
Fig. 1.
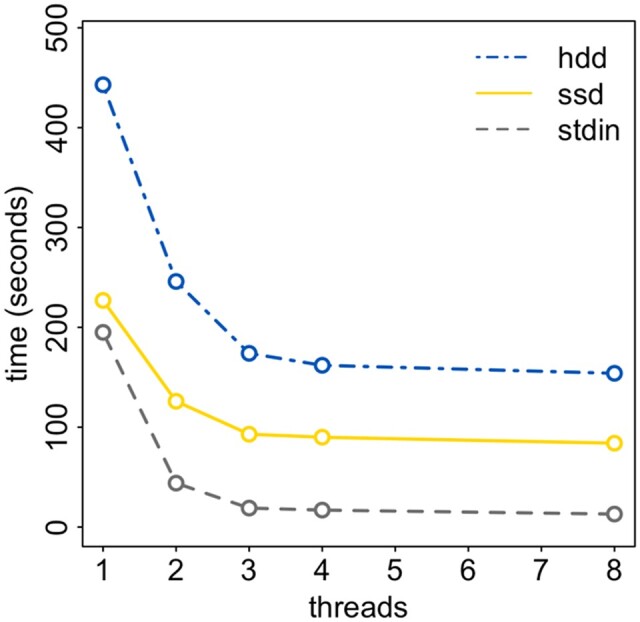
The additional time taken to convert a sam-format alignment of 1 million SARS-CoV-2 genomes to fasta format. Times are given for reading and writing from/to a rotating hard disk drive (dot-dashed line), from/to a solid state drive (solid line) and the additional time above minimap2’s runtime needed to write the fasta file to a hard disk drive while reading the sam file from standard in (dashed line). In each case, the sam file was generated on a server with 40 logical CPUs by running minimap2 with 32 threads and was ∼28 GB in size. gofasta was run with different numbers of threads (represented on the x-axis)