
Figure 1: Modes of programmed cell death that contribute to neurodegenerative disease.
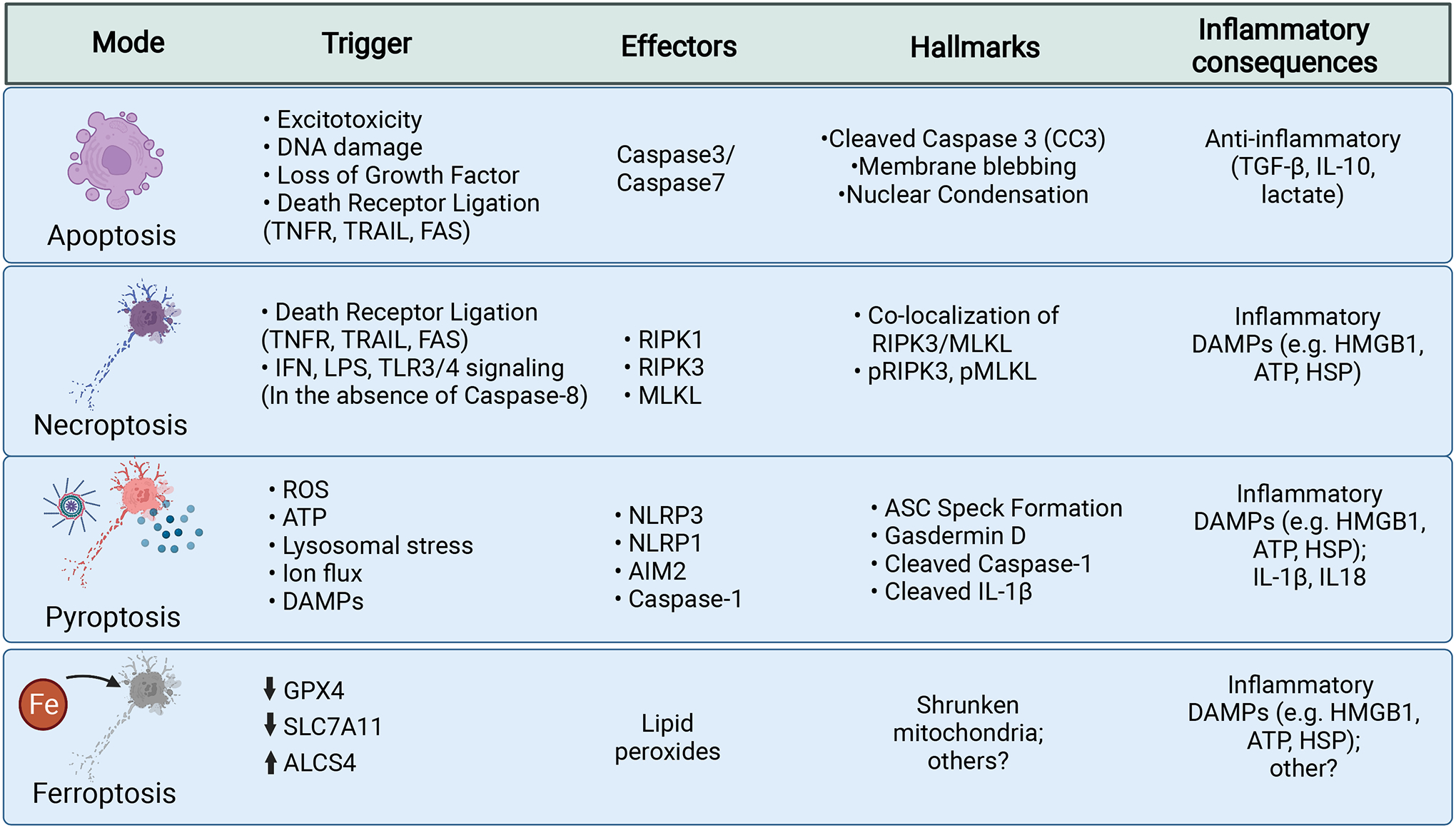
Cell death mechanisms are listed here including triggers for cell death, the molecular effectors, hallmarks of cell death, and the associated inflammatory consequences.
Apoptosis is a silent, or non-inflammatory, form of cell death that leads to cellular clearance without release of intracellular components. It is triggered by diverse stimuli and is enacted through two broad pathways: intrinsic and extrinsic apoptosis. These pathways converge on cleavage of Caspase-3. Cleaved Caspase-3 activates proteins to manifest the hallmarks of nuclear condensation (pyknosis), DNA fragmentation (karyorrhexis), cytoplasmic condensation, and membrane blebbing. Apoptosis leads to the release of TGF-β, IL-10, and lactate, which maintain homeostasis and provide anti-inflammatory signals.
Necroptosis can be initiated by several signals in the absence of Caspase-8. Canonically, necroptosis takes place downstream of death receptors (TNFR, TRAIL, FAS). These receptors recruit RIPK1, which is autophosphorylated and goes on to phosphorylate and activate RIPK3, which then acts to phosphorylate MLKL. pMLKL is then able to oligomerize into a pore like structure at the plasma membrane leading to loss of cell polarity and eventual lysis. Necroptosis is an inflammatory form of cell death that is associated with the release of intracellular contents that can act as DAMPs.
Pyroptosis similarly results in cell lysis via membrane pore formation. It is initiated by the integration of two signals. The first primes the inflammasome by producing inactive substrates. The second signal is generated by intracellular pattern recognition receptors (e.g. NLRP3, NLRP1, AIM2) that are activated by diverse triggers and go on to form inflammasome complexes. Activation of these sensors leads to recruitment of the adaptor ASC and the activation of Caspase-1 activation. Active Caspase-1 cleaves substrates including pro-IL-1β, pro-1L-18, and Gasdermin D to their active forms. Active Gasdermin D oligomerizes and forms pores in the plasma membrane, leading to release of cytokines and intracellular components, loss of membrane integrity, and eventual cell lysis. Pyroptosis is an inflammatory cell death that is associated with the release of proinflammatory cytokines and intracellular DAMPs.
Ferroptosis results from the build-up of toxic lipid hydroperoxides that form through lipids undergoing Fenton chemistry reactions with iron and interactions with other free-radicals, and also by the activity of lipoxygenase enzymes. Hydroperoxides lead to the generation of additional free-radical species that can damage lipids, proteins, and nucleic acids; although the ultimate executioner of cell death in unknown. GPX4 provides cellular defense against ferroptosis by neutralizing lipid hydroperoxides into lipid alcohols. Reductions in GPX4 or SLC7A11 activity, which provides reduced glutathione necessary for GPX4 activity, sensitizes cells to ferroptosis. Increases in the specific lipid substrates susceptible to peroxidation by enzymes such as ALCS4 can also sensitize cells. Ferroptosis is likely associated with release of intracellular contents that act as DAMPs. While, there are currently no widely used markers of ferroptosis, it has been noted that mitochondria in cells undergoing ferroptosis exhibit a fragmented/shrunken appearance, which can be evaluated through electron microscopy.