other character alone (Figure 3G). Additionally, the C-index of 0.742 for the nomogram was the highest among all factors determined.
Gene set enrichment analyses
GSEA analysis was implemented between the high-/low-risk score tumor samples and normal tissues in the TCGA cohort to investigate the underlying molecular mechanism of the 8-gene signature. Figure 4A and 4B showed the hallmarks with significant differences, and the top 5 hallmarks were listed in Table 3. Compared with normal tissues, the tumor samples from the two risk groups showed the up-regulation of “MYC targets V1” and the suppression of “epithelial-mesenchymal transition”. In addition, hallmarks “myogenesis”, “apoptosis”, and “TNF-α signaling via NF-κB” were also inhibited in the high-risk group, whereas “MYC targets V2” was highly expressed and “UV response DN” and “apical surface” were inhibited in the low-risk group compared to normal tissues (Table 3). These results revealed different biological features between the high- and low-risk groups.
Figure 4.
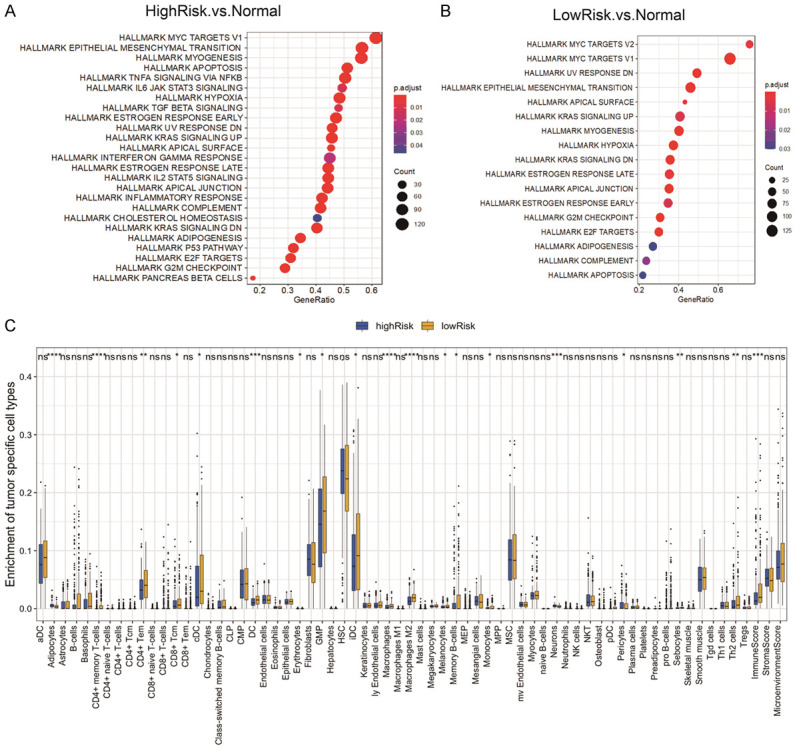
Gene set enrichment analyses, tumor immunity, and mRNA expression profiling analysis. A, B. Gene set enrichment analyses are conducted between the high-/low-risk tumor samples and normal tissues in the TCGA discovery cohort to identify enriched or depleted gene sets. C. Difference analysis of 64 immune cells infiltration between the high- and low-risk groups in the TCGA cohort using xCell algorithm. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Table 3.
Significantly enriched hallmarks in the TCGA cohort by GSEA
| NAME | SIZE | NES | NOM p-val | FDR q-val | Groups |
|---|---|---|---|---|---|
| HALLMARK_MYC_TARGETS_V1 | 200 | 1.699985701 | 0.000137665 | 6.09E-05 | HighRisk vs. Normal |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 200 | -2.167283953 | 1.67E-09 | 7.37E-10 | HighRisk vs. Normal |
| HALLMARK_MYOGENESIS | 199 | -2.766214294 | 1.67E-09 | 7.37E-10 | HighRisk vs. Normal |
| HALLMARK_APOPTOSIS | 160 | -1.910151921 | 1.10E-05 | 4.86E-06 | HighRisk vs. Normal |
| HALLMARK_TNFA_SIGNALING_VIA_NFKB | 200 | -2.209713985 | 1.67E-09 | 7.37E-10 | HighRisk vs. Normal |
| HALLMARK_MYC_TARGETS_V2 | 58 | 1.748083455 | 0.004336176 | 0.002738637 | LowRisk vs. Normal |
| HALLMARK_MYC_TARGETS_V1 | 200 | 1.936585623 | 2.67E-07 | 1.69E-07 | LowRisk vs. Normal |
| HALLMARK_UV_RESPONSE_DN | 144 | -1.873905197 | 0.000107974 | 6.82E-05 | LowRisk vs. Normal |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 200 | -1.808381186 | 0.000177091 | 0.000111847 | LowRisk vs. Normal |
| HALLMARK_APICAL_SURFACE | 44 | -1.875651113 | 0.003511655 | 0.002217887 | LowRisk vs. Normal |
Correlation with the infiltration levels of immune cells
Through the xCell algorithm, we investigated the relationship between this 8-gene signature and the immune infiltrate. Among the 64 subtypes of immune cells in tumors determined via xCell, the low-risk group was more infiltrated by multiple subtypes of immune cells, including memory B cells, T helper 2 cells, central memory CD8 T cells, CD4+ memory T cells, CD4+ naive T cells, activated dendritic cells, monocytes, as well as M2-polarized macrophages than the high-risk group (Figure 4C). Besides, the immune score is higher in the low-risk group than in the high-risk group (Figure 4C).
mRNA expression analysis
In view that the immunotherapeutic benefits were associated with immune checkpoint high expression, IFNγ pathway activation, T cell activation, and activated tumor microenvironment, we also evaluated the mRNA expression of common immune checkpoints, and representative genes of IFNγ pathway, effector T cell, T cell receptor, and the tumor microenvironment in the high- and low-risk patients of the TCGA dataset (Supplementary Figure 2A-D). In the tested immune checkpoints (PD-1, PD-L1, PD-L2, LAG3, CTLA-4, TIM3, and VTCN1), only the mRNA expression of PD-L1 and TIM3 was nearly significantly (P=0.0655) or significantly (P=0.0097) different between the two groups, both lower in the high-risk group than in the low-risk group. In terms of the activity of the IFNγ pathway, T cell, and TME, the high-risk group had significantly lower mRNA expression in some representing genes than the low-risk group, including GZMB, IFI16, IFI30, IRF1, CD3D, CD4, CD74, GRAP2, IL2RB, IL1B, IL6, PTGS2, and TNF, suggesting that high-risk score was associated with the immunosuppressive microenvironment (Supplementary Figure 3A-D). Additionally, the mRNA expression of DNA damage repair (DDR) genes was also analyzed. Among 106 DDR genes analyzed, the high-risk group had 11 up-regulated and 31 down-regulated genes compared with the low-risk group (Supplementary Figures 2E, 3E; Supplementary Table 3). These findings suggested that high-risk patients are more likely to have immunosuppressive microenvironment and DNA damage response than low-risk patients.
Discussion
PCa is one of the most deadly urinary tumors in males globally. Progression to BCR is a significant turning point in PCa development. Once BCR, the majority of PCa patients will metastasize, leading to ultimate death [21]. Early BCR accounts for nearly half of all tumor BCR following RP, and PCa patients with early BCR always suffer from dismal long-term outcomes compared with those without early BCR, although significant progress has been made in adjuvant therapy. Therefore, it is advisable to effectively stratify PCa with a high risk of early BCR after RP, so as to carry out early intervention before the progression of metastasis.
Herein, an 8-gene signature for predicting early BCR was established using gene expression and BFS data from the TCGA PCa cohort. In the light of the risk scores computed based on the expression level of 8 genes, patients in the discovery dataset can be separated into the high- and low-risk groups with significant differences in BFS and 5- and 10-year RMST. This gene classifier also effectively helped stratify patients with a PSA level of ≤10 or 20 ng/ml into different risk groups. Besides, it was successfully validated in three external datasets that this gene panel significantly discriminated between the high- and low-risk groups for BFS. This 8-gene classifier was also an independent predictor of BFS. A nomogram was developed via integrating the 8-gene classifier and clinicopathological indicators, which could precisely predict BFS. Additionally, it was noteworthy that the high-risk patients were more prone to the immunosuppressive microenvironment and DDR variations than the low-risk patients.
Among the eight genes included in this panel, six of them have been experimentally verified to be associated with cancer. HAS3, one family member of HSA, is responsible for producing both secreted and cell-associated forms of hyaluronan and has been widely reported to be linked to tumor progression, metastasis, and poor prognosis in multiple human cancers. It has been documented that the increase of HAS3 expression is associated with a dismal prognosis in oral cancer and breast cancer [22,23]. Up-regulation of HAS3 could enhance the proliferation of gastric cells and colon cancer cells [24,25]. Consistently, HAS3 down-regulation is also reported to be an indicator of poor outcome in urothelial carcinoma of the upper urinary tract and the bladder [26]. The anti-cancer effect of HAS3 remains controversial. Consistently, in PCa, HAS3 has been demonstrated to promote the growth of TSU PCa cells but inhibit the cell proliferation and migration of LNCap PCa cells [27,28]. ADPRHL1 is a pseudoenzyme expressed in the developing heart myocardium of all vertebrates. Wan et al. reported that ADPRHL1 might play a critical role in the recurrence of uveal melanoma [29]. But the role of ADPRHL1 in PCa is not revealed. TGM3, one of the critical enzymes contributing to the formation of protein polymers in the epidermis and the hair follicle, has been reported as a tumor suppressor involved in the modulation of cell proliferation, epithelial-to-mesenchymal transition, apoptosis in colorectal cancer, esophageal cancer, and hepatocellular carcinogenesis [30-33]. However, the association of TGM3 with PCa has not ever been reported. LGI3, a secreted protein that belongs to LGI/epitempin family, is highly expressed in the brain and regulates neuronal exocytosis and differentiation [34,35]. In the last few years, some studies documented that the high expression of LGI3 is associated with favorable outcomes in glioma and non-small cell lung cancer, which might be attributed to increased innate tumor immunity in the tumor microenvironment [36,37]. Hence, LGI3 might be a potential therapeutic target in PCa. SPTBN2 belonging to the spectrin family is widely reported to act as a structural carrier for stabilizing and activating membrane channels, receptors, and transporters involved in neurogenesis and degenerative diseases. Increasing evidence points out that SPTBN2 could serve as a crucial clinical parameter of tumor progression and survival for some cancers, such as ovarian cancer, bladder cancer, and colorectal cancer patients with m6A modifications [38,39]. Moreover, Wen et al. identified SPTBN2 as a signature gene, emerging in pathogenesis and pattern recognition of seven common cancers [40]. However, the exact role of SPTBN2 in PCa is still unknown. SLC25A27 encodes a neuronal mitochondrial uncoupling protein-4 (UCP4), which protects neurons against oxidative stress and mitochondrial dysfunction [41,42]. It has been discovered that the positivity of UCP4 is associated with classical prognostic factors such as lymph node metastases, p53 and Ki-67 in breast carcinomas. As yet it is not clear whether UCP4 is related to PCa. PCDHGA1, a member of the protocadherin family, mediates the formation and maintenance of specific synaptic connections [43]. LENG9 is a member of the leukocyte receptor complex [44]. They have never been documented associated with cancer to date. Further investigation of the role of above-mentioned genes in PCa are in our plan.
In comparison with several existing gene signatures, our multi-gene panel demonstrated sound performance in risk stratification of BCR, wherein its AUCs for 1-, 2-, and 3-year BFS in the discovery cohort were 0.737, 0.760, and 0.737, respectively, and remained stable in three external validation sets. For example, Yuan et al. developed a 4-gene signature, and its AUCs for 1- and 3-year BFS in the training set are 0.83 and 0.799, respectively, but its AUCs in the validation set are 0.638 and 0.723, respectively [7]. Long et al. also developed a 4-gene signature, and its AUCs for 1-, 2- and 3-year BFS are 0.827, 0.774 and 0.810 for the training set, and 0.718, 0.675, and 0.638 for the validation set [45]. Lv et al. established a 5-immune-related gene signature, and its AUC for 3-year BFS is 0.71 for the training dataset and 0.775 for the validation dataset [46]. In addition, our multi-gene panel had superior performance in predicting early BCR compared with three commercial panels in terms of AUC values for 1- and 2-year BFS prediction and C-index. Early BCR risk stratification tools have significant clinical value in personalized therapy. The patients with low-risk early BCR could be managed by active surveillance, while those with high-risk early BCR may require adjuvant therapies or/and closer clinical management. Given that this signature could be used for risk stratification in the subgroup with a PSA level of ≤10 or 20 ng/ml or a low TNM-stage, our signature may help identify low-risk patients more accurately, avoid unnecessary overtreatment, and select an optimal management strategy.
In addition, this signature might reflect the changes of TME from different aspects as shown by xCell and mRNA expression profiling, which offered convenience to monitor the infiltration of immune cells and the potentiality to assist rational diagnosis and individualized treatment. DNA damage is a primary driver of cancer initiation and progression, while loss-of-function alterations of DDR genes promote a more aggressive PCa phenotype [47,48]. A recent study has shown that the DDR alteration signature has a substantial prognostic value and may be utilized for risk stratification in PCa [45]. More instructively, DDR could help guide clinical decision-making in PCa. For example, DDR deficiency has been well-documented as a predictor of sensitivity to platinum agents in castration-resistant PCa [49-51]; and the FDA has approved olaparib for use in metastatic castration-resistant PCa patients with a pathogenic mutation in a homologous recombination repair gene and previous experience of enzalutamide or abiraterone therapy [52]. We consistently found that the high-risk score patients had a more severe DNA damage response than the low-risk score patients, which might provide treatment information for the patients with future relapse.
Some limitations still exist in this study. Firstly, this study is performed based on available retrospective data derived from public datasets, and thus it should be regarded as indicative for future research rather than conclusive. Secondly, most patients in these datasets were the European American population. Therefore extrapolating these results to the patients of other ethnicities should be done with caution. Thirdly, the underlying molecular mechanisms of the selected eight genes in early BCR of PCa remain to be further explored. To summarize, our study built an 8-gene signature that can reliably predict early BCR after RP, which will contribute to the personalized management of PCa patients.
Acknowledgements
We thank the patients and investigators who participated in TCGA, GEO and German Cancer Research Centre for providing data.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Shao N, Wang Y, Jiang WY, Qiao D, Zhang SG, Wu Y, Zhang XX, Wang JL, Ding Y, Feng NH. Immunotherapy and endothelin receptor antagonists for treatment of castration-resistant prostate cancer. Int J Cancer. 2013;133:1743–1750. doi: 10.1002/ijc.28162. [DOI] [PubMed] [Google Scholar]
- 3.Kessler B, Albertsen P. The natural history of prostate cancer. Urol Clin North Am. 2003;30:219–226. doi: 10.1016/s0094-0143(02)00182-9. [DOI] [PubMed] [Google Scholar]
- 4.Randall EC, Zadra G, Chetta P, Lopez BG, Syamala S, Basu SS, Agar JN, Loda M, Tempany CM, Fennessy FM. Molecular characterization of prostate cancer with associated Gleason score using mass spectrometry imaging. Mol Cancer Res. 2019;17:1155–1165. doi: 10.1158/1541-7786.MCR-18-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Roberts D, Takhar M, Erho N, Bibby BAS, Thiruthaneeswaran N, Bhandari V, Cheng WC, Haider S, McCorry AMB, McArt D, Jain S, Alshalalfa M, Ross A, Schaffer E, Den RB, Jeffrey Karnes R, Klein E, Hoskin PJ, Freedland SJ, Lamb AD, Neal DE, Buffa FM, Bristow RG, Boutros PC, Davicioni E, Choudhury A, West CML. Development and validation of a 28-gene hypoxia-related prognostic signature for localized prostate cancer. EBioMedicine. 2018;31:182–189. doi: 10.1016/j.ebiom.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein EA, Cooperberg MR, Magi-Galluzzi C, Simko JP, Falzarano SM, Maddala T, Chan JM, Li J, Cowan JE, Tsiatis AC, Cherbavaz DB, Pelham RJ, Tenggara-Hunter I, Baehner FL, Knezevic D, Febbo PG, Shak S, Kattan MW, Lee M, Carroll PR. A 17-gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur Urol. 2014;66:550–560. doi: 10.1016/j.eururo.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Yuan P, Ling L, Fan Q, Gao X, Sun T, Miao J, Yuan X, Liu J, Liu B. A four-gene signature associated with clinical features can better predict prognosis in prostate cancer. Cancer Med. 2020;9:8202–8215. doi: 10.1002/cam4.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Xu S, McClelland M, Rahmatpanah F, Sawyers A, Jia Z, Mercola D. An accurate prostate cancer prognosticator using a seven-gene signature plus Gleason score and taking cell type heterogeneity into account. PLoS One. 2012;7:e45178. doi: 10.1371/journal.pone.0045178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Y, Mei W, Gu Y, Lin X, He L, Zeng H, Wei F, Wan X, Yang H, Major P, Tang D. Construction of a set of novel and robust gene expression signatures predicting prostate cancer recurrence. Mol Oncol. 2018;12:1559–1578. doi: 10.1002/1878-0261.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu D, Jiang L, Luo S, Zhao X, Hu H, Zhao G, Tang W. Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J Transl Med. 2020;18:160. doi: 10.1186/s12967-020-02323-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu B, Li X, Li J, Jin H, Jia H, Ge X. Construction and validation of a robust cancer stem cell-associated gene set-based signature to predict early biochemical recurrence in prostate cancer. Dis Markers. 2020;2020:8860788. doi: 10.1155/2020/8860788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao N, Tang H, Mi Y, Zhu Y, Wan F, Ye D. A novel gene signature to predict immune infiltration and outcome in patients with prostate cancer. Oncoimmunology. 2020;9:1762473. doi: 10.1080/2162402X.2020.1762473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(NCCN) NCCN. NCCN Clinical practice guidelines in oncology. Prostate Cancer. (Version 2) 2021:2021. [Google Scholar]
- 14.Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, Heckmann D, Sidiropoulos N, Waszak SM, Hübschmann D. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell. 2018;34:996–1011. doi: 10.1016/j.ccell.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou T, Cai Z, Ma N, Xie W, Gao C, Huang M, Bai Y, Ni Y, Tang Y. A novel ten-gene signature predicting prognosis in hepatocellular carcinoma. Front Cell Dev Biol. 2020;8:629. doi: 10.3389/fcell.2020.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1. [PMC free article] [PubMed] [Google Scholar]
- 18.Farkas O, Héberger K. Comparison of ridge regression, partial least-squares, pairwise correlation, forward-and best subset selection methods for prediction of retention indices for aliphatic alcohols. J Chem Inf Model. 2005;45:339–346. doi: 10.1021/ci049827t. [DOI] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:1–14. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shipley WU, Seiferheld W, Lukka HR, Major PP, Heney NM, Grignon DJ, Sartor O, Patel MP, Bahary JP, Zietman AL. Radiation with or without antiandrogen therapy in recurrent prostate cancer. N Engl J Med. 2017;376:417–428. doi: 10.1056/NEJMoa1607529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo YZ, Fang WY, Huang CC, Tsai ST, Wang YC, Yang CL, Wu LW. Hyaluronan synthase 3 mediated oncogenic action through forming inter-regulation loop with tumor necrosis factor alpha in oral cancer. Oncotarget. 2017;8:15563. doi: 10.18632/oncotarget.14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auvinen P, Rilla K, Tumelius R, Tammi M, Sironen R, Soini Y, Kosma VM, Mannermaa A, Viikari J, Tammi R. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res Treat. 2014;143:277–286. doi: 10.1007/s10549-013-2804-7. [DOI] [PubMed] [Google Scholar]
- 24.Bai F, Jiu M, You Y, Feng Y, Xin R, Liu X, Mo L, Nie Y. miR-29a-3p represses proliferation and metastasis of gastric cancer cells via attenuating HAS3 levels. Mol Med Rep. 2018;17:8145–8152. doi: 10.3892/mmr.2018.8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teng BP, Heffler MD, Lai EC, Zhao YL, LeVea CM, Golubovskaya VM, Bullarddunn KM. Inhibition of hyaluronan synthase-3 decreases subcutaneous colon cancer growth by increasing apoptosis. Anticancer Agents Med Chem. 2011;11:620–628. doi: 10.2174/187152011796817655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang IW, Liang PI, Li CC, Wu WJ, Huang CN, Lin VC, Hsu CT, He HL, Wu TF, Hung CH. HAS3 underexpression as an indicator of poor prognosis in patients with urothelial carcinoma of the upper urinary tract and urinary bladder. Tumour Biol. 2015;36:5441–5450. doi: 10.1007/s13277-015-3210-z. [DOI] [PubMed] [Google Scholar]
- 27.Liu N, Gao F, Han Z, Xu X, Underhill CB, Zhang L. Hyaluronan synthase 3 overexpression promotes the growth of TSU prostate cancer cells. Cancer Res. 2001;61:5207–5214. [PubMed] [Google Scholar]
- 28.Czyrnik ED, Wiesehöfer M, Dankert JT, Wennemuth G. The regulation of HAS3 by miR-10b and miR-29a in neuroendocrine transdifferentiated LNCaP prostate cancer cells. Biochem Biophys Res Commun. 2020;523:713–718. doi: 10.1016/j.bbrc.2020.01.026. [DOI] [PubMed] [Google Scholar]
- 29.Wan Q, Tang J, Han Y, Wang D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp Eye Res. 2018;166:13–20. doi: 10.1016/j.exer.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Feng Y, Ji D, Huang Y, Ji B, Zhang Y, Li J, Peng W, Zhang C, Zhang D, Sun Y. TGM3 functions as a tumor suppressor by repressing epithelial-to-mesenchymal transition and the PI3K/AKT signaling pathway in colorectal cancer. Oncol Rep. 2020;43:864–876. doi: 10.3892/or.2020.7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W, Zhang Z, Zhao W, Han N. Transglutaminase 3 protein modulates human esophageal cancer cell growth by targeting the NF-κB signaling pathway. Oncol Rep. 2016;36:1723–1730. doi: 10.3892/or.2016.4921. [DOI] [PubMed] [Google Scholar]
- 32.Wu X, Wang R, Jiao J, Li S, Yu J, Yin Z, Zhou L, Gong Z. Transglutaminase 3 contributes to malignant transformation of oral leukoplakia to cancer. Int J Biochem Cell Biol. 2018;104:34–42. doi: 10.1016/j.biocel.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 33.Hu JW, Yang ZF, Li J, Hu B, Luo CB, Zhu K, Dai Z, Cai JB, Zhan H, Hu ZQ, Hu J, Cao Y, Qiu SJ, Zhou J, Fan J, Huang XW. TGM3 promotes epithelial-mesenchymal transition and hepatocellular carcinogenesis and predicts poor prognosis for patients after curative resection. Dig Liver Dis. 2020;52:668–676. doi: 10.1016/j.dld.2019.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Park WJ, Lee SE, Kwon NS, Baek KJ, Kim DS, Yun HY. Leucine-rich glioma inactivated 3 associates with syntaxin 1. Neurosci Lett. 2008;444:240–244. doi: 10.1016/j.neulet.2008.08.044. [DOI] [PubMed] [Google Scholar]
- 35.Park WJ, Lim YY, Kwon NS, Baek KJ, Kim DS, Yun HY. Leucine-rich glioma inactivated 3 induces neurite outgrowth through Akt and focal adhesion kinase. Neurochem Res. 2010;35:789–796. doi: 10.1007/s11064-010-0136-0. [DOI] [PubMed] [Google Scholar]
- 36.Kim DS, Kwon NS, Yun HY. Leucine rich repeat LGI family member 3: integrative analyses reveal its prognostic association with non-small cell lung cancer. Oncol Lett. 2019;18:3388–3398. doi: 10.3892/ol.2019.10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon NS, Kim DS, Yun HY. Leucine-rich glioma inactivated 3: integrative analyses support its prognostic role in glioma. Onco Targets Ther. 2017;10:2721. doi: 10.2147/OTT.S138912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Wang Q, Zhang M, Zhang W, Zhao L, Yang C, Wang B, Jiang K, Ye Y, Shen Z. Comprehensive analysis of the transcriptome-wide m6A methylome in colorectal cancer by MeRIP sequencing. Epigenetics. 2021;16:425–435. doi: 10.1080/15592294.2020.1805684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng P, Ge Z, Guo Z, Lin L, Yu Q. A comprehensive analysis of the downregulation of miRNA-1827 and its prognostic significance by targeting SPTBN2 and BCL2L1 in ovarian cancer. Front Mol Biosci. 2021;8:687576. doi: 10.3389/fmolb.2021.687576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen JX, Li XQ, Chang Y. Signature gene identification of cancer occurrence and pattern recognition. J Comput Biol. 2018;25:907–916. doi: 10.1089/cmb.2017.0261. [DOI] [PubMed] [Google Scholar]
- 41.Liu D, Chan SL, de Souza-Pinto NC, Slevin JR, Wersto RP, Zhan M, Mustafa K, de Cabo R, Mattson MP. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 2006;8:389–413. doi: 10.1385/NMM:8:3:389. [DOI] [PubMed] [Google Scholar]
- 42.Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH, Ho SL. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012;2:468–478. doi: 10.1002/brb3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999;97:779–790. doi: 10.1016/s0092-8674(00)80789-8. [DOI] [PubMed] [Google Scholar]
- 44.Wende H, Volz A, Ziegler A. Extensive gene duplications and a large inversion characterize the human leukocyte receptor cluster. Immunogenetics. 2000;51:703–713. doi: 10.1007/s002510000187. [DOI] [PubMed] [Google Scholar]
- 45.Long G, Ouyang W, Zhang Y, Sun G, Gan J, Hu Z, Li H. Identification of a DNA repair gene signature and establishment of a prognostic nomogram predicting biochemical-recurrence-free survival of prostate cancer. Front Mol Biosci. 2021;8:66. doi: 10.3389/fmolb.2021.608369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lv D, Wu X, Chen X, Yang S, Chen W, Wang M, Liu Y, Gu D, Zeng G. A novel immune-related gene-based prognostic signature to predict biochemical recurrence in patients with prostate cancer after radical prostatectomy. Cancer Immunol Immunother. 2021;70:3587–3602. doi: 10.1007/s00262-021-02923-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bancroft EK, Page EC, Castro E, Lilja H, Vickers A, Sjoberg D, Assel M, Foster CS, Mitchell G, Drew K. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: results from the initial screening round of the IMPACT study. Eur Urol. 2014;66:489–499. doi: 10.1016/j.eururo.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrovics G, Price DK, Lou H, Chen Y, Garland L, Bass S, Jones K, Kohaar I, Ali A, Ravindranath L. Increased frequency of germline BRCA2 mutations associates with prostate cancer metastasis in a racially diverse patient population. Prostate Cancer Prostatic Dis. 2019;22:406–410. doi: 10.1038/s41391-018-0114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol. 2016;69:992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pomerantz MM, Spisák S, Jia L, Cronin AM, Csabai I, Ledet E, Sartor AO, Rainville I, O’Connor EP, Herbert ZT. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–3539. doi: 10.1002/cncr.30808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mota JM, Barnett E, Nauseef JT, Nguyen B, Stopsack KH, Wibmer A, Flynn JR, Heller G, Danila DC, Rathkopf D. Platinum-based chemotherapy in metastatic prostate cancer with DNA repair gene alterations. JCO Precis Oncol. 2020;4:355–366. doi: 10.1200/PO.19.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–2102. doi: 10.1056/NEJMoa1911440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.