
Extended Data Fig. 14. Integrated model of TCR (see also Movie 2).
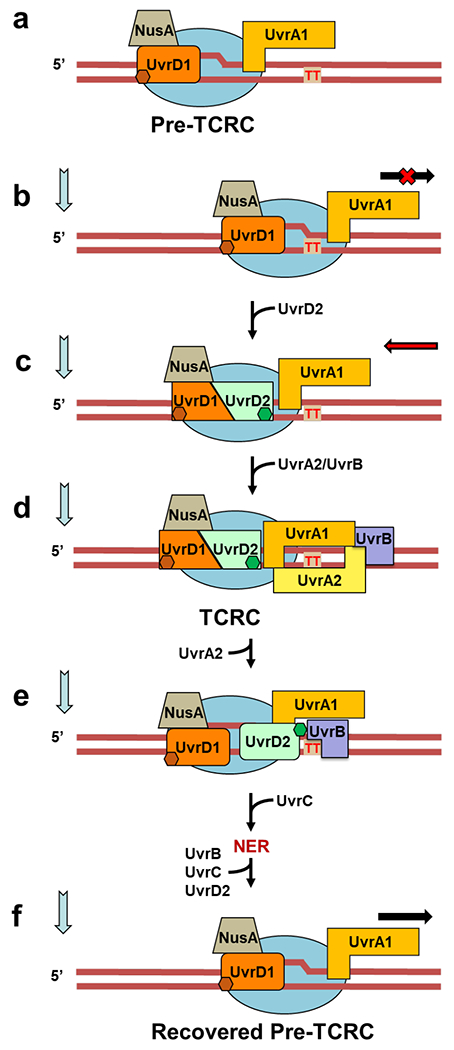
Based on the in vivo and in vitro data presented, we propose a structure-functional model of NER in E. coli in which the elongating RNAP functions as the primary lesion scanner and platform for the assembly of active NER complexes. a, A subpopulation of elongating RNAPs persistently interacts with UvrD1 and UvrA1, as shown in the structural model of Fig. 2b. The in vivo RNAP pulldowns and XLMS demonstrate that such surveillance pre-TCRCs can form even before the genotoxic stress. b,c, Upon stalling at the DNA lesion in the template strand (CPD is marked as red “TT”), the pre-TCRC recruits UvrD2 to form a helicase competent UvrD dimer. UvrD2-CTD (green hexagon) interacts with a RNAP βi4 domain to stabilize the UvrD dimer. UvrD12 pulls TCRC backward, thereby exposing a CPD to the NER enzymes15. ppGpp contributes at this stage by rendering RNAP backtracking-prone9. d, TCRC recruits UvrA2/UvrB to initiate the lesion processing. While a single UvrB monomer is sufficient for lesion verification and UvrC recruitment27,62,63, the second UvrB molecule may be recruited as well26,61,64. In vitro (Extended Data Fig. 3) and in vivo XLMS (Fig. 2c,d) are consistent with a single UvrB monomer model. This UvrB can interact with the CTD of UvrD2 (Extended Data Fig. 5), thereby displacing UvrD2 from RNAP (Movie 2). The release of UvrD2 that occurs coincidentally with the UvrA2B recruitment (Fig. 1b,c) supports such a sequence of events in vivo. UvrD2 displacement would abrogate any further UvrD-mediated backtracking, e, The pre-incision TCRC recruits UvrC and releases UvrA2B followed by the NER execution step2,62. Once repair has been completed, the backtracked pre-TCRC is promptly recovered by the anti-backtracking factors (GreB, Mfd, and a leading ribosome) to resume elongation.