

Abstract
Clinical use of doxorubicin (Dox) is limited by cumulative myelo- and cardiotoxicity. This research focuses on the detailed characterization of PhAc-ALGP-Dox, a targeted tetrapeptide prodrug with a unique dual-step activation mechanism, designed to circumvent Dox-related toxicities and is ready for upcoming clinical investigation. Coupling Dox to a phosphonoacetyl (PhAc)-capped tetrapeptide forms the cell-impermeable, inactive compound, PhAc-ALGP-Dox. After extracellular cleavage by tumor-enriched thimet oligopeptidase-1 (THOP1), a cell-permeable but still biologically inactive dipeptide-conjugate is formed (GP-Dox), which is further processed intracellularly to Dox by fibroblast activation protein-alpha (FAPα) and/or dipeptidyl peptidase-4 (DPP4). In vitro, PhAc-ALGP-Dox is effective in various 2D- and 3D-cancer models, while showing improved safety toward normal epithelium, hematopoietic progenitors, and cardiomyocytes. In vivo, these results translate into a 10-fold higher tolerability and 5-fold greater retention of Dox in the tumor microenvironment compared with the parental drug. PhAc-ALGP-Dox demonstrates 63% to 96% tumor growth inhibition in preclinical models, an 8-fold improvement in efficacy in patient-derived xenograft (PDX) models, and reduced metastatic burden in a murine model of experimental lung metastasis, improving survival by 30%. The current findings highlight the potential clinical benefit of PhAc-ALGP-Dox, a targeted drug-conjugate with broad applicability, favorable tissue biodistribution, significantly improved tolerability, and tumor growth inhibition at primary and metastatic sites in numerous solid tumor models.
Introduction
Conventional chemotherapy is currently indispensable in most oncologic treatments. However, clinical application is often limited by dose-related side effects and therefore, reducing systemic toxicity and improving therapeutic index remains an important challenge. With that purpose, different formulations have been optimized to decrease drug clearance, increase therapeutic efficacy, and/or modulate systemic toxicity. Alternatively, strategies masking cytotoxicity and selectively delivering payloads into cancer cells, such as antibody–drug conjugates (ADC), have been applied. However, ADCs remain hampered by uncontrolled payload release, immunogenicity, poor tumor penetration, and resistance (1).
Another elegant therapeutic approach are protease-activated conjugates (PAC). Here, the payload is attached to a protease-cleavable linker, making the molecule inactive prior to cleavage by the target protease. This reduces the size of the molecule compared with ADCs, potentially improving tumor penetration and reducing immunogenicity. Moreover, efficient hydrolysis and selective payload activation are guaranteed by swift recognition of the peptidic moiety by tumor-specific proteases.
Dox is one of the most effective anticancer drugs used for treatment of a broad range of solid tumors. However, clinical applicability has long been limited due to severe dose-dependent cardio- and myelotoxicity. DTS-201 (CPI0004Na) consists of the tetrapeptide N-succinyl-β-alanyl-L-leucyl-L-alanyl-L-leucine covalently linked to Dox. DTS-201 is stable in blood but cleavable by specific peptidases present in the tumor environment (2, 3). DTS-201 demonstrated good efficacy and lower toxicity compared with Dox in preclinical models (4, 5). In patients, DTS-201 was well tolerated up to 400 mg/m2;, corresponding to 3.75-fold the standard dose of Dox and showed encouraging results in clinical trials (6).
Following the same rationale, PhAc-ALGP-Dox was developed as an advanced tetrapeptidic conjugate of Dox. The payload is coupled to a phosphonoacetyl-capped L-Alanyl-L-Leucyl-L-glycyl-L-prolyl peptide, rendering it inactive and preventing cellular uptake. Once in the tumor vicinity, PhAc-ALGP-Dox is metabolized to a cell-permeable but biologically inactive intermediate, GP-Dox, which is further cleaved intracellularly by FAPα and/or DPP4 exopeptidases, liberating Dox exclusively at the tumor (7).
In this study, the unique mode of activation, preclinical evidences of the in vitro potency, and selectivity of PhAc-ALGP-Dox are unraveled, together with the antitumor efficacy in clinically relevant xenograft and PDX models spanning a variety of indications.
Materials and Methods
Drugs, recombinant enzymes, and inhibitors
Doxorubicin-HCl was acquired from LC-Labs (D-4000–500 mg). PhAc-ALGP-Dox was synthetized by WuXi AppTec (China). Following recombinant human enzymes were purchased from R&D Systems (Bio-techne): THOP1 (3439-ZNC, 0.1 μg/mL), CD10 (1182-ZNC, 0.08 μg/mL), NLN (4308-SE, 0.8 μg/mL), FAPα (3715-SE, 0.04 μg/mL), and DPP4 (9168-SE, 0.04 μg/mL). Following inhibitors were used to block activity of endogenous peptidases: THOP1 was specifically inhibited with Cpp-AAF-pAB (100 μmol/L Bachem), THOP1 and CD10 were blocked with JMV-390 (10 μmol/L, Tocris #2575), DPP4 and FAPα was constrained using talabostat (=PT-100, 10 μmol/L, Tocris #3719), whereas FAPi (MedChemExpress, HY100684) was used to specifically inhibit FAPα.
Peptidase cleavage assay (PCA) and LC/MS-MS
Two micromolar of PhAc-ALGP-Dox or DTS-201 were dissolved in PCA buffer (150 mmol/L NaCl, 50 mmol/L Tris, 1% BSA, pH 7.5, 37°C). The reaction was initiated through addition of recombinant enzymes at equi-active concentrations (t = 0). Samples were snap frozen in liquid nitrogen at t = 1, 5, 15, 30, 60, 240, and 960 minutes and stored at −80°C. Prior to analysis, samples were defrosted and mixed with ice cold MeOH/CH3CN containing 13C-Dox (1:5) to remove salts, proteins, and stop enzymatic reactions. Samples were vortexed and centrifuged (12,000 rpm, 10 minutes at 4°C). Supernatants were transferred to a matrix tube for LC/MS-MS using an Acquity Class I Xevo TQS micro (Waters Corporation). Five millimolar of ammonium formate (pH 3.75) and 5% aqueous CH3CN 5 mmol/L ammonium formate (pH 3.75) were used as mobility phase. Electron spray ionization and subsequent MS/MS analysis was performed using the Xevo TQ-S at a source temperature of 150°C and cone gas flow of 50 L/h. Desolvation gas flow was 1,200 L/h. Half-life (t1/2) was calculated using the Xlfit software (IBDS Ltd.). Hundred percent of parent compound was extrapolated from the curve-fit to determine time zero concentrations.
Cell lines
Human cell lines and rat cardiomyoblasts (H9C2) were acquired from ATCC (LGC Standards s.a.r.l.). Murine cell lines were purchased from European Collection of Authenticated Cell Culture (ECACC, Sigma-Aldrich). E0771-AKA-Luc2 cells were kindly provided by prof. Massimiliano Mazzone. Following cell lines were used and RRID; year of acquisition are mentioned between brackets; A-172 (CVCL_00131; 2020), A2058 (CVCL_01059; 2020), A2780 (CVCL_0134; 2020), A2780 CpR (CVCL_H745; 2020), A549 (CVCL_0023; 2020), H9C2 (CVCL_0286; 2015), E0771 (CVCL_GR23; 2013), HC-11 (CVCL_0288; 2013), HCT-116 (CVCL_0291; 2019), HME-1 (CVCL_3383; 2015/2020), HUVEC (CVCL_2959; 2015), LS 174T (CVCL_1384; 2015), MDA-MB-231 (CVCL_0062; 2015), MDA-MB-468 (CVCL_0419; 2015), MIA PaCa-2 (CVCL_0428; 2019), NCI-1299 (CVCL_0060; 2020), NCI-292 (CVCL_0455; 2019), PANC-1 (CVCL_0480; 2019), and U-87 MG (CVCL_0022; 2020). Upon arrival, cells were expanded and aliquots were frozen at low passage number to limit deviation from their original identity, as authenticated by the source via STR profiling and CO1 assay. Cells were cultured according to manufacturer's recommendations and propagated at 37°C, 5% CO2. Experiments were performed within 10 weeks after defrosting at consistent passage number (<20). Mycoplasma testing was performed routinely using the MycoAlert PLUS Mycoplasma Detection Kit (LT07–705, Lonza). Cell lines used for in vivo studies were screened for rodent pathogens (IMPACT Mouse I package; IDEXX BioAnalytics).
THOP1 ELISA
Cellular or extracellular/secreted protein levels of THOP1 were determined on total protein extracts and conditioned media (20× concentrated), respectively, using The RayBio Human Thimet Oligopeptidase ELISA Kit (ELH-THOP1; Raybiotech). To concentrate secreted proteins in cell-conditioned media, serumfree medium was collected after being conditioned overnight and concentrated using Amicon ultra-15 Centrifugal Filter Units with 50 kDa cut-off (30 minutes, 4,000 × g, 4°C). Following centrifugation, supernatant was diluted to reach a final concentration factor of 20 (starting volume/assay volume) and was analyzed for THOP1 protein levels. In parallel, cells were placed on ice and intracellular proteins were extracted in RIPA buffer with Pierce protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Ten micrograms of total proteins was used for quantification of cellular THOP1 protein expression.
Peptidase activity assay
THOP1 activity in conditioned media was assessed using a custom fluorogenic substrate resembling PhAc-ALGP-Dox (Pepscan B.V.). ALGP was used to couple quencher (2,4-dinitrophenol) and fluorophore ß-(7-methoxy-coumarin-4-yl)-Ala-OH, generating a specific substrate that only yields signal after cleavage of the tetra-sequence. Medium from either normal (HME-1) or tumor cells (LS 174 T) was conditioned overnight at 37°C, 5% CO2. Next, the substrate (10 μmol/L) was dissolved in conditioned medium and fluorescence was monitored every 15 minutes for 6 hours (Ex 325 nm/Em 392 nm). Relative fluorescence units (RFU) were obtained from duplicate wells after background subtraction. THOP1 activity in tumor conditioned media was blocked by preincubation with JMV-390 (10 μmol/L) for 15 minutes.
In vitro 2D cytotoxicity and selectivity
Cells were seeded in 96-well plates at optimal cell densities (ranging from 7 to 15 × 103 cells/well). After overnight attachment, cells were exposed to a serial dilution of PhAc-ALGP-Dox decreasing from 100 μmol/L in 1:3 increments. Dox was used as a reference compound, at concentration 10 times lower. After 72 hours, cells were rinsed and RPMI1640 phenol red-free medium containing WST-1 reagent (Roche) was added to asses cell. Absolute cytotoxicity was calculated on the basis ofviability normalized to nontreated conditions. Absolute IC50 (IC50) is the concentration required to reduce cell viability with 50%. Relative IC50 (rIC50) is the concentration required to reach 50% of the maximal effect. Maximal toxicity was calculated on the basis of potency at highest concentration tested.
To define safety towards tumor cells, a selectivity index (SI) was defined as the ratio of absolute IC50 in normal (HME-1) versus tumor cells (8).
Triplicate values were plotted as mean ± SEM and each experiment was repeated three to five times. GraphPad Prism 7.0 (RRID:SCR_002798) was used for nonlinear fitting according to the sigmoidal-4PL model. Outliers were automatically excluded and Log(IC50) values were extrapolated. Similarly, rIC50 and maximal toxicity (100% viability – lowest viability) were deducted from the nonlinear regression.
In vitro 3D cytotoxicity
Cells, magnetized with NanoShuttle-PL (Greiner Bio-One), were seeded in 96-well ULA plates at 5 × 103 cells/well and spheroids were allowed to form for 72 hours (37°C, 5% CO2). For MDA-MB-231 spheroids, addition of 2.5% Matrigel at day 1 was required to drive condensation. To prevent aspiration or disturbance of the spheroids, a magnetic spheroid drive (Greiner Bio-One) was used during manipulation. Next, spheroids were stimulated with different concentrations of Dox or PhAc-ALGP-Dox for 72 hours. Every 3 to 4 days, culture medium was refreshed. Brightfield microscopy images were taken to calculate spheroid volume using the following formula:
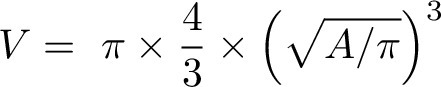 |
where A is the area (Fiji ImageJ 2.1.0/1.53 RRID:SCR_003070). Spheroids with a volume exceeding the field of view, were not further quantified (=cut-off). To visualize growth over time, sizes were normalized to their respective volume at 72 hours in culture (pretreatment).
Tissue distribution and pharmacokinetics
Levels of PhAc-ALGP-Dox or Dox in mouse tissues after intravenous administration at 92 μmol/kg in tumor bearing mice were determined by LC/MS-MS (Acquity I-Class - Xevo TQS micro; Waters Corporation). Potential intermediates (ALGP-Dox, LGP-Dox, GP-Dox, P-Dox) and Doxorubicinol were similarly monitored. Briefly, tissue homogenate was transferred in a microtube containing the same volume of an AgNO3 solution (66% w/v; second dilution factor = 2) and extracted with ice-cold acetonitrile/methanol (50:50) containing 1 μmol/L internal standard (Doxorubicin 13C D3, dilution factor = 5). After centrifugation, supernatant was transferred to matrix tubes for LC/MS-MS analysis as described above. LOD and LLOQ were respectively 0.076 and 0.26 μg/g of tissue for PhAc-ALGP-Dox, 0.0066 and 0.022 μg/g for ALGP-Dox, 0.0061 and 0.020 μg/g for LGP-Dox, 0.0052 and 0.017 μg/g for GP-Dox, 0.0048 and 0.016 μg/g for P-Dox, 0.0081 and 0.027 μg/g for Dox, and 0.0041 and 0.014 μg/g for Doxorubicinol. Bioanalytical analyses were performed at Laboratoire Chimie Générale, Faculté des Sciences Pharmaceutiques et Biologiques.
In vivo hematotoxicity
As described by Till and McCulloch (9), sublethally irradiated C57BL6 mice (9.5 Gy) were intravenously injected with 1 × 107 hematopoietic stem cells, extracted from the bone marrow of donor mice treated with 1,026 mg/kg PhAc-ALGP-Dox or 58 mg/kg Dox. Treatment was performed by means of osmotic minipumps (Alzet 1007D) implanted intraperitoneally. Blood count on donor mice was performed using Abbott Cell-Dyn 3700 hematology analyzer, while in dept characterization of subpopulations was performed via FACS analysis (B cells = CD45R+; CTL = CD3+ CD8a+; CD4+ = CD3+ CD4+; NK = NK1.1+).
In vivo efficacy studies
Studies were conducted in 6- to 8-week-old nu/nu NMRI or C57BL6 female mice (Janvier Laboratories). For orthotopic models, 1 × 106 E0771 cells (50 μL PBS) were injected into the mammary fat pad of syngeneic C57BL6 mice. For subcutaneous models, single-cell suspensions of 5 × 106 MDA-MB-231 or MDA-MB-468 cells (200 μL PBS:Matrigel, 1:1) were injected subcutaneously into the right posterior flank. Similarly, 5 × 106 U-87 MG (200 μL PBS:Matrigel, 1:1), 3 × 106 A2780 CpR, 5 × 106 A549, 5 × 106 NCI-H292, and 1 × 107 HCT-116 (200 μL PBS) were injected subcutaneously into the right posterior flank of NMRI nude mice.
Tumor size was measured every 2 days using a caliper and volume was estimated using V = 4/3p × (d/2)2 × D/2, where d indicates minor and D major tumor axis. Tumor growth inhibition (TGI%) was calculated as follows: %TGI = (1 − [Tt/T0/Ct/C0]/1 − [C0/Ct]) × 100, where Tt and T0 are the individual volume of treated animal X at time t and 0 respectively, Ct and C0 are mean volume of control group at time t and 0, respectively.
E0771 lung metastatic nodules were contrasted after intratracheal injection of 15% India ink and assessed under a stereomicroscope. MDA-MB-468 lung metastasis was quantified by qPCR measuring the ratio between the human GAPDH expression over murine ribosomal subunit 18s.
PDX models: establishment and maintenance
PDX models were established and maintained via bilateral subcutaneous transplantation of fresh human tumor fragment into the flank of 6- to 7-week-old female nu/nu NMRI mice (Janvier Laboratories). Animals bearing the original patient tissue were denominated “passage 0” (p0). To maintain PDX models, p0 mice were sacrificed once tumor growth reached a volume of ∼200 mm3. Tumors were collected and cut in smaller fragments, part of which were immediately retransplanted to next-generation mice (p1, n = 2/model). This process was repeated multiple times and tumor tissue of every passage was characterized histologically and immunohistochemically. Models were considered established after stable characteristics for at least two passages.
Study approval
All procedures were conducted in accordance to the ethical standards for experiments on animals established and approved by the Animal Ethics Committee of KU Leuven (ECD numbers: P064/2014, P082/2017, and P102/2019). Xenografting of tumor tissue from consenting patients was approved by the Medical Ethical Committee of UZ Leuven (S53483) Model establishment and subsequent usage for in vivo studies was approved by the Ethics Committee for Laboratory Animals of KU Leuven (P175–2015). All experiments with human material were executed in accordance with the Declaration of Helsinki and in accordance with applicable regulatory requirements and written confirmed consent, approved by UZ Leuven Ethics Committee (S60218).
Results
PhAc-ALGP-Dox is sequentially activated via a tumor-specific two-step cleavage, initiated by extracellular THOP1 and further processed by FAPα/DPP4
To identify critical enzymes responsible for the dual-step activation of PhAc-ALGP-Dox (Supplementary Fig. S1A), the formation of potential intermediates was monitored upon exposure to recombinant human CD10, neurolysin (NLN), or THOP1, three related endopeptidases (EC 3.4.24 family) with highly conserved metal-binding motif and substrate similarities (10). Both THOP1 and CD10 were previously suggested to play an important role in the activation of DTS-201 (3). THOP1 demonstrated improved activity towards PhAc-ALGP-Dox compared with DTS-201, resulting rapid decrease of the full-length molecule (t1/2 = 82 minutes) in favor of the predicted intermediate (GP-Dox) whereas no free Dox was detected (Fig. 1A; Supplementary Fig. S1B). Where CD10 effectively activated DTS-201 (Supplementary Fig. S1C), neither NLN nor CD10 were involved in PhAc-ALGP-Dox activation (t1/2 > 960 minutes). Indeed, only neglectable amounts of LGP-Dox were detected after exposure to CD10 (2.65% at 16 hours), whereas no intermediates were identified in the presence of NLN (Supplementary Figs. S1D and S1E). Similarly, incubation with FAPα or DPP4 alone did not generate free Dox or any intermediate, confirming the protective role of the capping group against exopeptidase activity (Supplementary Figs. S1F and S1G).
Figure 1.

PhAc-ALGP-Dox is activated sequentially by extracellular THOP1 and cytoplasmic DPP4/FAPα in the tumor microenvironment. A–C, Generation of PhAc-ALGP-Dox cleavage products in the presence of recombinant enzymes (THOP1, 0.1 μg/mL; FAPα, 0.04 μg/mL; or DPP4, 0.04 μg/mL). Data are represented as mean ± SD of triplicate analysis, LoQ, 3 nmol/L. D, Quantification of Dox autofluorescence following exposure to 20 μmol/L PhAc-ALGP-Dox for 5 hours. Nuclear/cytoplasmic fluorescence was assessed in HUVEC cells in the presence of recombinant THOP1 (0.1 μg/mL) alone or together with either recombinant FAPα (0.04 μg/mL) or PT-100 (300 nmol/L), an inhibitor of FAPα/DPP4. Bars represent mean + SD of n = 8–12. Asterisks indicate significant differences versus no enzyme or as specified by brackets. ***, P < 0.001, ****, P < 0.0001 as defined by one-way ANOVA corrected for multiple comparisons (Holm–Sidak). E–L, Representative images of HUVEC cells stimulated with PhAc-ALGP-Dox alone (E, F), together with THOP1 (G, H), THOP1 and FAPα (I–J) or THOP1, and PT-100 (K, L). Top panels are merged images of cytoplasmic (Agglutinin, green), nuclear (DAPI, blue), or Dox (autofluorescence, red). Bottom panels are black-white images of Dox autofluorescence. Scale bar, 20 μm. M, Quantification of ELISA data of human THOP1 protein levels in cell lysates and conditioned media of cancer cells (MDA-MB-231 and LS 174T) compared with normal (HME-1) cells. Bars are mean + SD of three independent ELISAs run in duplicate. Asterisks indicate significant differences versus no enzyme or as specified by brackets. ***, P < 0.001, ****, P < 0.0001 as defined by one-way ANOVA corrected for multiple comparisons (Holm–Sidak). N, Kinetic activation of Dnp(k)-ALGP-CouAla by extracellular peptidases in normal (HME-1, blue) or tumor (LS 174T, purple) conditioned medium. Dashed purple line represents the signal in tumor conditioned media, preincubated with a THOP1 inhibitor (THOPi, 10 μmol/L). O, Quantification of Dnp(k)-ALGP-CouAla conversion after 180 minutes. Bars are mean + SD, pooled from three independent experiments (n = 6). **, P < 0.01 as defined by one-way ANOVA.
Next, in the presence of THOP1, both FAPα and DPP4 were able to complete the second step and finalize prodrug activation. Although GP-Dox is being generated by THOP1 activity and peaks after 4 hours incubation, free Dox is produced simultaneously from this intermediate in the presence of either DPP4 or FAPα with similar efficiency (57% and 49% conversion at 16 hours respectively, Fig. 1B and C).
To better reflect clinical applicability, RNA expression of PhAc-ALGP-Dox processing enzymes was assessed in biopsies of patient diagnosed with stage IV TNBC. Both THOP1 and FAPα were significantly upregulated compared with normal adjacent tissue (NAT), but not in whole blood from the same patients, excluding the risk of ectopic activation (Supplementary Figs. S1H–S1K).
PhAc-ALGP-Dox activation is sequential and spatially controlled
To demonstrate that sequential activation of PhAc-ALGP-Dox by tumor enriched peptidases is controlled both temporally and spatially, enzymatic activity towards ALGP and subsequent cellular localization of Dox was monitored. First, results from parallel artificial membrane permeability assay indicate that PhAc-ALGP-Dox is cell impermeable (Peff < 0.017 nm/s). It is only after cleavage by THOP1 that GP-Dox diffuses into the cell. This biologically inactive intermediate is then cleaved intracellularly by FAPα and/or DPP4, generating active Dox. To validate this principle, normal cells that are not responsive towards the prodrug (HUVECs), were exposed to 20 μmol/L PhAc-ALGP-Dox for 5 hours and autofluorescence of Dox was monitored. Although no intracellular signal was observed in the absence of recombinant enzymes, administration of THOP1 increased intracellular localization, signifying the requirement of extracellular THOP1 activity for entering the cell. Subsequently, Dox autofluorescence was significantly confined to the nucleus as soon as FAPα was added in addition to THOP1, while blocking FAPα/DPP4 activity prevented nuclear localization (Fig. 1D–L).
To correlate our findings to the tumor-targeting mode of action of PhAc-ALGP-Dox, THOP1 protein levels in tumor cells were compared with normal epithelial cells (HME-1). Quantification reveals a modest increase in intracellular protein expression in MDA-MB-231 and LS 174T compared with normal cells (1.6- and 4.3-fold, respectively), but significantly higher abundancy in conditioned media (14.2- and 34.3-fold increase; Fig. 1M). Finally, using a mimetic of PhAc-ALGP-Dox, featuring a quencher and fluorophore separated by ALGP, only tumor-conditioned medium effectively activates the substrate, while blocking THOP1 abrogated cleavage (Fig. 1N and O).
PhAc-ALGP-Dox is a highly selective drug conjugate with promising anticancer efficacy
To define potency in clinically relevant indications, cell viability was assessed in murine (E0771) and human (MDA-MB-231 and MDA-MB-468) models for TNBC and colon adenocarcinoma (LS 174T). Consistent with the dual-step activation of PhAc-ALGP-Dox, potency was shifted from the parental compound, both in E0771 (IC50 = 0.35 μmol/L vs. 0.02 μmol/L) and human cancer cell lines (MDA-MB-231: IC50 = 14.89 μmol/L vs. 0.31 μmol/L, MDA-MB-468: 2.07 μmol/L vs. 0.11 μmol/L, and LS 174T: 0.31 μmol/L vs. 0.02 μmol/L). When compared with DTS-201 however, potency of PhAc-ALGP-Dox was superior in all models (Supplementary Figs. S2A and S2B). Moreover, selectivity of PhAc-ALGP-Dox towards tumor cells was significantly higher, both in murine and human cell lines (Fig. 2; Supplementary Fig. S2C). Selectivity index, defined as the ratio of IC50 in normal versus tumor cells, was 9- to 129-fold higher than Dox and even 6- to 46-fold superior to DTS-201 (Supplementary Table S1). In line with enhanced selectivity, improved safety of PhAc-ALGP-Dox was further highlighted by reduced maximal toxicity in normal compared with tumor cells (Fig. 2A–E). To assess the broader applicability, additional cell lines spanning eight different cancer indications were tested. After validation of abundant THOP1 expression, 14/15 investigated cell lines were effectively targeted by PhAc-ALGP-Dox. Potency (IC50) ranged from 311 nmol/L to 34.25 μmol/L, whereas HME-1 cells confirmed to be resilient (IC50 > 100 μmol/L; Supplementary Table S1).
Figure 2.

PhAc-ALGP-Dox is highly potent and selective toward in vitro cancer models. A–E, Dose–response curves after 72 hours' exposure to Dox (blue) or PhAc-ALGP-Dox (purple) in 2D monolayers for (A) murine TNBC (E0771), (B) normal murine epithelium (HC-11), (C) human TNBC (MDA-MB-231 and MDA-MB-468), (D) human CrC (LS 174T), or (E) normal human epithelium (HME-1). Graphs are presented as sigmoidal nonlinear fittings, normalized to nontreated controls. F–H, Growth of MDA-MB-231 spheroids after exposure to different concentrations of (F) Dox or (G) PhAc-ALGP-Dox. H, Representative bright field images of spheroids at day 17. I–K, Growth of LS 174T spheroids treated with (I) Dox or (J) PhAc-ALGP-Dox. K, Representative bright field images of LS 174T spheroids at day 14. Data are represented as mean ± SEM of three to five experiments, run in triplicate (n = 9–15). ##, P < 0.01 and ****, P < 0.0001 versus. controls as defined by two-way ANOVA. Scale bar, 200 μm. CrC, colorectal carcinoma; NT, nontreated; TNBC, triple-negative breast cancer.
Although 2D monolayers are relevant for screening purposes, they poorly resemble tumor complexity, with significant differences in drug exposure, cellular organization, growth kinetics, and gene expression (11). Especially when considering peptidase-activated prodrugs such as PhAc-ALGP-Dox, results are worthwhile confirming in 3D-spheroids to better anticipate in vivo response. Exposing MDA-MB-231 spheroids to PhAc-ALGP-Dox dose-dependently reduced spheroid growth (Fig. 2F–H). These findings were confirmed using LS 174T spheroids, with equipotent concentrations being approximately 10-fold higher compared with free Dox (Fig. 2I–K). Together, these data highlight the potential of PhAc-ALGP-Dox to target tumor cells in a 3D architecture.
Proof-of-concept: PhAc-ALGP-Dox activation by peptidases in vitro
To confirm the dual-step activation in a cellular context, two elegant approaches were applied. First, the tumor microenvironment was mimicked by exposing normal HME-1 cells to excess recombinant peptidases (gain-of-function). Alternatively, endogenous expression was inhibited in tumor cells (loss-of-function). In line with the in vitro kinetics (Fig. 1), PhAc-ALGP-Dox cytotoxicity improved in the presence of excess THOP1, resulting in a significant shift of IC50 from ∼373.3 to 2.06 μmol/L (Supplementary Fig. S2D). Exogenous presence of FAPα or DPP4, on the other hand, had no noteworthy effect (Supplementary Figs. S2E and S2F). Similarly, exposure of tumor cells to a dose titration of PhAc-ALGP-Dox in the presence of exogenous THOP1, but not FAPα or DPP4 shifted the cytotoxicity near the response towards Dox, while blocking endogenous THOP1 or FAPα/DPP4 activity protected against PhAc-ALGP-Dox cytotoxicity (Supplementary Figs. S2G–S2R). In addition, enhanced activity of THOP1 towards PhAc-ALGP-Dox compared with DTS-201 was confirmed using a knockdown approach and a second, more selective THOP1 inhibitor, Cpp-AAF-pAb. Because of the limited window of effective knockdown and short stability of the inhibitor respectively, drug exposure was limited to 16 hours and cell viability assessed 72 hours later. Although ∼50% to 70% reduction in THOP1 protein revealed a modest effect on PhAc-ALGP-Dox activation, the specific inhibition with Cpp-AAF-pAb confirmed the greater THOP1-dependency of PhAc-ALGP-Dox compared with DTS-201 in both cell lines investigated (Supplementary Fig. S3).
Tissue distribution and pharmacokinetics
Pharmacokinetic properties were evaluated after intravenous injection in healthy mice. PhAc-ALGP-Dox, as well as the free Dox derived from PhAc-ALGP-Dox, show favorable linearity within the dose range investigated (Supplementary Fig. S4). At the highest concentration of 849.5 mg/kg, PhAc-ALGP-Dox rapidly distributed with free Dox, GP-Dox and LGP-Dox being the most abundant metabolites in decreasing order (t1/2 = 6.5–8.5 minutes, Fig. 3A). Compared with administration of Dox (13.3 mg/kg), exposure to free Dox increased only 1.9-fold, although molar concentration of PhAc-ALGP-Dox was 36-fold higher. When comparing equimolar concentrations, free Dox was 19-fold lower following administration of PhAc-ALGP-Dox compared with Dox (Fig. 3B).
Figure 3.

Plasma levels and tissue distribution of PhAc-ALGP-Dox and Dox. A, The plasma levels of PhAc-ALGP-Dox, its potential intermediates, and Dox after intravenous administration of 849.5 mg/kg PhAc-ALGP-Dox in healthy mice (n = 5). B, The plasma levels of Dox after intravenous administration of equimolar doses PhAc-ALGP-Dox (23.6 mg/kg) or Dox (13.3 mg/kg) in healthy animals (n = 5). C, Tissue imaging by MALDI-FITCR MSI depicting the distribution of Dox in E0771 orthotopically implanted tumor-bearing mouse whole body sections at 24 and 48 hours after intravenous administration with PhAc-ALGP-Dox (849.5 mg/kg) or Dox (23 mg/kg). D, H&E-staining (left) and overlapping imaging by MALDI-FITCR MSI (right) depicting the distribution of GP-Dox, Dox, and PhAc-ALGP-Dox in E0771 orthotopically implanted tumor sections at 15 minutes after intravenous administration with PhAc-ALGP-Dox (849.5 mg/kg).
To determine whether PhAc-ALGP-Dox treatment led to a preferential release of Dox in tumor tissue, E0771 tumor-bearing mice were injected with equimolar concentration of PhAc-ALGP-Dox or Dox (94.4 or 53.4 mg/kg, respectively) and plasma pharmacokinetic parameters are summarized in Table 1. Most abundant metabolite in plasma was GP-Dox, followed by free Dox and LGP-Dox. As observed in healthy animals, free Dox exposure was 15-fold lower when compared with equimolar dosage of Dox, whereas intratumoral concentration was only 2.8-fold lower. In addition to free Dox, the only other metabolite detectable in the tumor, as expected after THOP1 cleavage, was GP-Dox. Taken together, PhAc-ALGP-Dox specifically accumulated in the tumor (Cmax = 25.2 μmol/L at 5 minutes) before efficient metabolization (2.5 μmol/L after 1 hour), whereas Dox presented slower accumulation in tumor (Cmax = 21.5 μmol/L at 4 hours) and elimination (7.3 μmol/L at 72 hours).
Table 1.
Pharmacokinetic properties and tissue distribution of Dox, DTS-201, and PhAc-ALGP-Dox and intermediates in E0771 tumor-bearing mice.
| Tumor-bearing mice (92 μmol/kg) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Tissue | Drug | Metabolites | t max | C max | AUC | t 1/2α | t 1/2β | CL |
| Heart | PhAc-ALGP-Dox | PhAc-ALGP-Dox | 5 | 19 | 3.7 | 0.17 | — | — |
| GP-Dox | 5 | 1.65 | 2.3 | 0.3 | 7 | — | ||
| Dox | 30 | 4.12 | 94 | 8.2 | 28 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
| Doxorubicin | Dox | 30 | 115 | 1,933 | 5.6 | 34 | — | |
| DoxOL | 240 | 0.86 | 54 | 47 | — | — | ||
| DTS-201a | Succ-βALAL-Dox | 5 | 35.9 | 15.1 | 0.2 | 0.5 | — | |
| Dox | 60 | 10 | 266 | 8 | 39 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
| Kidney | PhAc-ALGP-Dox | PhAc-ALGP-Dox | 5 | 530 | 388 | 0.1 | 9 | — |
| LGP-Dox | 5 | 5.6 | 3.7 | 0.1 | 3.3 | — | ||
| GP-Dox | 5 | 54 | 288 | 0.2 | 12.7 | — | ||
| Dox | 5 | 80 | 584 | 1 | 16 | — | ||
| DoxOL | 30 | 1.96 | 20.8 | 1 | 19.7 | — | ||
| Doxorubicin | Dox | 30 | 339 | 6,025 | 2.7 | 36 | — | |
| DoxOL | 30 | 6.8 | 168 | 2.5 | 49 | — | ||
| DTS-201a | Succ-βALAL-Dox | 15 | 811 | 771 | 0.4 | 10.7 | — | |
| Dox | 240 | 93 | 1,983 | — | 17.6 | — | ||
| DoxOL | 240 | 1.48 | 49.7 | — | 27.1 | — | ||
| Liver | PhAc-ALGP-Dox | PhAc-ALGP-Dox | 5 | 69 | 152.5 | 0.7 | 16.8 | — |
| GP-Dox | 5 | 5.3 | 72.2 | 1.4 | 14.8 | — | ||
| Dox | 30 | 16 | 235 | 2.1 | 15 | — | ||
| DoxOL | 240 | 0.44 | 10.6 | — | 21.7 | — | ||
| Doxorubicin | Dox | 5 | 189 | 2,184 | 2.1 | 26 | — | |
| DoxOL | 1 | 6 | 104 | 2.2 | 33.3 | — | ||
| DTS-201a | Succ-βALAL-Dox | 15 | 49 | 48 | 0.6 | 20.5 | — | |
| Dox | 60 | 14 | 449 | 25.2 | — | — | ||
| DoxOL | 120 | 0.67 | 30.8 | — | 33.4 | — | ||
| Tumor | PhAc-ALGP-Dox | PhAc-ALGP-Dox | 5 | 25.2 | 16.9 | 0.3 | 2.6 | — |
| GP-Dox | 15 | 20.85 | 59.6 | 0.5 | 10 | — | ||
| Dox | 240 | 6.72 | 459 | — | 46 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
| Doxorubicin | Dox | 120 | 21.4 | 1,327 | — | 53 | — | |
| DoxOL | 2,880 | 0.12 | 8 | — | — | — | ||
| DTS-201a | Succ-βALAL-Dox | 5 | 56.5 | 25.6 | 0.4 | — | — | |
| Dox | 720 | 13.3 | 988 | — | 36 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
| Plasma | PhAc-ALGP-Dox | PhAc-ALGP-Dox | 5 | 560 | 156.5 | 0.21 | — | — |
| LGP-Dox | 5 | 0.74 | 0.133 | 0.15 | — | — | ||
| GP-Dox | 5 | 3.7 | 1.49 | 0.2 | — | — | ||
| Dox | 5 | 1.2 | 0.74 | 0.2 | 9 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
| Doxorubicin | Dox | 5 | 7.09 | 11.3 | 0.2 | 13 | 135 | |
| DoxOL | — | <LoQ | — | — | — | — | ||
| DTS-201a | Succ-βALAL-Dox | 5 | 243 | 1,21.3 | 0.25 | — | 12.6 | |
| Dox | 30 | 0.404 | 1.41 | 0.6 | 16.7 | — | ||
| DoxOL | — | <LoQ | — | — | — | — | ||
aFor DTS-201, only the parent molecule and Dox, but no intermediates, were measured.
In systemic tissues, GP-Dox and free Dox were the major metabolites, generally reaching Cmax within 5 to 30 minutes after injection, and interestingly, lowest in heart. Consistent with the expected clearance and metabolization, liver and kidney were most exposed to free Dox. Both intact drug-conjugate and the two metabolites remained highest in kidney. Of note, direct comparison of DTS-201 and PhAc-ALGP-Dox revealed a safer pharmacokinetic profile of the latter, considering that exposure to free Dox resulting from DTS-201 activation was 2- to 3-fold higher in normal organs (Table 1).
Next, MALDI-FTICR imaging was used to give a more general view on tissue biodistribution of PhAc-ALGP-Dox and its metabolites in E0771 tumor-bearing mice. Where administration of Dox (23 mg/kg) dispersed homogenously throughout the body, PhAc-ALGP-Dox (849.5 mg/kg) distributed faster, particularly to excretion and well-perfused organs such as kidney, lung, and liver, which could be expected for a stable molecule unable to enter healthy cells. Importantly, activated PhAc-ALGP-Dox accumulated in the tumor within 2 hours after injection and the presence of free Dox remained stable at least up to 48 hours. On the contrary, the majority of other organs showed negligible amounts of free Dox with the exception of kidney, where the molecule underwent metabolic and renal elimination. Consequently, free Dox from activated PhAc-ALGP-Dox could be detected, but its presence rapidly decreased over time (Fig. 3C and D).
PhAc-ALGP-Dox circumvents systemic toxicity associated with Dox treatment in vitro and in vivo
To address the increased safety of PhAc-ALGP-Dox, acute toxicity was assessed in mice. Median lethal dose (LD50) of PhAc-ALGP-Dox was 780 mg/kg compared with 17 mg/kg for Dox, highlighting improved tolerability and allowing to administer 27 times more payload (780 mg/kg = 459 mg Dox/kg). Next, the effects of PhAc-ALGP-Dox on human hematopoietic progenitor cells and cardiomyocytes were further analyzed. In contrast to significant toxicity of Dox, PhAc-ALGP-Dox exhibits better safety profiles towards total myeloid (IC50 = 0.07 μmol/L vs. 0.01 μmol/L for Dox) and erythroid derived cells (IC50 = 0.05 μmol/L vs. 0.02 μmol/L for Dox; Fig. 4A and B). In-depth characterization of different human progenitor-derived colonies confirmed the safer nature of PhAc-ALGP-Dox (Supplementary Figs. S5A–S5G).
Figure 4.

PhAc-ALGP-Dox shows reduced hematotoxicity and cardiotoxicity as well as improved MTD with tumor sequestration in tumor-bearing mice. A and B, Dose–response curves for the effect of continuous exposure (14 days) to PhAc-ALGP-Dox on human erythroid (A) and myeloid (B) progenitor proliferation using MethoCult GF H84434. C, Kaplan–Meier plot describing the survival rate of recipient mice after BMT of vehicle, Dox (58 mg/kg/week) or PhAc-ALGP-Dox (1,026 mg/kg/week) treated donors. Mice were monitored for 41 days following BMT (n = 6). Gehan–Breslow–Wilcoxon test, P = 0.03. D–F, Blood count of donor mice treated with Dox (58 mg/kg/week) or PhAc-ALGP-Dox (1,026 mg/kg/week) by means of osmotic minipumps; whole blood was analyzed at the time of bone marrow collection. Data represent mean ± SD (n = 8). ****, P < 0.0001 versus control as defined by two-way ANOVA. G–L, Impedance-based cardiotoxicity of Dox (blue curves) and PhAc-ALGP-Dox (purple curves) in hiPSC-CMs on base impedance, contraction amplitude, and beat rate. Data of quadruplicate values are represented as mean ± SD. M–Q, PhAc-ALGP-Dox dose–response in healthy versus MDA-MB-468 tumor-bearing mice. Body weight (M, O), survival rate (N, P), and tumor volume (Q) were periodically monitored during and after the treatment (as indicated by arrows). Data represent mean ± SD (n = 5). ****, P < 0.0001 versus control (saline) as defined by two-way ANOVA; Gehan–Breslow–Wilcoxon test, P < 0.0001 (N) and P = 0.0013 (P). BMT, bone marrow transplantation; hiPSC-CM, human induced pluripotent stem cell derived cardiomyocyte.
To investigate the biological impact of PhAc-ALGP-Dox treatment on bone marrow (BM) long-term repopulation ability, in vivo transplantation experiments have been performed. Donor mice were treated continuously for 7 days via osmotic minipumps containing Dox (58 mg/kg/week) or PhAc-ALGP-Dox (1,026 mg/kg/week; Supplementary Fig. S5J). Two days after treatment, blood samples and BM were collected from donor mice. Although no alteration was noticed in neutrophil and monocyte count, whole blood count revealed a drop in white blood cells both in Dox and PhAc-ALGP-Dox treated animals, mainly attributed to a decrease in lymphocytes (Fig. 4D–G). FACS analysis confirmed a reduction in B-cells, whereas CD4+ and NK cells were unaffected. In contrast to Dox, PhAc-ALGP-Dox treated mice had 15% less circulating cytotoxic T lymphocytes (CTL; Supplementary Fig. S5L). Despite these changes, BM from PhAc-ALGP-Dox, but not from Dox treated donors, maintained high repopulation abilities following transplantation. In fact, 80% of mice receiving BM from PhAc-ALGP-Dox treated donors survived throughout the study (day 41 post-transplantation), compared with only 30% of animals receiving BM from Dox-treated mice (Fig. 4C). Considering that PhAc-ALGP-Dox donors received 10 times higher Dox-equivalent concentration, these data provide important evidences on reduced hematopoietic cytotoxicity, confirming in vitro results.
In addition, safety of PhAc-ALGP-Dox towards cardiomyocytes was evaluated. In contrast to nanomolar potency of Dox in mitotic and differentiated H9C2 rat cardiomyocytes, mitotic cells were considerably less affected by PhAc-ALGP-Dox whereas differentiated cardiomyocytes were essentially resilient (Supplementary Figs. S5H and S5I). Clinically more relevant, human iPSCs-derived cardiomyocytes were analyzed for signs of acute or chronic toxicity based on cellular impedance, revealing a notably safer profile for PhAc-ALGP-Dox (Fig. 4H–M). While Dox-mediated cardiotoxicity was already evident shortly after exposure and increased to 100% by 72 hours, PhAc-ALGP-Dox induced alterations only at concentrations exceeding equipotency of Dox in cancer cells (>5 μmol/L) and were delayed in time and reduced in effect size. Similarly, changes in contractility were only observed following exposure to high concentrations of PhAc-ALGP-Dox. Moreover, maximal toxicity did not further increase with higher concentrations and tended to reach a plateau, highlighting the lack of cumulative toxicity. Finally, Dox induced significant arrythmia, signified by an initial incline of beating rate, rapidly followed by cardiac arrest. Although PhAc-ALGP-Dox triggered increased beating rates at higher concentrations, these effects were reversible and normalized within 72 hours. Taken together, these results emphasize the significantly improved safety of PhAc-ALGP-Dox.
Finally, to define the most appropriate PhAc-ALGP-Dox dose and regimen for preclinical efficacy studies, MTD was calculated (Fig. 4N–Q). Increasing concentrations of PhAc-ALGP-Dox (103, 206, and 309 mg/kg) were dosed weekly in healthy or MDA-MB-468 tumor bearing mice. Confirming the hypothesis that tumors sequester PhAc-ALGP-Dox and reduce systemic exposure, toxicity (when observed) tended to be more severe and mortality was higher in healthy compared with tumor bearing mice. Focusing on tumor bearing mice, four cycles of 206 mg/kg PhAc-ALGP-Dox revealed mild but reversible body weight loss (15%) and a temporary decrease of lymphocyte and neutrophil count, whereas TGI was approximately 140%. On the basis of these results, the highest tolerated dose of a PhAc-ALGP-Dox was defined at 154 mg/kg (every weeks × 4), corresponding to 87 mg/kg Dox equivalent.
PhAc-ALGP-Dox shows evidence of superior efficacy in clinically relevant xenograft and PDX models
Efficacy of PhAc-ALGP-Dox was compared with Dox in several tumor models of murine and human origin, including TNBC and STS. Both in a syngeneic orthotopic model (E0771) and a human xenograft model (MDA-MB-231) for TNBC, PhAc-ALGP-Dox significantly improved the therapeutic index of Dox, following superior tolerability while maintaining equipotency in terms of TGI and metastatic dissemination. Histologic characterization revealed that PhAc-ALGP-Dox significantly reduced tumor necrosis (Fig. 5A–F; Supplementary Figs. S6A and S6B). Interestingly, mice treated with equimolar concentrations of DTS-201 (155 mg/kg) displayed signs of toxicity, while well-tolerated concentrations resulted in suboptimal efficacy (Supplementary Fig. S6C). These data highlight the increased therapeutic index of PhAc-ALGP-Dox compared with equimolar concentrations of DTS-201 or equipotent concentrations of Dox and revealed consistent reduction of tumor necrosis.
Figure 5.

In vivo efficacy of PhAc-ALGP-Dox. A–D, The tumor volume (A) and survival rate (C) of E0771 TNBC orthografts implanted in C57/Bl6 mice treated with Dox (4.4 mg/kg) or PhAc-ALGP-Dox (154 mg/kg). Mice were treated with intravenous bolus injection twice a week for a total of four cycles as indicated by arrows. The number of lung metastatic nodules of each individual group (B) and histological quantification of tumor necrosis in H&E-stained tumor sections (D) were determined at the day of sacrifice. Data represent average + SD (n = 8). *, P = 0.011; **, P < 0.01 ****, P < 0.0001 versus vehicle as defined by one-way (D) or two-way (A) ANOVA; Gehan–Breslow–Wilcoxon test, P < 0.0001 (C). E–H, The tumor volume of MDA-MB-231 TNBC xenografts implanted in NMRI nude mice treated with (E) bi-weekly IV bolus infusion of Dox (4.4 and 8.8 mg/kg) or (G) PhAc-ALGP-Dox (63 and 154 mg/kg) for five cycles in total or by means of osmotic minipumps intraperitoneally implanted delivering 58 mg/kg/week Dox or 103 or 1026 mg/kg/week PhAc-ALGP-Dox for 1 week. At the end of the study, tumor necrosis in intravenously injected (F) and in osmotic minipump treated (H) mice was quantified in H&E-stained tumor sections. Data represent average + SD (n = 8). *, P < 0.03; ***, P < 0.0004 ****, P < 0.0001 versus control or #P < 0.01; ##P < 0.001 versus Dox (58 mg/kg) as defined by one-way (F, H) or two-way (E, G) ANOVA. I–K, The tumor volume (I) of MDA-MB-468 TNBC xenografts implanted in NMRI nude mice treated by means of osmotic minipumps intraperitoneally implanted delivering 58 mg/kg/week Dox or 103 or 1,026 mg/kg/week PhAc-ALGP-Dox for 1 week. In brackets, the number of mice died during the study. At the end of the study, lung metastatic dissemination was determined by qPCR measuring human GAPDH copy number in murine lung tissue (J) and tumor necrosis (K) was quantified in H&E-stained tumor sections. Data represent average + SD (n = 9). *, P < 0.03; **, P = 0.006 ***, P < 0.0009 ****, P < 0.0001 versus control or ##P = 0.0015 versus Dox (58 mg/kg) as defined by one-way (J, K) or two-way (I) ANOVA. L and M, The plot quantifies the relative metastatic growth of E0771 experimental lung colonies (L) and survival rate (M) treated with corresponding different concentration of Dox, or PhAc-ALGP-Dox. Arrows indicate when mice were treated. Data were normalized toward value at day of randomization (d21) to homogenize the variability related to extravasation and lung colonization. Mice received the treatment via TV injection at the time indicated by the arrows. Data represent median of bioluminescence signal (n = 9 per group; *, P = 0.0179; ****, P < 0.0001 vs. control; ##, P = 0.0064 vs. Dox); Gehan–Breslow–Wilcoxon test, P = 0.0034 (M). N and O, Tumor volume assessment during treatment of two different models of patient-derived leiomyosarcoma xenografted in NMRI nude mice. Data are presented as relative tumor volume (%) compared with the baseline. Mice received the weekly treatment via TV injection at the time indicated by the arrows. In brackets, the number of mice died during the study. All data points are shown as mean of relative tumor volume ± SD (n = 10); ***, P = 0.0002; ****, P < 0.0001 versus control or ####P < 0.0001 versus Dox (5 mg/kg) as defined by two-way ANOVA.
Next, the MDA-MB-231 xenograft model was used to investigate whether optimizing route of administration increased efficacy of PhAc-ALGP-Dox. Continuous delivery of PhAc-ALGP-Dox via osmotic minipumps decreased tumor volume in a dose-dependent manner (Fig. 5G). At 1,026 mg/kg, PhAc-ALGP-Dox treatment stabilized tumor growth with a TGI of 98%, whereas in Dox treated animals TGI did not exceed 84%. Mild, but reversible, body weight loss was observed after administration of Dox, as well as with the highest dose of PhAc-ALGP-Dox (1,026 mg/kg; Supplementary Fig. S6D). Importantly, where Dox therapy failed to prevent necrosis, PhAc-ALGP-Dox significantly reduced tumor necrosis at therapeutic concentrations (Fig. 5H). These results were confirmed using a second model for TNBC (MDA-MB-468), where PhAc-ALGP-Dox treatment not only induced important inhibition of tumor growth, but also reduced metastatic dissemination (Fig. 5I and J). Moreover, therapeutic concentrations of PhAc-ALGP-Dox resulted in superior TGI compared with Dox (151% vs. 70%). Again, reduction of tumor volume corresponds to a significant decrease in necrosis following treatment with 1,026 mg/kg/wk PhAc-ALGP-Dox, without changes in body weight (Fig. 5K; Supplementary Fig. S6E).
The potential of PhAc-ALGP-Dox in targeting metastatic cells in vivo was evaluated in a model for experimental lung metastasis. E0771-AKA-Luc2 cells were injected into the lateral tail vein to mimic metastatic homing and growth into the lung. Mice receiving PhAc-ALGP-Dox treatment, exhibited significantly lower metastatic burden without signs of toxicity, resulting in clear improvements of overall survival (Fig. 5L and M; Supplementary Figs. S6F–S6H). To emphasize the broad applicability of PhAc-ALGP-Dox, efficacy was demonstrated in numerous additional human xenograft models, including colorectal cancer, glioblastoma, non–small cell lung cancer (NSCLC), and cisplatin-resistant ovarian cancer. A summary is shown in (Supplementary Table S1). Recently, patient-derived xenografts (PDX) are favored over cell-line derived xenografts, as they better reflect the heterogeneity and properties of the original tumor. Therefore, PhAc-ALGP-Dox efficacy was confirmed using two PDX models of leiomyosarcoma. In both models, TGI following PhAc-ALGP-Dox treatment was superior when compared with Dox (Fig. 5N and O).
Altogether, these preclinical results emphasize improved safety of PhAc-ALGP-Dox and potential superiority compared with Dox at well-tolerated concentrations, leading to an increased therapeutic index and promising clinical applicability.
Discussion
Although Dox remains a cornerstone of many anticancer therapies, its potential remains limited by cumulative toxicities, despite various attempts to improve the therapeutic index. In this study, PhAc-ALGP-Dox has proven to be a valid alternative with broad applicability, favorable tissue distribution and improved therapeutic index due to its tumor-targeting nature. The unique mode of activation is mediated by a sequential and spatially controlled activation. Where DTS-201 activation is suggested to be mediated by both THOP1 and CD10 (2, 3), PhAc-ALGP-Dox cleavage is more effectively and more exclusively driven by THOP1. Indeed, similar oligopeptidases, such as CD10 and NLN, or more general matrix-metalloproteases involved in tumor biology (e.g., MMP2 and MMP9), were excluded as potential activators of PhAc-ALGP-Dox. Intriguingly, the expression of THOP1 is upregulated in many tumor types and has been repeatedly correlated with prognosis (12–16). On the other hand, expression of CD10 is less tumor specific and, although indisputable, its role in cancer remains controversial (17). In addition, hematopoietic cells rely on CD10 for differentiation and maturation (18, 19), increasing the risk of DTS-201 related myelosuppression, the main toxicity observed in a Phase I clinical trial (6). Although the presence of THOP1 in normal tissues has been described, detection is confined to the intracellular space (20), while extracellular THOP1 activity is considerably more restricted to the tumor microenvironment. Indeed, we demonstrated that PhAc-ALGP-Dox is cell impermeable before being activated by extracellular THOP1 (Fig. 1). Moreover, tumor specificity could be twofold, since not only THOP1 expression is upregulated, but also pathways responsible for its secretion are more active in cancer cells (21–26). THOP1 secretion occurs via the non-canonical pathway and is enhanced following protein kinase A (PKA) mediated phosphorylation and association with 14–3-3ε (27–30). The role of PKA and 14–3-3ε in tumor progression, vascularization and metastasis has been well described (14, 26, 31, 32), and could increase the pool of extracellular THOP1 during progression. Moreover, activated immune cells could also contribute and sustain THOP1 within the tumor microenvironment (12, 33), maintaining PhAc-ALGP-Dox activation. Altogether, these factors mediate tumor specific activation of PhAc-ALGP-Dox to a cell permeable intermediate, which rapidly enters tumor cells where it is further processed to active Dox in a self-sustainable way, limiting the chance of enzyme exhaustion and subsequent resistance.
The importance of activating enzymes was further demonstrated by blocking endogenous enzymes in tumor cell cultures. Inhibiting the activity of either THOP1 or FAP/DPP4 abrogated PhAc-ALGP-Dox cytotoxicity, while ∼50% to 70% reduction in THOP1 protein expression provided a modest but apparent shift in potency (Supplementary Figs. S2 and S3). Indeed, the residual THOP1 could still be responsible for activating PhAc-ALGP-Dox and causing cytotoxicity. However, and in line with higher THOP1-dependency, the effects on cytotoxicity following either limited knockdown or specific THOP1 inhibition were more pronounced for PhAc-ALGP-Dox as compared with DTS-201. Furthermore, mimicking the tumor secretome in normal cell cultures by adding exogenous recombinant enzymes resulted in activation of PhAc-ALGP-Dox. Similarly, enriching the extracellular presence of THOP1 in tumor cells increased the potency to a similar level as Dox alone, signifying complete activation (Supplementary Fig. S2). Because the unique mode of activation hijacks pathways used by tumor cells to progress, PhAc-ALGP-Dox is applicable to a broad variety of solid tumors as demonstrated by in vitro potency and in vivo efficacy in several preclinical models (Supplementary Table S1). Somewhat intriguing, the mere expression of THOP1 was required, but not sufficient, to predict in vivo response. Indeed, although all THOP1-expressing models were responsive to PhAc-ALGP-Dox, potency nor efficacy correlates with THOP1 expression per se, although a positive trend towards maximal efficacy could be observed in vitro. Most likely, increased expression in tumor cells, combined with the significantly higher extracellular activity underlies the rapid activation and targeted delivery of large quantities of payload. This concept was further highlighted by the favorable tissue distribution of PhAc-ALGP-Dox (Fig. 3). Although one could argue that introduction of a linear peptide moiety favors renal clearance, we demonstrated that, despite the short half-life of PhAc-ALGP-Dox, large quantities of active payload are delivered specifically to the tumor site. Tumor requisitioning of PhAc-ALGP-Dox was further highlighted by the differential MTD in healthy versus tumor bearing mice, where systemic exposure was potentially reduced due to sequestration within the tumor (Fig. 4M–P). Consistent with the expected metabolization and clearance, intact prodrug and metabolites were detected in kidney and to a lesser extend in liver. However, passage was transient without significant conversion to Dox, and therefore expected to be cleared. Indeed, no signs of nephrotoxicity were observed. Extra attention was given to the heart, since cardiotoxicity is a major concern for Dox-based therapies. Encouragingly, neither Dox nor its toxic metabolite, doxorubicinol, were detected following PhAc-ALGP-Dox administration. Improved safety towards cardiomyocytes was further demonstrated in hiPSC-CMs, where changes in cellular impedance, contractility or beating rate were only observed at concentrations exceeding equipotency in tumor cells. Even at supraphysiologic concentrations, alterations in cardiac function were delayed in time, less pronounced as compared with Dox and reversible (Fig. 4G–L). Similarly, in-depth analysis of hematotoxicity, the second common dose-limiting toxicity of Dox, revealed transient effects of PhAc-ALGP-Dox on blood parameters with a significant drop in lymphocytes, similar to what was reported for Dox (34–36). However, survival was significantly improved when using bone marrow explants from treated donors to repopulate sublethally irradiated recipient mice. This indicates that, in contrast to Dox, the reduction in blood cells following PhAc-ALGP-Dox treatment is reversible without underlying toxicity on hematopoietic progenitor cells.
Despite its limitations, anthracyclines remain one of the most used drug classes in clinic to treat breast cancer with an ORR of 30% to 50%. However, their use in metastatic setting is often limited due to concerns that cumulation from adjuvant chemotherapy raises the risk of cardiotoxicity (37). The favorable profile of PhAc-ALGP-Dox leads to improved inhibition of primary tumor growth in multiple models, secondary to high tumor retention of activated Dox (Figs. 3 and 5; Supplementary Table S1). This resulted in decreased tumor necrosis when compared with Dox treatment, indicating a possible switch from the canonical pro-apoptotic pathway, to a “softer” antiproliferative and death-inducing signal cascade. This phenotype is currently under investigation and reveals similarities to mitotic catastrophe.
In addition to effectively targeting the primary tumor in a variety of models, these results also indicate a suppressive effect on metastatic dissemination in a xenograft model for breast cancer (Fig. 5B). However, the intrinsic limitation of preclinical models does not allow to discriminate between a direct impact of PhAc-ALGP-Dox on metastatic behavior or simply a consequence to significant primary TGI following the treatment. Using a murine model for experimental lung metastasis, the direct effect of PhAc-ALGP-Dox on metastatic growth was demonstrated to be superior to Dox. Although metastatic cells are difficult to target, their pro-metastatic properties also render them more susceptible to PhAc-ALGP-Dox treatment. Mechanisms used by cancer cells to detach from the primary lesion, survive in circulation and colonize distant sites are far from elucidated, but include proteolytic activity during multiple stages of metastatic dissemination (38–40). Although the role of zinc(II)-dependent endopeptidases herein has been well-studied (41, 42), the specific contribution of THOP1 to distant colonization is less understood. Nevertheless, it is expected that secretory pathways are significantly upregulated in metastatic cells to orchestrate the proteolytic activity required for invasion and colonization. As such, THOP1 secretion is potentially stimulated as well, increasing PhAc-ALGP-Dox activation. Intriguingly, THOP1 expression has been repeatedly linked to metastasis in NSCLC (12), renal clear cell carcinoma (43), and HCC (15). Whether, and how, THOP1 expression correlates to metastasis and disease prognosis remains a matter of debate and should be further investigated (10). Nevertheless, we have demonstrated that, in addition to efficacy on the primary tumor, PhAc-ALGP-Dox is superior to Dox in targeting metastatic growth.
In a clinical setting, Dox remains the first-line standard-of-care treatment in patients with STS with locally or advanced metastasis, despite poor response rates (<15%), underscoring the high medical need (44). The potential of PhAc-ALGP-Dox as a new effective therapy was demonstrated in PDX models derived from patients with grade III leiomyosarcoma. These data corroborate the superior and statistically significant antitumor activity of PhAc-ALGP-Dox as compared with Dox and anticipate the improved therapeutic potential in a larger setting of STS (7, 45). Although STS could be an interesting indication for clinical development, the extensive applicability of PhAc-ALGP-Dox in a variety of solid tumors was apparent. Compared with other targeted strategies, such as ADCs or PDCs, PhAc-ALGP-Dox is not limited to the presence of specific epitopes to be effective. In contrast, it requires an extracellular pool of THOP1 to initiate intracellular entrapment of the payload, which has proven to be highly tumor specific and applicable to a broad range of solid tumors.
Taken together, the unique sequential and spatially controlled two-step activation of PhAc-ALGP-Dox, driven by extracellular THOP1 and cytoplasmic FAPα and/or DPP4 respectively, results in a molecule with superior therapeutic value compared with Dox. In addition to the tumor-favoring biodistribution, these findings show an improved therapeutic index in multiple preclinical models for solid tumors, including TNBC, colorectal cancer, glioblastoma, NSCLC, and cisplatin-resistant ovarian cancer. Moreover, the superiority compared with Dox was highlighted using clinically relevant PDX models of STS, emphasizing the extensive clinical potential of PhAc-ALGP-Dox.
Authors' Disclosures
A. Casazza reports grants from Scientific Research Flanders during the conduct of the study; also has a patent for EP21155657.6 and PCT/EP2021/087374 pending. L. Van Helleputte reports grants from Fund for Scientific Research Flanders during the conduct of the study; also has a patent for WO2014102312 issued, a patent for EP21155657.6 pending, and a patent for PCT/EP2021/087374 pending. B. Van Renterghem reports grants from CoBioRes during the conduct of the study. M. De Petrini reports grants from Fund for Scientific Research Flanders during the conduct of the study. T. Janssens reports grants from Funt for Scientific Research Flanders and Scientific Research Flanders during the conduct of the study. M. Pellens reports grants from Fund for Scientific Research Flanders during the conduct of the study. M. Diricx reports grants from Fund for Scientific Research Flanders during the conduct of the study. A. Wozniak reports grants from CoBioRes, Belgium, during the conduct of the study. G. Reyns reports grants from Fund for Scientific Research Flanders (Vlaio) during the conduct of the study; also has a patent for EP21155657.6 and PCT/EP2021/087374 pending. N. Kindt reports grants from Fund for Scientific Research Flanders (Vlaio) during the conduct of the study; also has a patent for EP21155657.6 and PCT/EP2021/087374 pending. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We would like to express our gratitude to Em. Prof. Dr. Désiré Collen for his comprehensive support for this program. Our appreciation goes out to Dr. Catherine Piveteau from the University of Lille for the services she provided related to mass spectrometry. The authors thank David Bonnel and Mathieu Gaudin at Imabiotech for the results regarding PhAc-ALGP-Dox mass spectrometry imaging and tissue distribution. This work is in memory of professor Andrè Trouet (1936–2014). This work was supported by grants from the Fund for Scientific Research Flanders (HBC-2016-0887 and HBC-2017-1005).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Contributions
A. Casazza: Conceptualization, data curation, supervision, funding acquisition, investigation, writing–original draft, project administration, writing–review and editing. L. Van Helleputte: Conceptualization, data curation, formal analysis, supervision, validation, investigation, writing–original draft, writing–review and editing. B. Van Renterghem: Data curation, formal analysis. P. Pokreisz: Conceptualization, supervision, investigation. N. De Geest: Data curation, formal analysis. M. De Petrini: Data curation, formal analysis. T. Janssens: Data curation, formal analysis. M. Pellens: Data curation, formal analysis. M. Diricx: Data curation, formal analysis. C. Riera-Domingo: Data curation. A. Wozniak: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft. M. Mazzone: Conceptualization, supervision, investigation, writing–original draft. P. Schöffski: Conceptualization, supervision, investigation, writing–original draft. O. Defert: Conceptualization, supervision, investigation, project administration. G. Reyns: Conceptualization, supervision, investigation, visualization, project administration. N. Kindt: Conceptualization, formal analysis, supervision, visualization, writing–original draft, project administration, writing–review and editing.
References
- 1. Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody-drug conjugates: the last decade. Pharmaceuticals 2020;13:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan C, Cardarelli PM, Nieder MH, Pickford LB, Gangwar S, King DJ, et al. CD10 is a key enzyme involved in the activation of tumor-activated peptide prodrug CPI-0004Na and novel analogues: implications for the design of novel peptide prodrugs for the therapy of CD10+ tumors. Cancer Res 2003;63:5526–31. [PubMed] [Google Scholar]
- 3. Dubois V, Nieder M, Collot F, Negrouk A, Nguyen TT, Gangwar S, et al. Thimet oligopeptidase (EC 3.4.24.15) activates CPI-0004Na, an extracellularly tumour-activated prodrug of doxorubicin. Eur J Cancer 2006;42:3049–56. [DOI] [PubMed] [Google Scholar]
- 4. Dubois V, Dasnois L, Lebtahi K, Collot F, Heylen N, Havaux N, et al. CPI-0004Na, a new extracellularly tumor-activated prodrug of doxorubicin: in vivo toxicity, activity, and tissue distribution confirm tumor cell selectivity. Cancer Res 2002;62:2327–31. [PubMed] [Google Scholar]
- 5. Ravel D, Dubois V, Quinonero J, Meyer-Losic F, Delord J, Rochaix P, et al. Preclinical toxicity, toxicokinetics, and antitumoral efficacy studies of DTS-201, a tumor-selective peptidic prodrug of doxorubicin. Clin Cancer Res 2008;14:1258–65. [DOI] [PubMed] [Google Scholar]
- 6. Schoffski P, Delord JP, Brain E, Robert J, Dumez H, Gasmi J, et al. First-in-man phase I study assessing the safety and pharmacokinetics of a 1-hour intravenous infusion of the doxorubicin prodrug DTS-201 every 3 weeks in patients with advanced or metastatic solid tumours. Eur J Cancer 2017;86:240–7. [DOI] [PubMed] [Google Scholar]
- 7. Cornillie J, Wozniak A, Pokreisz P, Casazza A, Vreys L, Wellens J, et al. In vivo antitumoral efficacy of PhAc-ALGP-Doxorubicin, an enzyme-activated doxorubicin prodrug, in patient-derived soft tissue sarcoma xenograft models. Mol Cancer Ther 2017;16:1566–75. [DOI] [PubMed] [Google Scholar]
- 8. Badisa RB, Darling-Reed SF, Joseph P, Cooperwood JS, Latinwo LM, Goodman CB. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res 2009;29:2993–6. [PMC free article] [PubMed] [Google Scholar]
- 9. Till JE, McCulloch EA. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. 1961. Radiat Res 2012;178:AV3–7. [DOI] [PubMed] [Google Scholar]
- 10. Ferro ES, Gewehr MCF, Navon A. Thimet oligopeptidase biochemical and biological significances: past, present, and future directions. Biomolecules 2020;10:1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pinto B, Henriques AC, Silva PMA, Bousbaa H. Three-dimensional spheroids as in vitro preclinical models for cancer research. Pharmaceutics 2020;12:1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qi L, Li SH, Si LB, Lu M, Tian H. Expression of THOP1 and its relationship to prognosis in non-small cell lung cancer. PLoS One 2014;9:e106665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu H, Zhao H. Prognosis related miRNAs, DNA methylation, and epigenetic interactions in lung adenocarcinoma. Neoplasma 2019;66:487–93. [DOI] [PubMed] [Google Scholar]
- 14. Paschoalin T, Carmona AK, Rodrigues EG, Oliveira V, Monteiro HP, Juliano MA, et al. Characterization of thimet oligopeptidase and neurolysin activities in B16F10-Nex2 tumor cells and their involvement in angiogenesis and tumor growth. Mol Cancer 2007;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nomoto S, Hishida M, Inokawa Y, Takano N, Kanda M, Nishikawa Y, et al. Expression analysis of THOP1 in background liver, a prognostic predictive factor in hepatocellular carcinoma, extracted by multiarray analysis. Ann Surg Oncol 2014;21:S443–50. [DOI] [PubMed] [Google Scholar]
- 16. Liu Y, Bruce LA, Wolfson AJ. Extracellular thimet oligopeptidase is carried by cell membrane microvesicles of prostate cancer cells. FASEB J 2013;27. [Google Scholar]
- 17. Maguer-Satta V, Besancon R, Bachelard-Cascales E. Concise review: neutral endopeptidase (CD10): a multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011;29:389–96. [DOI] [PubMed] [Google Scholar]
- 18. Bene MC. Immunophenotyping of acute leukaemias. Immunol Lett 2005;98:9–21. [DOI] [PubMed] [Google Scholar]
- 19. Hystad ME, Myklebust JH, Bo TH, Sivertsen EA, Rian E, Forfang L, et al. Characterization of early stages of human B cell development by gene expression profiling. J Immunol 2007;179:3662–71. [DOI] [PubMed] [Google Scholar]
- 20. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 21. Yang X, Cao W, Lin H, Zhang W, Lin W, Cao L, et al. Isoform-specific expression of 14–3-3 proteins in human astrocytoma. J Neurol Sci 2009;276:54–9. [DOI] [PubMed] [Google Scholar]
- 22. Liang S, Xu Y, Shen G, Liu Q, Zhao X, Xu Z, et al. Quantitative protein expression profiling of 14–3-3 isoforms in human renal carcinoma shows 14–3-3 epsilon is involved in limitedly increasing renal cell proliferation. Electrophoresis 2009;30:4152–62. [DOI] [PubMed] [Google Scholar]
- 23. Liang S, Shen G, Liu Q, Xu Y, Zhou L, Xiao S, et al. Isoform-specific expression and characterization of 14–3-3 proteins in human glioma tissues discovered by stable isotope labeling with amino acids in cell culture-based proteomic analysis. Proteomics Clin Appl 2009;3:743–53. [DOI] [PubMed] [Google Scholar]
- 24. Qi W, Liu X, Qiao D, Martinez JD. Isoform-specific expression of 14–3-3ε proteins in human lung cancer tissues. Int J Cancer 2005;113:359–63. [DOI] [PubMed] [Google Scholar]
- 25. Gong X, Yan L, Gu H, Mu Y, Tong G, Zhang G. 14–3-3epsilon functions as an oncogene in SGC7901 gastric cancer cells through involvement of cyclin E and p27kip1. Mol Med Rep 2014;10:3145–50. [DOI] [PubMed] [Google Scholar]
- 26. Sapio L, Di Maiolo F, Illiano M, Esposito A, Chiosi E, Spina A, et al. Targeting protein kinase A in cancer therapy: an update. EXCLI J 2014;13:843–55. [PMC free article] [PubMed] [Google Scholar]
- 27. Tullai JW, Cummins PM, Pabon A, Roberts JL, Lopingco MC, Shrimpton CN, et al. The neuropeptide processing enzyme EC 3.4.24.15 is modulated by protein kinase A phosphorylation. J Biol Chem 2000;275:36514–22. [DOI] [PubMed] [Google Scholar]
- 28. Carreno FR, Goni CN, Castro LM, Ferro ES. 14–3-3 epsilon modulates the stimulated secretion of endopeptidase 24.15. J Neurochem 2005;93:10–25. [DOI] [PubMed] [Google Scholar]
- 29. Russo LC, Asega AF, Castro LM, Negraes PD, Cruz L, Gozzo FC, et al. Natural intracellular peptides can modulate the interactions of mouse brain proteins and thimet oligopeptidase with 14–3-3ε and calmodulin. Proteomics 2012;12:2641–55. [DOI] [PubMed] [Google Scholar]
- 30. Ferro ES, Carreno FR, Goni C, Garrido PA, Guimaraes AO, Castro LM, et al. The intracellular distribution and secretion of endopeptidases 24.15 (EC 3.4.24.15) and 24.16 (EC 3.4.24.16). Protein Pept Lett 2004;11:415–21. [DOI] [PubMed] [Google Scholar]
- 31. Ko BS, Chang TC, Hsu C, Chen YC, Shen TL, Chen SC, et al. Overexpression of 14–3-3ε predicts tumour metastasis and poor survival in hepatocellular carcinoma. Histopathology 2011;58:705–11. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Kong Q, Wang J, Jiang Y, Hua H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp Hematol Oncol 2020;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Santos NBD, Franco RD, Camarini R, Munhoz CD, Eichler RAS, Gewehr MCF, et al. Thimet oligopeptidase (EC 3.4.24.15) key functions suggested by knockout mice phenotype characterization. Biomolecules 2019;9:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Panchuk RR, Skorokhyd NR, Kozak YS, Lehka LV, Moiseenok AG, Stoika RS. Tissue-protective activity of selenomethionine and D-panthetine in B16 melanoma-bearing mice under doxorubicin treatment is not connected with their ROS scavenging potential. Croat Med J 2017;58:171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhinge KN, Gupta V, Hosain SB, Satyanarayanajois SD, Meyer SA, Blaylock B, et al. The opposite effects of doxorubicin on bone marrow stem cells versus breast cancer stem cells depend on glucosylceramide synthase. Int J Biochem Cell Biol 2012;44:1770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tahover E, Segal A, Isacson R, Rosengarten O, Grenader T, Gips M, et al. Dexrazoxane added to doxorubicin-based adjuvant chemotherapy of breast cancer: a retrospective cohort study with a comparative analysis of toxicity and survival. Anticancer Drugs 2017;28:787–94. [DOI] [PubMed] [Google Scholar]
- 37. Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer 2016;10:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 2014;16:876–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol 2011;21:228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park KC, Dharmasivam M, Richardson DR. The role of extracellular proteases in tumor progression and the development of innovative metal ion chelators that inhibit their activity. Int J Mol Sci 2020;21:6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue H, Lu B, Lai M. The cancer secretome: a reservoir of biomarkers. J Transl Med 2008;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duffy MJ. The role of proteolytic enzymes in cancer invasion and metastasis. Clin Exp Metastasis 1992;10:145–55. [DOI] [PubMed] [Google Scholar]
- 43. Meng T, Huang R, Zeng Z, Huang Z, Yin H, Jiao C, et al. Identification of prognostic and metastatic alternative splicing signatures in kidney renal clear cell carcinoma. Front Bioeng Biotechnol 2019;7:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Judson I, Verweij J, Gelderblom H, Hartmann JT, Schoffski P, Blay JY, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 2014;15:415–23. [DOI] [PubMed] [Google Scholar]
- 45. Van Renterghem B, Tarantola L, Wozniak A, Wellens J, Nysen M, Vanleeuw U, et al. Promising activity of an enzyme-activated doxorubicin prodrug in a panel of patient-derived xenograft models of soft tissue sarcoma. In Proceedings (Abstr. P 045) of the Connective Tissue Oncology Society Virtual 25th Annual Meeting; 2020 Nov 18–21.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.