

Abstract
Objective
Ischemic stroke is a leading cause of human mortality and long‐term disability worldwide. As one of the main forms of regulator of calcineurin 1 (RCAN1), the contribution of RCAN1.4 in diverse biological and pathological conditions has been implicated. But the role of RCAN1.4 in ischemic stroke progression remains elusive. This study is to explore the expression changes and roles of RCAN1.4 in ischemic stroke as well as the underlying mechanisms for these changes and effects of RCAN1.4 in ischemic stroke.
Methods
Middle cerebral artery occlusion model in C57BL/6J mice and oxygen–glucose deprivation (OGD) model in primary astrocytes were performed to induce the cerebral ischemic stroke. The expression pattern of RCAN1.4 was assessed using real‐time quantitative PCR and western blotting in vivo and in vitro. Mechanistically, the underlying mechanism for the elevation of RCAN1.4 in the upstream was investigated. Lentiviruses were administrated, and the effect of RCAN1.4 in postischemic inflammation was clearly clarified.
Results
Here we uncovered that RCAN1.4 was dramatically increased in mouse ischemic brains and OGD‐induced primary astrocytes. HIF1α, activated upon OGD, significantly upregulated RCAN1.4 gene expression through specifically binding to the RCAN1.4 promoter region and activating its promoter activity. The functional hypoxia‐responsive element (HRE) was located between −254 and −245 bp in the RCAN1.4 promoter region. Moreover, elevated RCAN1.4 alleviated the release of pro‐inflammatory cytokines TNFα, IL1β, IL6 and reduced expression of iNOS, COX2 in primary astrocytes upon OGD, whereas RCAN1.4 silencing has the opposite effect. Of note, RCAN1.4 overexpression inhibited OGD‐induced NF‐κB activation in primary astrocytes, leading to decreased degradation of IκBα and reduced nuclear translocation of NF‐κB/p65.
Interpretation
Our results reveal a novel mechanism underscoring the upregulation of RCAN1.4 by HIF1α and the protective effect of RCAN1.4 against postischemic inflammation, suggesting its significance as a promising therapeutic target for ischemic stroke treatment.
Introduction
Stroke is a leading cause of human death and long‐term disability worldwide. 1 , 2 Ischemic stroke, the most common type of stroke, is a consequence of sudden obstruction of cerebral arteries, which leads to ischemia and hypoxia in part of brain tissue. 3 It is generally accepted that the postischemic inflammation induced by ischemic stroke contributes to brain damage and that astrocytes and microglia play vital roles in modulating inflammatory responses after ischemic stroke. 2 , 4 Although great effort has been made in the clinical therapies targetting the postischemic inflammation for ischemic stroke, 5 , 6 the prognosis is still unsatisfactory. When an ischemic stroke occurs, astrocytes become activated and secrete several kinds of pro‐inflammatory cytokines and pro‐inflammatory mediators, impairing viability and promoting apoptosis in neurons. 7 , 8 , 9 Therefore, a thorough understanding of the underlying mechanisms of ischemic stroke in astrocytes is urgent, and a more potent treatment scheme targeting astrocytic inflammation for ischemic stroke is needed.
The regulator of calcineurin 1 gene (RCAN1) was first identified as the Down syndrome critical region 1 (DSCR1) gene on chromosome 21q22, consisting of two major isoforms: the RCAN1 isoform 1 (RCAN1.1) and the RCAN1 isoform 4 (RCAN1.4). 10 , 11 , 12 RCAN1.1 has been proven to be increased in the cortex and hippocampus of Down syndrome and Alzheimer's disease, leading to neuronal apoptosis and death. 13 , 14 , 15 , 16 RCAN1.4 has been recognized as playing important roles in diverse biological and pathological conditions. According to our previous studies, RCAN1.4 suppresses lymphoma growth in mice. 17 In addition, RCAN1.4, which is downregulated in hepatocellular carcinoma, prevents the proliferation, migration, and invasion activity of cancer cells and the growth of orthotopic tumors by inhibiting nuclear translocation of NFAT1. 18 Moreover, methylation of RCAN1.4 enhanced hepatic stellate cell activation and liver fibrogenesis through calcineurin/NFAT3 signaling. 19 Xu et al. 20 reported that RCAN1.4 displays antiangiogenic properties via an NFATc1‐dependent pathway in ischemic retinal ganglion cells and that it is upregulated in atherosclerotic human tissues and contributes to atherosclerosis development. 21 Nonetheless, the expression and distribution of RCAN1.4 in the brain have not been characterized in detail, and whether it plays a role in ischemic stroke has yet to be clarified.
In this study, we investigated the expression and biological function of RCAN1.4 in ischemic stroke. We found that RCAN1.4 was markedly upregulated in mouse ischemic brain and oxygen–glucose deprivation (OGD)‐induced primary astrocytes. Mechanistically, we proved that RCAN1.4 transcription was specifically activated by HIF1α under OGD conditions through a specific hypoxia‐responsive element (HRE) in the RCAN1.4 promoter. The functional HRE was located between −254 and −245 bp in the RCAN1.4 promoter region. Moreover, RCAN1.4 exerts protective effects in astrocytes by alleviating the inflammatory response via inhibition of the NF‐κB signaling pathway after OGD treatment. These results may indicate that RCAN1.4 could be a potential therapeutic target for the treatment of ischemic stroke.
Materials and Methods
Cell culture
HEK293 cells were obtained from ATCC and cultured in high‐glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS), 100 units/mL penicillin, and 0.1 mg/mL streptomycin. Primary astrocytes were isolated from neonatal Wistar rats (0–2 days postnatal) and cultured as previously described. 22 Neonatal Wistar rats were obtained from the experimental animal center of Shandong University. Rats were anesthetized with Pentobarbital sodium before decapitation to minimize animal suffering. Briefly, rat primary astrocytes were obtained from cortical tissues and cultured in DMEM‐F12 (Gibco, containing 25 mmol/L d‐glucose) containing 10% (vol/vol) FBS in coated T25 flasks. Nonastrocytic cells (microglia and neurons) were detached from the flasks by shaking at 250 rpm for 18 h and removed by changing the medium. After 17–20 days in culture, immunofluorescence analysis and flow cytometry plots indicated that approximately 92% of the cells in the culture were GFAP (a specific marker for astrocytes) positive.
OGD of primary astrocytes
Primary astrocytes were washed twice with phosphate buffered saline (PBS, pH 7.4) and incubated in glucose‐free DMEM. Primary astrocytes were placed in a hypoxia chamber with 95% N2/5% CO2/1% O2 for indicated times at 37°C. Nontreated cells were incubated in normal DMEM for the same period of time.
Focal cerebral ischemic stroke model of mice
Male C57BL/6J mice (20–25 g, 6–8 weeks) were purchased from the Experimental Animal Center of Shandong University (Shandong, China). All the procedures were approved by the Institutional Animal Care and Use Committee of Shandong University according to the guidelines of the National Institutes of Health on the care and use of animals. Middle cerebral artery occlusion (MCAO) surgery was performed following previously established protocols 23 and the suture was carefully removed after 1.5 h of occlusion. The animals were awake during the occlusion. The Sham group underwent all surgical procedures but without the suture insertion. Successful occlusion was confirmed by the laser speckle contrast imaging (LSCI) and 2, 3, 5‐triphenyltetrazolium chloride (TTC) staining. The mice were anesthetized and killed at indicated times (12 and 24 h) after MCAO surgery. The brains were harvested and stored for the following studies.
Plasmid construction, transfection, and infection
The HIF1α expression plasmid pHA‐HIF1α, RCAN1.4 expression plasmid pcDNA3.1‐RCAN1.4‐6myc, and silencing plasmid were constructed as previously described. 17 , 24 A fragment of the 5′ upstream region of RCAN1.4 was amplified by PCR using the primers 5′‐CCGCTCGAGCATCGCAGAGCACTTCTC‐3′ and 5′‐CACAAGCTTGTGAAAGCGCTACAGACC‐3′ and cloned into the pGL3‐basic vector to generate pRCAN1.4 luc‐Long. Deletion plasmids pRCAN1.4 luc‐B, ‐C, and ‐D were created from pRCAN1.4 luc‐Long by utilization of restriction enzyme sites, as previously described. 25 These constructs were all cloned into pGL3‐basic as well. All constructed plasmids were confirmed by DNA sequencing. Transient transfections were performed with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Lentiviruses and infection
Recombinant lentiviruses Lenti‐RCAN1.4‐GFP, Lenti‐shRCAN1.4‐GFP, and their control viruses were purchased from Vigene Bio. Inc. (Shandong, China). Primary astrocytes were infected at 10 MOI after 5 days of culture in vitro. The medium was replaced with a fresh medium within 24 h, and the cells were incubated for another 5 days before experiments.
siRNA assay
HIF1α siRNA (siHIF1α; 5′CCAGCAGACUCAAAUACAATT‐3′ and 5′‐UUGUAUUUGAGUCUGCUGGTT‐3′) or its negative control (siCON) was transfected into HEK293 cells with Lipofectamine 2000 transfection reagent, and the cells were harvested at 48 h after transfection to detect HIF1α protein by western blotting to determine the efficiency of HIF1α siRNA. pRCAN1.4 luc‐Long was cotransfected with siHIF1α or siCON into HEK293 cells using Lipofectamine 2000 according to the manufacturer's instructions. At 48 h after transfection, the cells were treated with or without OGD for 6 h. Dual luciferase assay was performed to assess promoter activity.
Dual luciferase assay
A dual luciferase assay was performed at 48 h after transfection using a dual luciferase reporter assay system (Promega, Madison, WI, USA), as previously described. 26
EMSA and ChIP
Electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) were performed as previously described. 27 The sense sequences for RCAN1.4‐HRE, RCAN1.4‐HRE mut, consensus HRE (cons. HRE), and consensus HRE mut (cons. HREmut) oligonucleotides were 5′‐ GGTGTTGACGTCACCTCTTT‐3′, 5′‐GGTGTTGAAAACACCTCTTT‐3′, 5′‐AGCTTGCCCTACGTGCTGTCTCAGA‐3′, and 5′‐AGCTTGCCCTAAAAGCTGTCTCAG A‐3′, respectively. RCAN1.4‐HRE and cons. HRE oligonucleotides were both end‐labeled with IRDye 800 to generate double‐stranded probes. The binding of HIF1α to the RCAN1.4 promoter in nuclear extracts was confirmed using a ChIP assay kit (#17–371, Millipore, Burlington, MA, USA) following the manufacturer's protocol. HEK293 cells transfected with the HIF1α expression plasmid pHA‐HIF1α were cross‐linked with formaldehyde (final concentration of 1%) for 10 min at 37°C and then washed twice with cold PBS. The cells were lysed in 1% SDS lysis buffer and sheared by sonication. Proteins and DNA were pulled down with an anti‐HIF1α monoclonal antibody (sc‐13515, Santa Cruz Biotechnology, Dallas, TX, USA); anti‐RNA polymerase II and normal mouse IgG were used as positive and negative controls, respectively. The primers used for ChIP‐PCR were 5′‐CGGCCTTAAAGGGGCCAC‐3′ and 5′‐TGTCAGCAGTCTCCCAGCG‐3′.
Real‐time quantitative PCR
Total RNA was isolated from astrocytes using an RNA fast extraction kit (Cat# 220010, Fastagen, Shanghai, China). Real‐time amplification was achieved using the ABI 7900HT Fast real‐time PCR system (Applied Biosystems, Foster City, CA, USA). TaqMan probes for RCAN1.4 were obtained from Applied Biosystems (TaqMan Gene Expression Assays). The mRNA levels of other genes were quantified by SYBR® Green‐based gene expression analysis. The primers employed for real‐time quantitative PCR (RT‐qPCR) were as follows: rat TNFα (5′‐GTAGCCCACGTCGTAGCAAA‐3′ and 5′‐GGTGAGGAGCACGTAGTCG‐3′), rat IL1β (5′‐GACTTCACCATGGAACCCGT‐3′ and 5′‐CAGGGAGGGAAACACACGTT‐3′), rat IL6 (5′‐TCCTACCCCAACTTCCAATGC‐3′ and 5′‐GGTCTTGGTCCTTAGCCACT‐3′), and rat β‐actin (5′‐CGCGAGTACAACCTTCTTGC‐3′ and 5′‐CGTCATCCATGGCGAACTGG‐3′).
Western blotting and antibodies
Western blotting was performed as previously described. 28 Primary antibodies against HIF1α (sc‐13515, Santa Cruz Biotechnology), RCAN1.4 (DCT3), COX2 (#12282, Cell Signaling, Danvers, MA, USA), iNOS (18985‐1‐AP, Proteintech, Wuhan, China) pIKKβ (#2694, Cell Signaling), IKKβ (#8943, Cell Signaling), pIκBα‐S32/36 (#9246, Cell Signaling), IκBα (#4814, Cell Signaling), NF‐κB/p65 (#8242, Cell Signaling), and β‐actin (A5441, Sigma‐Aldrich, Shanghai, China) were used according to the manufacturer's instructions. Detection and quantification were achieved with the LI‐COR odyssey imaging system (LI‐COR, Lincoln, NE, USA) and Image J software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
Immunofluorescence analysis of cultured primary astrocytes was performed as previously described. 28 Rabbit anti‐GFAP mAb (#80788, Cell Signaling) and CoraLite488‐conjugated Affinipure goat antirabbit IgG (SA00013‐2, Proteintech) were applied to detect GFAP. Rabbit anti‐NF‐κB/p65 mAb (#8242, Cell Signaling) and CoraLite594‐conjugated Affinipure goat antirabbit IgG (SA00013‐4, Proteintech) were applied to detect NF‐κB/p65. Images were captured with an LSM 880 fluorescence microscope (Carl Zeiss, Jena, Germany) and analyzed with ZEN software.
ELISA
Cultured primary astrocytes were treated with or without OGD for 6 h, and the cell culture supernatant was harvested 12 h later. TNFα, IL1β, and IL6 levels in the primary astrocyte supernatant were analyzed with ELISA kits (Thermo Fisher, Shanghai, China) according to the manuals provided by the manufacturer.
Data analysis
Data are presented as means ± SEM from three to five independent experiments. Differences between two groups were evaluated by Student's t test, and those among more than two groups were assessed with one‐way or two‐way ANOVA with Bonferroni's multiple comparison post hoc test. Differences were considered statistically significant at p < 0.05. All analyses were performed with Prism (GraphPad Software, Inc., San Diego, CA, USA).
Results
RCAN1.4 was sharply upregulated in mouse ischemic brain and OGD‐induced primary astrocytes
Previous reports have shown that RCAN1.4 expression in human tissues occurs predominantly in the heart and muscle. 29 However, the expression and function of RCAN1.4 in the brain have not been characterized in detail. According to previous studies, RCAN1 was mainly in neurons and astrocytes in the brain. 30 Thus, to investigate the cellular localization of RCAN1.4 in the brain, we examined its expression in cultured primary neurons and astrocytes. Using a modified protocol, we purified primary astrocytes, obtaining approximately 92% purity, as depicted in the representative GFAP immunofluorescence and flow cytometry plots (Fig. S1A and B). RT‐qPCR and western blotting results showed RCAN1.4 to be mainly expressed in astrocytes, compared with that in primary neurons (Fig. 1A and B). To evaluate whether RCAN1.4 is regulated in mouse ischemic brain in vivo, the ischemic mice model of MCAO was performed and the time course of RCAN1.4 expression after MCAO (12 and 24 h) was determined. The LSCI and TTC staining were detected to confirm the MCAO model of ischemic stroke (Fig. 1C and D). The mRNA and protein levels of RCAN1.4 were detected by RT‐qPCR and western blotting. The results showed that the mRNA level of RCAN1.4 was sharply increased to 252.8 ± 48.2% at 12 h and 551.8 ± 48.2% at 24 h after MCAO, compared with the Sham group (Fig. 1E; p = 0.0393 and 0.0002, respectively). Similar results were obtained in the protein level of RCAN1.4. RCAN1.4 proteins were significantly elevated to 256.3 ± 15.3% at 12 h and 274.6 ± 15.3% at 24 h after MCAO, compared with the Sham group (Fig. 1F and G; p = 0.0001 and p < 0.0001, respectively). These results indicated that the mRNA and protein expressions of RCAN1.4 are associated with ischemic stroke in vivo.
Figure 1.
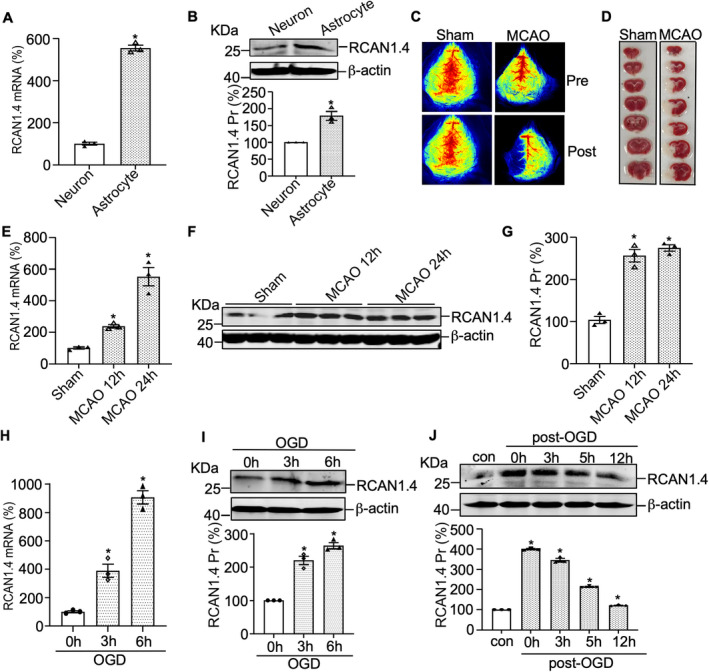
RCAN1.4 is upregulated in mouse ischemic brain and OGD‐induced primary astrocytes. (A and B) RCAN1.4 mRNA and protein in primary astrocytes and primary neurons were detected by RT‐qPCR and western blotting. 18S was used as the loading control for RT‐qPCR. β‐actin was used as the loading control for western blotting. (C and D) Male C57BL/6J mice were subjected to MCAO surgery for indicated times (12 and 24 h) and LSCI and TTC staining were performed to confirm the MCAO model of ischemic stroke. (E–G) The mRNA and protein levels of RCAN1.4 were determined in the brain tissues from different MCAO (12 and 24 h) and Sham groups. 18S was used as the loading control for RT‐qPCR. β‐actin was used as the loading control for western blotting. (H and I) Primary astrocytes were treated with OGD for the indicated times (0–6 h), and the mRNA and protein levels of RCAN1.4 were detected by RT‐qPCR and western blotting. 18S was used as the loading control for RT‐qPCR. β‐actin was used as the loading control for western blotting. (J) Primary astrocytes were treated with OGD for 6 h and then moved to normal conditions for the indicated times. RCAN1.4 protein levels were examined by western blotting, and β‐actin was used as the loading control. Values represent means ± SEM (n = 3), *p < 0.05, as calculated by Student's t test (for A and B) or one‐way ANOVA with Bonferroni's multiple comparison post hoc test (for E and G–J). OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1; RT‐qPCR, real‐time quantitative PCR; MCAO, middle cerebral artery occlusion; LSCI, laser speckle contrast imaging.
To further confirm the RCAN1.4 expression in ischemic stroke in vitro, the cellular model of OGD in primary astrocytes was performed. The time course of RCAN1.4 expression under OGD conditions (OGD at 0, 3, and 6 h) was determined, and the results showed that RCAN1.4 mRNA was significantly elevated under OGD conditions in primary astrocytes (Fig. 1H, 389.7 ± 39.2% of control at OGD 3 h, p = 0.0358 and 906.9 ± 44.1% of control at OGD 6 h, p = 0.0060, respectively). Similar results were obtained for RCAN1.4 protein levels in primary astrocytes (Fig. 1I, 220.1 ± 12.5% of control at OGD 3 h, p = 0.0213 and 264.2 ± 9.2% of control at OGD 6 h, p = 0.0063, respectively). To further assess the change in RCAN1.4 after OGD treatment, we treated primary astrocytes with OGD for 6 h and then moved the cells to normal conditions at various times. The protein levels of RCAN1.4 peaked at OGD 6 h and rapidly declined to basal levels after returning to normal conditions (Fig. 1J). Taken together, these results demonstrated the significant upregulation of RCAN1.4 by an ischemic stroke in vivo and in vitro.
HIF1α specifically activates the RCAN1.4 promoter
According to previous studies, OGD promotes HIF1α expression in astrocytes, and several target genes of HIF1α under OGD conditions have been reported. 24 , 31 , 32 , 33 To uncover the molecular mechanism of RCAN1.4 gene transcription under OGD conditions, a 1200‐bp fragment from the 5′ UTR of RCAN1.4 (pRCAN1.4 luc‐Long) was amplified by PCR, cloned into the pGL3‐basic vector and functionally analyzed. The RCAN1.4 ATG translation start codon was set as +1. The cloned fragment was transfected into HEK293 cells, which were subjected to OGD for 0, 3, and 6 h after 48 h transfection. HIF1α protein expression was upregulated to 333.9 ± 19.7% of control at OGD 3 h and 571.0 ± 36.8% of control at OGD 6 h (Fig. 2A; p = 0.0142 and p = 0.0122, respectively), and dual luciferase assay revealed that the relative luciferase activity of pRCAN1.4 luc‐Long was also increased by OGD, consistent with results of HIF1α protein (Fig. 2B, from 24.1 RLU to 53.2 RLU at OGD 3 h, p = 0.0094 and from 24.1 RLU to 88.7 RLU at OGD 6 h, p = 0.0043 respectively). To confirm the relationship between HIF1α and RCAN1.4 gene transcription, pRCAN1.4 luc‐Long was cotransfected into HEK293 cells together with siHIF1α or siCON, and a dual luciferase assay was performed with or without OGD at 48 h after transfection. The RLU of pRCAN1.4 luc‐Long was significantly decreased compared with that of siCON (Fig. 2C, from 69.6 RLU to 44.5 RLU, p = 0.0098) and the knockdown of siHIF1α was confirmed by western blotting (Fig. 2D; p < 0.0001). The above results suggest that HIF1α specifically activates the RCAN1.4 promoter under OGD conditions.
Figure 2.
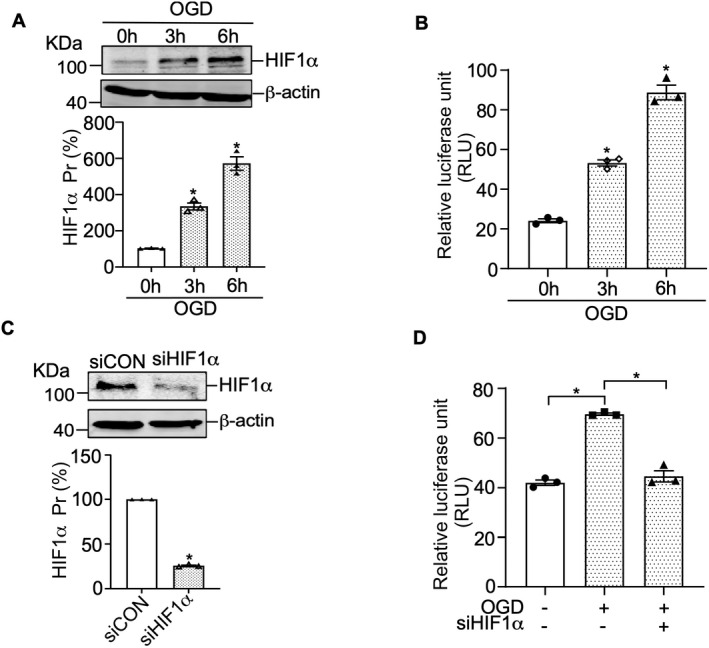
HIF1α specifically activates the RCAN1.4 promoter. (A) HEK293 cells were subjected to OGD for the indicated times, and HIF1α protein levels were examined by western blotting using β‐actin as the loading control. (B) The RCAN1.4 promoter constructs pRCAN1.4 luc‐Long, containing the 5′‐UTR fragment from human RCAN1.4, was transfected into HEK293 cells. The cells were subjected to the indicated duration of OGD, and dual luciferase activity was measured with a luminometer. (C) HEK293 cells were transfected with siHIF1α or negative control (siCON) and harvested at 48 h after transfection to detect HIF1α protein levels by western blotting using β‐actin as the loading control. (D) The RCAN1.4 promoter constructs pRCAN1.4 luc‐Long was cotransfected with siHIF1α or siCON into HEK293 cells. At 48 h after transfection, the cells were treated with or without OGD for 6 h, and dual luciferase activity was measured. Values represent means ± SEM (n = 3), *p < 0.05, as calculated by Student's t test (for C) or one‐way ANOVA with Bonferroni's multiple comparison post hoc test (for A, B, and D). OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
Identification of HRE site in the RCAN1.4 promoter
The HIF1α protein is a member of the hypoxia‐inducible factor 1 (HIF1) transcription factor family and regulates target genes by binding to hypoxia‐responsive elements (HREs). 24 , 32 To further investigate the location of the HRE in the RCAN1.4 promoter, a series of truncation plasmids were constructed by deletion of pRCAN1.4 luc‐Long, as follows: pRCAN1.4 luc‐B (−860 to −15 bp), pRCAN1.4 luc‐C (−576 to −15 bp), and pRCAN1.4 luc‐D (−315 to −15 bp) (Fig. 3A). Each deletion construct was transfected into HEK293 cells with the HIF1α expression plasmid, and a dual luciferase assay was performed at 48 h after transfection. According to the results, HIF1α significantly increased the RCAN1.4 promoter activity of the pRCAN1.4 luc‐Long, pRCAN1.1 luc‐B, pRCAN1.1 luc‐C, and pRCAN1.1 luc‐D constructs (Fig. 3B), suggesting that region −315 to −15 bp of the RCAN1.4 promoter contains the HRE site. Computer‐based sequence analysis of this −315 to −15 bp fragment showed a putative HRE site at −254 to −245 bp. To verify this result, the mutation of pRCAN1.4 luc‐D (pRCAN1.4 luc‐Dmut) were constructed by substitution of the HRE wild‐type sequence with an HRE mutant sequence (Fig. 3C). Compared with the empty vector, the RLU of pRCAN1.4 luc‐D, but not that of pRCAN1.4 luc‐D mut, was significantly elevated by HIF1α (Fig. 3C, from 38.4 RLU to 68.9 RLU for pRCAN1.4 luc‐D, p < 0.0001). These results demonstrate that the HRE at −254 to −245 bp is the functional HRE site in the RCAN1.4 promotor.
Figure 3.
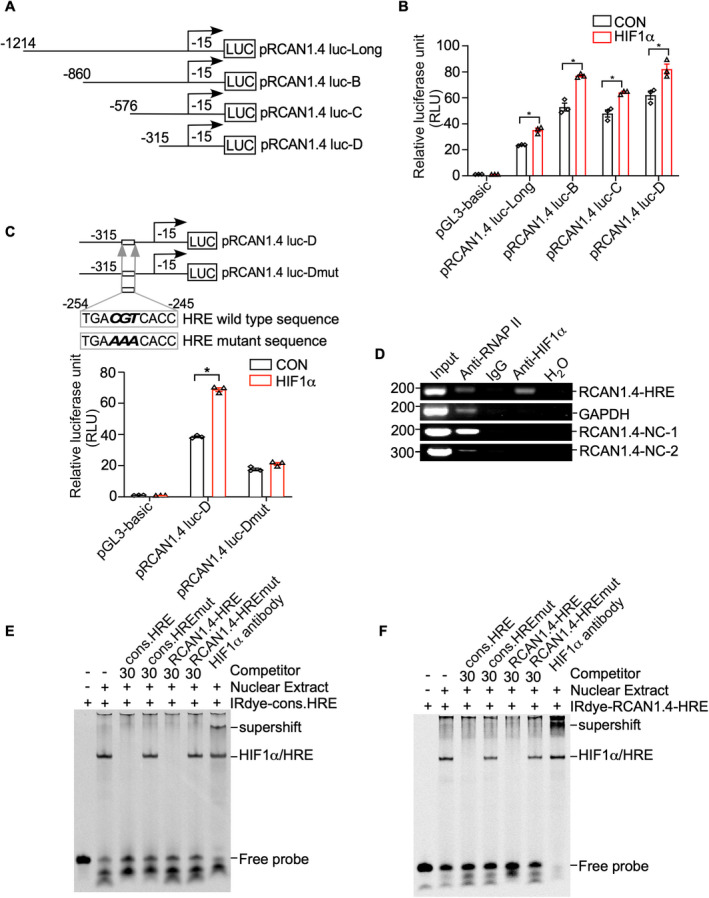
Identification of the HRE site in the RCAN1.4 promoter fragment. (A) Schematic diagrams of human RCAN1.4 promoter truncation constructs. Four different lengths of the 5′‐flanking region of the human RCAN1.4 promoter fragment were cloned into the pGL3‐basic plasmid upstream of the luciferase reporter gene (Luc). The arrow indicates the direction of transcription. The numbers represent the end points of each construct. (B) RCAN1.4 promoter truncation constructs were transfected into HEK293 cells with either the HIF1α expression plasmid (pHA‐HIF1α) or an empty vector. A dual luciferase assay was performed 48 h after transfection. pGL3‐basic was used as the negative control. (C) Comparison of the mutant and wild‐type sequences of the putative RCAN1.4‐HRE site in the RCAN1.4 promotor region. pRCAN1.4 luc‐D and pRCAN1.4 luc‐Dmut were transfected with the HIF1α expression vector or empty vector into HEK293 cells. pGL3‐basic was used as a negative control. A dual luciferase assay was performed 48 h after transfection. (D) DNA and protein from HEK293 cells were cross‐linked and immunoprecipitated with anti‐RNA polymerase II antibody (anti‐RNAPII), anti‐HIF1α antibody, and antimouse IgG, respectively. The nonimmunoprecipitated sample was used as an input. PCR was performed with the input, precipitated chromatin samples, and H2O to amplify a short DNA fragment spanning the putative RCAN1.4‐HRE site in the RCAN1.4 promoter region (panel 1), a short fragment in the GAPDH gene (panel 2), a nonspecific DNA fragment upstream RCAN1.4‐HRE (RCAN1.4‐NC‐1, panel 3) and a nonspecific DNA fragment downstream RCAN1.4‐HRE (RCAN1.4‐NC‐2, panel 4). Input and anti‐RNA polymerase II antibodies (anti‐RNAPII) were used as positive controls. IgG and H2O were used as negative controls. (E) EMSA was carried out with IRDye800‐labeled consensus HRE oligonucleotides (IRDye‐cons. HRE). The free probe is shown in lane 1. The addition of the nuclear extract shifted the probe band (lane 2). Competition assays were performed with a 30‐fold excess of unlabeled consensus HRE (cons. HRE, lane3), mutant consensus HRE (cons. HREmut, lane 4), putative RCAN1.4 promoter HRE (RCAN1.4‐HRE, lane 5), and mutant putative RCAN1.4 promoter HRE (RCAN1.4‐HREmut, lane 6). A supershift assay was carried out with an anti‐HIF1α antibody (lane 7). (F) IRDye800‐labeled HRE site in the human RCAN1.4 promoter (IRDye‐RCAN1.4‐HRE) was employed. The free probe is shown in lane 1. The addition of nuclear extract shifted the probe band (lane 2). Competition assays were carried out with a 30‐fold excess of unlabeled consensus HRE (cons. HRE, lane3), mutant consensus HRE (cons. HREmut, lane 4), and putative RCAN1.4 promoter HRE (RCAN1.4‐HRE, lane 5), and mutant putative RCAN1.4 promoter HRE (RCAN1.4‐HREmut, lane 6). A supershift assay was carried out with an anti‐HIF1α antibody (lane 7). Values represent means ± SEM (n = 3), *p < 0.05, as calculated by two‐way ANOVA with Bonferroni's multiple comparison post hoc test. HRE, hypoxia‐responsive element; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
ChIP assay was then conducted to further address whether HIF1α binds to the putative HRE site. The cross‐linked HIF1α‐DNA complex was immunoprecipitated by anti‐HIF1α antibody; IgG and H2O were used as negative controls and an anti‐RNA polymerase II antibody as a positive control. The ChIP‐PCR results showed that the RCAN1.4‐HRE fragment was amplified in the presence of the anti‐HIF1α and anti‐RNA polymerase II antibodies but not IgG or H2O (Fig. 3D). Two nonspecific sequences located at approximately 1 kb upstream and 1 kb downstream RCAN1.4‐HRE were named RCAN1.4‐NC‐1 and RCAN1.4‐NC‐1, respectively. Neither of them was amplified by HIF1α antibody immunoprecipitation (Fig. 3D, lane 4 of panels 3–4), indicating the specific binding of HIF1α/RCAN1.4‐HRE complex. These results further demonstrate that HIF1α binds to the HRE at −254 to −245 bp in the RCAN1.4 promoter region.
EMSA was performed to further confirm binding between HIF1α and −254 to −245 bp in the RCAN1.4 promoter fragment. On DNA polyacrylamide gel electrophoresis (PAGE) gel, free probe was the consensus HRE oligonucleotides, which was synthesized and labeled with IRDye800 (IRDye‐cons.HRE) (Fig. 3E, lane 1). The band was shifted above after adding a nuclear extract of HEK293 cells transfected with HIF1α expression plasmid due to the binding of the free probe and transcript factor (Fig. 3E, lane 2). To ensure the specificity of the HIF1α/HRE interaction, unlabeled oligonucleotides of the consensus HRE sequence (cons. HRE) and mutant consensus HRE sequence (cons. HREmut) were performed. As expected, unlabeled consensus HRE oligonucleotides at a 30‐fold excess successfully competed with the labeled probe (Fig. 3E, lane 3), whereas mutant consensus HRE oligonucleotides did not (Fig. 3E, lane 4). In addition, to confirm the binding between HIF1α and this putative HRE located in the RCAN1.4 gene promoter, unlabeled oligonucleotides of RCAN1.4‐HRE and mutant RCAN1.4‐HRE (RCAN1.4‐HREmut) were conducted with a 30‐fold excess of labeled probe. The results suggested that RCAN1.4‐HRE, but not RCAN1.4‐HREmut, successfully competed out the shifted HIF1α/ HRE complex band (Fig. 3E, lanes 5 and 6). Anti‐HIF1α antibody was conducted to further confirm the specific binding of HIF1α/RCAN1.4‐HRE complex (Fig. 3E, lane 7).
Similar results were obtained when using IRDye800‐labeled RCAN1.4‐HRE oligonucleotides (IRDye800‐RCAN1.4‐HRE) as the probe (Fig. 3F), which shifted after incubation with the nuclear extract of HEK293 cells transfected with the HIF1α expression plasmid, as shown in Figure 3F (lane 2). The intensity of the shifted band was markedly reduced after the addition of unlabeled consensus HRE oligonucleotides and unlabeled RCAN1.4‐HRE (Fig. 3F, lanes 3 and 5). However, no obvious competition was observed with the addition of either the mutant consensus HRE or mutant RCAN1.4‐HRE (Fig. 3F, lanes 4 and 6). As above, a further shift of the band was observed with the application of the anti‐HIF1α antibody (Fig. 3F, lane 7). These results demonstrate that HIF1α binds to the HRE in the RCAN1.4 promoter region. Taken together, our EMSA and ChIP results clearly confirm that HIF1α binds to the specific HRE site (−254 to −245 bp) in the RCAN1.4 promoter region.
RCAN1.4 alleviates the inflammatory response after OGD treatment in primary astrocytes
Astrocytes activated by ischemia and anoxia secrete many pro‐inflammatory factors and pro‐inflammatory mediators, such as TNFα, IL1β, and IL6, which affect neuronal dysfunction and death. 9 , 34 To investigate the functional role of RCAN1.4 in ischemic stroke, we infected primary astrocytes with Lenti‐RCAN1.4‐GFP, Lenti‐shRCAN1.4‐GFP, and their negative controls for 5 days. The efficiency of viral infection in primary astrocytes was confirmed based on the fluorescence of Lenti‐RCAN1.4‐GFP/Lenti‐shRCAN1.4‐GFP (Fig. S2A and C) and western blotting of the RCAN1.4 protein (Fig. S2B and D). In primary astrocytes, the mRNA levels of TNFα, IL1β, and IL6 increased significantly at 12 h after OGD treatment, and these effects were markedly alleviated by RCAN1.4 overexpression (Fig. 4A, lane 4 vs 3; p = 0.0041 for TNFα, p = 0.0032 for IL1β, and p = 0.0180 for IL6, respectively) but exacerbated by RCAN1.4 knockdown (Fig. 4D, lane 4 vs 3; p = 0.0343 for TNFα, p = 0.0148 for IL1β, and p = 0.0039 for IL6, respectively). Furthermore, we determined the effect of RCAN1.4 on the presence of TNFα, IL1β, and IL6 in the cell culture supernatant. The results showed elevated secretion of TNFα, IL1β, and IL6 in the supernatant at 12 h after OGD treatment, which was inhibited by overexpression of RCAN1.4 (Fig. 4B, lane 4 vs 3; p = 0.0229 for TNFα, p = 0.0136 for IL1β, and p = 0.0218 for IL6, respectively) but was further increased by knockdown of RCAN1.4 in OGD‐stimulated primary astrocytes (Fig. 4E, lane 4 vs 3; p = 0.0117 for TNFα, p = 0.0278 for IL1β, and p = 0.0015 for IL6, respectively). To further confirm the influence of RCAN1.4 on ischemic stroke progression, expression of COX2 and iNOS in unstimulated and OGD‐treated primary astrocytes was detected by western blotting. The results showed that under OGD conditions, expression of COX2 and iNOS in Lenti‐RCAN1.4‐GFP‐treated astrocytes was much lower than that in control (Lenti‐CON‐GFP)‐treated astrocytes (Fig. 4C, lane 4 vs 3; p = 0.0021 for COX2 and p = 0.0037 for iNOS), and Lenti‐shRCAN1.4‐GFP‐treated astrocytes displayed significantly higher levels of COX2 and iNOS than control (Lenti‐shCON‐GFP)‐treated cells (Fig. 4F, lane 4 vs 3; p = 0.0041 for COX2 and p = 0.0025 for iNOS). Taken together, these results indicate that RCAN1.4 alleviates the inflammatory response induced by OGD in primary astrocytes.
Figure 4.
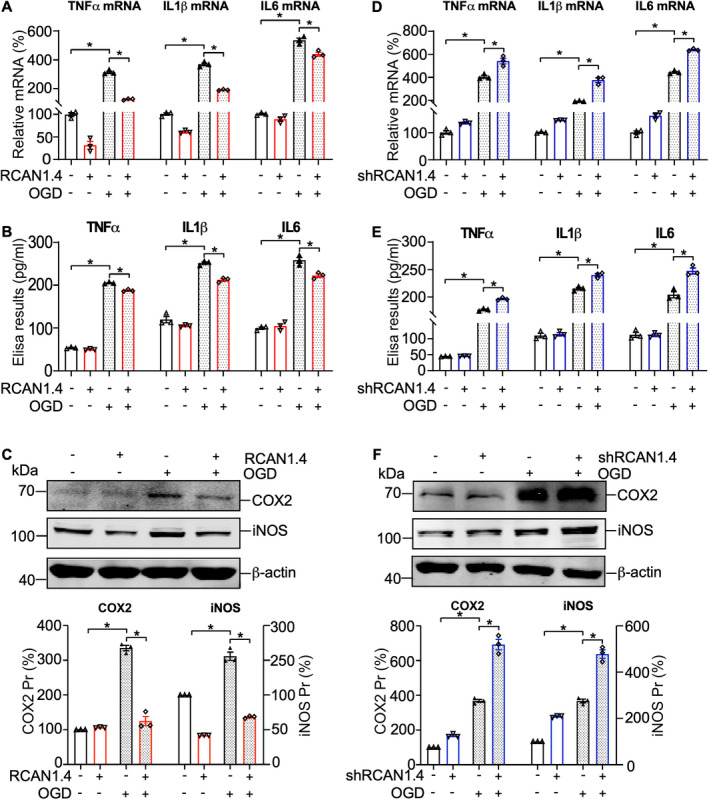
RCAN1.4 alleviates the inflammatory response after OGD treatment in primary astrocytes. (A and D) Lenti‐RCAN1.4‐GFP or Lenti‐shRCAN1.4‐GFP‐infected astrocytes were treated with OGD for 6 h and harvested 12 h later. The mRNA levels of TNFα, IL1β, and IL6 in astrocytes were determined by RT‐qPCR. (B and E) Secreted TNFα, IL1β, and IL6 levels in the cell culture supernatant of Lenti‐RCAN1.4‐GFP or Lenti‐shRCAN1.4‐GFP‐infected astrocytes were measured using ELISA kits. (C and F) COX2 and iNOS proteins were detected by western blotting in primary astrocytes after different treatments. β‐actin was used as a loading control. Values represent means ± SEM (n = 3), *p < 0.05, as calculated by one‐way ANOVA with Bonferroni's multiple comparison post hoc test. OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
RCAN1.4 inhibits NF‐κB/p65 nuclear translocation by elevating the expression of IκBα in primary astrocytes
Nuclear factor κ‐light‐chain‐enhancer of activated B cells (NF‐κB) is a key transcription factor that is activated in ischemic stroke, accompanied by the release of inflammatory cytokines and chemokines. 35 , 36 To elucidate the molecular mechanism of RCAN1.4's effect on the inflammatory response, we analyzed the impact of RCAN1.4 on the NF‐κB signaling in primary astrocytes. The results showed that the canonical NF‐κB signaling pathway was significantly activated by OGD treatment, indicated by the upregulated protein levels of pIKKβ and pIκBα‐S32/36 and enhanced degradation of IκBα (an inhibitor protein of NF‐κB/p65) (Fig. 5A and E). Overexpression of RCAN1.4 inhibited the OGD‐upregulated pIKKβ and pIκBα‐S32/36, leading to decreased degradation of IκBα in primary astrocytes (Fig. 5A–D, lane 4 vs 3; p = 0.0117 for pIKKβ/IKKβ, p = 0.0326 for pIκBα‐S32/36, and p = 0.0365 for IκBα, respectively) and the opposite effect was observed when RCAN1.4 was knocked down (Fig. 5E–H, lane 4 vs 3; p = 0.0298 for pIKKβ/IKKβ, p = 0.0008 for pIκBα‐S32/36, and p = 0.0390 for IκBα, respectively). In addition, Liu et al. 17 have demonstrated that in the H2O2‐induced HEK293 cells, RCAN1.4 overexpression increased the protein of IκBα by decreasing pIκBα‐Y42, but not pIκBα‐S32/S36. To further clarify the underlying mechanism under OGD conditions, we also detected the pIκBα‐Y42 in the OGD‐induced astrocytes. The results showed increased pIκBα‐Y42 under OGD conditions and RCAN1.4 overexpression inhibited this increase by OGD (Fig. S3A, lane 4 versus 3; p = 0.0289), while RCAN1.4 knockdown exacerbated this increase (Fig. S3B, lane 4 versus 3; p = 0.0384). These results imply that RCAN1.4 inhibits the phosphorylation of IκBα at both pIκBα‐S32/S36 and pIκBα‐Y42 under OGD conditions. Importantly, nuclear NF‐κB/p65 was sharply increased after OGD treatment, compared with that in the control group (non‐OGD treatment) (Fig. 6A and B, lane 7 versus 5) and RCAN1.4 overexpression inhibited the NF‐κB/p65 nuclear translocation in astrocytes subjected to OGD (Fig. 6A, lane 8 versus 7; p = 0.0022), while RCAN1.4 knockdown promoted the NF‐κB/p65 nuclear translocation (Fig. 6B, lane 8 versus 7; p = 0.0009). To confirm the function of RCAN1.4 on NF‐κB/p65, dual luciferase activity controlled by NF‐κB was assessed under normal and OGD conditions, and similar results were obtained (Fig. 6C and D). Additionally, immunofluorescence analysis of NF‐κB/p65 in primary astrocytes was also performed to consolidate the conclusion. The results showed that nuclear NF‐κB/p65 was significantly inhibited by RCAN1.4 overexpression (Fig. 6E, lane 4 versus 3; p = 0.0023), while RCAN1.4 knockdown exacerbated the nuclear NF‐κB/p65 (Fig. 6F, lane 4 versus 3; p = 0.0050). Therefore, RCAN1.4 alleviates the OGD‐induced inflammatory response by inhibiting the NF‐κB/p65 nuclear translocation in primary astrocytes (Fig. 7).
Figure 5.
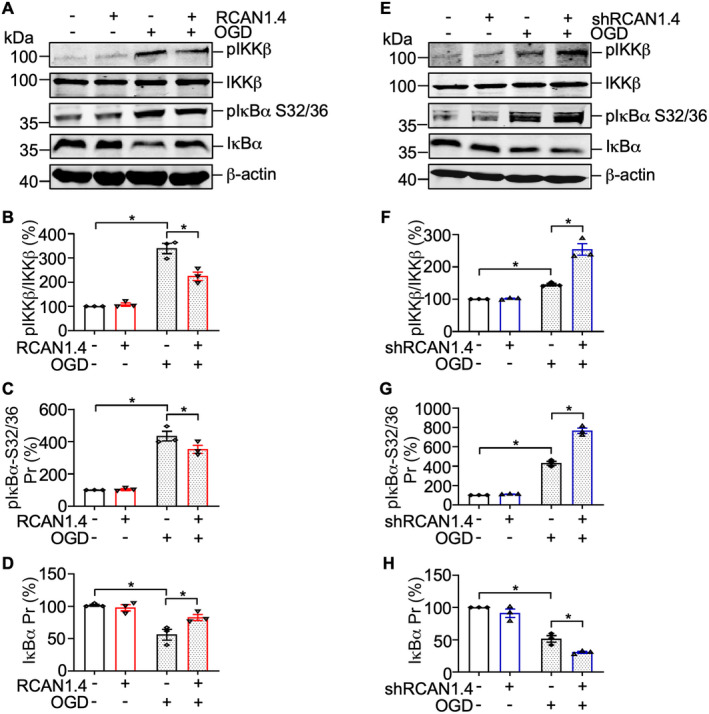
RCAN1.4 inhibits the degradation of IκBα in OGD‐induced primary astrocytes. (A) Cultured primary astrocytes infected with Lenti‐CON‐GFP or Lenti‐RCAN1.4‐GFP for 5 days were treated with or without OGD for 6 h. The pIKKβ, IKKβ, pIκBα‐S32/36, and IκBα were detected by western blotting. β‐actin was used as a loading control. (B–D) The quantification of A. (E) Cultured primary astrocytes infected with Lenti‐shCON‐GFP or Lenti‐shRCAN1.4‐GFP for 5 days were treated with or without OGD for 6 h. The pIKKβ, IKKβ, pIκBα‐S32/36, and IκBα were detected by western blotting. β‐actin was used as a loading control. (F–H) The quantification of E. Values represent means ± SEM (n = 3), *p < 0.05, as calculated by one‐way ANOVA with Bonferroni's multiple comparison post hoc test. OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
Figure 6.
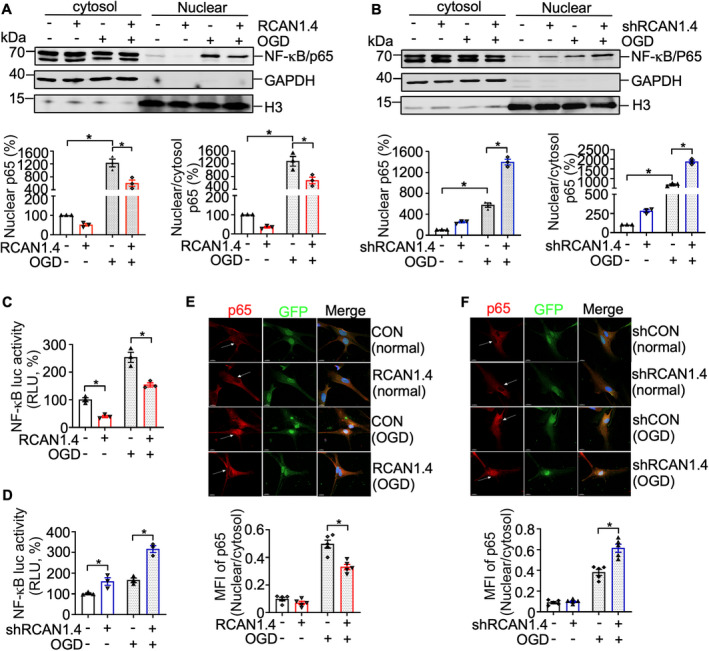
RCAN1.4 suppresses OGD‐induced NF‐κB/p65 nuclear translocation in primary astrocytes. (A and B) Cultured primary astrocytes infected with Lenti‐RCAN1.4‐GFP or Lenti‐shRCAN1.4‐GFP for 5 days were treated with or without OGD for 6 h. Nuclear proteins were extracted from cells infected with Lenti‐RCAN1.4‐GFP or Lenti‐shRCAN1.4‐GFP. NF‐κB/p65 proteins were detected using an anti‐p65 antibody. GAPDH and H3 were used as a loading control for the cytosolic and nuclear fractions, respectively. (C and D) The RCAN1.4 expression construct or silencing plasmid was cotransfected with pNF‐κBLuc into HEK293 cells. A dual luciferase assay was performed at 12 h after OGD treatment. (E and F) Cultured primary astrocytes infected with Lenti‐RCAN1.4‐GFP or Lenti‐shRCAN1.4‐GFP for 5 days were treated with or without OGD for 6 h. The cells were fixed and permeated, then stained with anti‐p65 (Alexa Fluor 594, red) and DAPI for the nucleus (blue). The image was captured with Leica confocal microscopy (LSM880). Scale bar 10 μm. Values represent means ± SEM (n = 3), *p < 0.05, as calculated by one‐way ANOVA with Bonferroni's multiple comparison post hoc test. MFI, mean fluorescence intensity; OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
Figure 7.
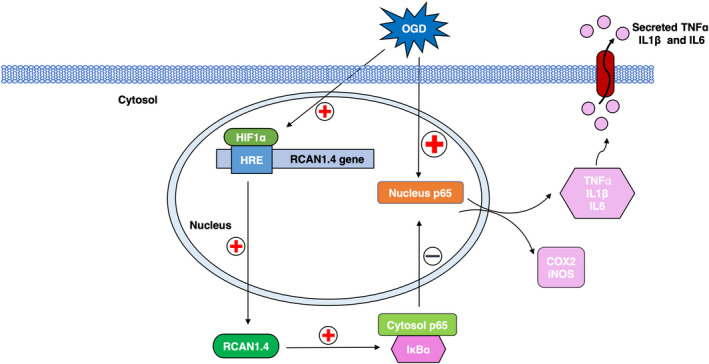
Schematic diagram of enhanced RCAN1.4 by HIF1α in astrocytes under OGD conditions. RCAN1.4 inhibits postischemic inflammation in astrocytes via inhibition of the NF‐κB signaling pathway. Activation of OGD results in upregulation of HIF1α in the nucleus, and HIF1α interacts with the specific HRE in the RCAN1.4 promoter, initiating its transcription. By inhibiting the phosphorylation and degradation of IκBα protein, RCAN1.4 suppresses NF‐κB/p65 nuclear translocation, subsequently alleviating postischemic inflammation in astrocytes. OGD, oxygen–glucose deprivation; RCAN1.4, the isoform 4 of regulator of calcineurin 1.
Discussion
Ischemic stroke, accounting for about 6.5 million deaths every year, has been one of the most common causes of adult death and disability in the world. 37 Although great breakthroughs have been made in the diagnosis technology and new clinical therapies for ischemic stroke, the prognosis is still unsatisfactory and the underlying mechanisms of ischemic stroke are controversial. RCAN1 consists of two major isoforms: RCAN1.1 and RCAN1.4. According to our previous studies, RCAN1.1 has been proved to be upregulated in the brain of Down syndrome and Alzheimer's disease, inducing neuronal apoptosis. 16 Recently, we reported that RCAN1.1 is a novel RNA‐binding protein, increasing the adenine nucleotide translocator 1 mRNA, to regulate mitochondrial dysfunctions and neuronal apoptosis. 26 , 38 However, the distribution and function of RCAN1.4 in the brain have not been characterized in detail, and the effect of RCAN1.4 on ischemic stroke pathogenesis has not been clearly clarified.
In the present study, we demonstrated that RCAN1.4 was significantly upregulated in the infarct region of the MCAO model of ischemic stroke. Cho et al. also reported the elevation of RCAN1 in ischemic stroke and RCAN1 was distributed both in neurons and astrocytes. 30 However, due to the lack of a specific antibody for RCAN1.4, the distribution of RCAN 1.4 in the brain was not been clearly clarified. We purified primary neurons and primary astrocytes and found that the expression of RCAN1.4 in primary astrocytes was much more than that in primary neurons in the brain. Furthermore, we proved that RCAN1.4 was also sharply increased in the OGD‐induced primary astrocytes. Why was RCAN1.4 so markedly elevated in ischemic stroke? In ischemic stroke pathogenesis, the sudden obstruction of criminal arteries leads to the ischemia and hypoxia of involved brain tissue. Hypoxia regulates gene transcription by stabilizing HIF1 (hypoxia‐inducible factor 1) and HIF1α is the functional subunit, which binds to the specific HRE in the promoter region of the target genes. 24 Several target genes of HIF1α involved in central nervous system diseases have been reported. 31 , 33 , 39 Our previous study found that HIF1α positively regulates the expression of BACE1 under hypoxic conditions and facilitates Alzheimer's disease pathogenesis. 24 Furthermore, VEGF expression induced by HIF1α causes vascular leakage in the brain. 39 In ischemic stroke, Lipocalin‐2 is upregulated by hypoxia, contributing to the inflammatory response in astrocytes. 31 In this study, we speculate that RCAN1.4 is a novel target gene of HIF1α and is significantly upregulated by HIF1α under OGD conditions. To further confirm this, we investigated the specific HRE in the RCAN1.4 promoter and found that the functional HRE was located between −254 and −245 bp in the RCAN1.4 promoter region.
Emerging evidence suggests that the inflammatory response, mediated by microglia and astrocytes, plays a vital role in ischemic stroke and has been considered to be one of the most important targets for new therapies for stroke. 2 , 4 , 40 According to previous studies, astrocytes are the most abundant cell type in the brain, providing structural support and releasing beneficial factors that maintain brain cell development and extracellular environment homeostasis. 34 , 40 , 41 Astrocytes remain in a resting state under normal conditions; but under pathological conditions, such as ischemic stroke, these cells are activated and secrete inflammatory factors such as TNFα, IL1β, and IL6 into the lesions, directly or indirectly contributing to brain damage aggravation, neuron dysfunction, microglial activation, and peripheral immune cell recruitment. 9 , 34 , 40 , 42 Previous studies have demonstrated that RCAN1.4 helps to maintain a more fused mitochondrial network in cardiomyocytes during ischemia/reperfusion (I/R) damage. 43 According to our data, overexpression of RCAN1.4 attenuates the release of pro‐inflammatory cytokines, such as TNFα, IL1β, and IL6, as well as pro‐inflammatory mediators (COX2 and iNOS), in astrocytes after OGD treatment, indicating a protective role in ischemic stroke. Nevertheless, further research needs to investigate the effect of RCAN1.4 on postischemic inflammation, long‐term survival, and functional recovery outcomes in an animal model of ischemic stroke.
It is well established that NF‐κB is activated in the pathological process of ischemic stroke, 35 , 36 regulating the transcriptional expression of a panel of inflammatory cytokines and chemokines, such as TNFα, IL1β, and IL6. 44 , 45 The nuclear translocation of the NF‐κB/p65 subunit in the canonical pathway is one of the most direct and appropriate indicators for evaluating the activation and functional status of the NF‐κB pathway. 46 Thus, to further elucidate the mechanism responsible, the canonical NF‐κB signaling was detected in the OGD‐induced primary astrocytes and we found that OGD activated the canonical NF‐κB signaling via increased pIKKβ and pIκBα‐S32/S36, while RCAN1.4 inhibited the OGD‐upregulated pIKKβ and pIκBα‐S32/S36, leading to decreased degradation of IκBα and negative regulation of NF‐κB/p65 nuclear translocation in primary astrocytes. Moreover, according to our previous study, Liu et al. 17 have demonstrated that in the H2O2‐induced HEK293 cells, RCAN1.4 overexpression increased the protein of IκBα by decreasing pIκBα‐Y42, but not pIκBα‐S32/S36. To further clarify the underlying mechanism under OGD conditions, the pIκBα‐Y42 in the OGD‐induced astrocytes was also detected and the results showed that OGD significantly increased the pIκBα‐Y42 and RCAN1.4 overexpression inhibited this increase by OGD, implying that RCAN1.4 inhibits the phosphorylation of IκBα at both pIκBα‐S32/S36 and pIκBα‐Y42 under OGD conditions. However, further investigation would be needed to elucidate the molecular mechanism between OGD and NF‐κB signaling as well as how RCAN1.4 interferes with this interaction. In addition, our previous study has demonstrated that the transcription of RCAN1.4 is upregulated by NF‐κB through an NF‐κB responsive element in the RCAN1.4 promoter, 25 thereby forming a negative feedback loop of RCAN1.4 and NF‐κB. Taken together, the upregulated RCAN1.4 by HIF1α inhibits the NF‐κB signaling, and the NF‐κB signaling upregulates RCAN1.4 expression, further amplifying the negative function of RCAN1.4 on the inflammatory response in OGD‐induced astrocytes.
According to previous reports, RCAN1.4 not only can suppress calcineurin/NFAT signaling pathway 47 but also can be activated by the target genes of calcineurin/NFAT pathway such as calcium ionophore, VEGF, angiotensin II, TNFα, and so on, 48 , 49 forming another negative feedback loop for RCAN1.4 regulation. We also detect the NFAT activity after RCAN1.4 overexpression or knockdown using the pIL2Luc containing NFAT responsive elements, and the results showed that the NFAT activity was sharply decreased after RCAN1.4 overexpression under OGD conditions, which was confirmed by RCAN1.4 knockdown (Fig. S4A and B). These results imply that under OGD conditions, the calcineurin/NFAT may be another underlying pathway that has important functions in OGD‐induced inflammation. It is acknowledged that activated astrocytes display spatiotemporally dynamic Ca2+ signals including amplitude, duration, and frequency. 50 , 51 The difference in the activation of NF‐κB and NFAT signaling can be explained by the amplitude and duration of intracellular Ca2+ concentration, leading to different transcriptional activation. 52 , 53 , 54 NFAT activation requires lower intracellular Ca2+ concentration than NF‐κB, but this low intracellular Ca2+ concentration rise needs to be present for an extended time. However, NF‐κB is selectively activated by one or a few transient large Ca2+ rises.52, 53, 54 Remarkably, a single Ca2+ “spike” is sufficient to initiate IκBα degradation and NF‐κB/p65 nuclear translocation to activate transcription of its target genes. Taken together, the two negative feedback loops in RCAN1.4‐NF‐κB and RCAN1.4‐NFAT indicate the vital role of RCAN1.4 in inflammatory response (Fig. S5), and further investigations in vivo are badly needed to clarify the effect and underlying mechanism of RCAN1.4 regulation in ischemic stroke.
In summary, our study demonstrates that RCAN1.4 is significantly upregulated in both cellular and animal models of ischemic stroke. The upregulation of RCAN1.4 inhibits the NF‐κB signaling pathway under OGD conditions and subsequently alleviates the postischemic inflammation. Furthermore, RCAN1.4 is a novel target gene of HIF1α and HIF1α activates RCAN1.4 expression by binding to the specific HRE (−254 and −245 bp) in the RCAN1.4 promoter under OGD conditions. Our research suggests a novel mechanism of RCAN1.4 regulation in ischemic stroke and further highlights the potential use of RCAN1.4 in interventions for ischemic stroke. Further research using animal models of ischemic stroke will provide a more detailed view of the vital effects of RCAN1.4 in astrocytes, especially with regard to postischemic inflammation, long‐term survival, and functional recovery outcomes.
Conflict of Interest
The authors declare that they have no conflict of interest with the contents of this article.
Author Contributions
X. Y. and X. S. conceived and designed the study. X. Y. and Y. Y. performed the study. X. S. and P. W. revised the article for intellectual content. X. Y. and J. Z. performed data statistics and analysis. X. Y. wrote the article. All authors read and approved the final manuscript.
Supporting information
Figure S1. Representative immunofluorescence images and flow cytometry plots indicated the purity of astrocytes.
Figure S2. Representative immunofluorescence images indicated the infection efficiency of lentiviruses and RCAN1.4 protein expression was confirmed by western blotting.
Figure S3. RCAN1.4 inhibits the protein level of pI κ B α‐Y42 in primary astrocytes under OGD conditions.
Figure S4. RCAN1.4 inhibits the NFAT activity under OGD conditions.
Figure S5. Schematic diagram of the two negative feedback loops of RCAN1.4 regulation.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant 91849130).
Annals of Clinical and Translational Neurology 2022;9(8): 1224–1240
Funding Information
This work was supported by the National Natural Science Foundation of China (grant 91849130).
Funding Statement
This work was funded by National Natural Science Foundation of China grant 91849130.
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiulian Sun (xiulians@gmail.com).
References
- 1. Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14(11):1363‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest. 2020;130(6):2777‐2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barker‐Collo S, Starkey N, Lawes C, Feigin V, Senior H, Parag V. Neuropsychological profiles of 5‐year ischemic stroke survivors by Oxfordshire stroke classification and hemisphere of lesion. Stroke. 2012;43(1):50‐55. [DOI] [PubMed] [Google Scholar]
- 4. Famakin BM. The immune response to acute focal cerebral ischemia and associated post‐stroke immunodepression: a focused review. Aging Dis. 2014;5(5):307‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lapchak PA, Zhang JH. The high cost of stroke and stroke cytoprotection research. Transl Stroke Res. 2017;8(4):307‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54(1):34‐66. [DOI] [PubMed] [Google Scholar]
- 7. Giffard R, Swanson R. Ischemia‐induced programmed cell death in astrocytes. Glia. 2005;50(4):299‐306. [DOI] [PubMed] [Google Scholar]
- 8. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiese S, Karus M, Faissner A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol. 2012;3:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fuentes JJ, Pritchard MA, Planas AM, Bosch A, Ferrer I, Estivill X. A new human gene from the Down syndrome critical region encodes a proline‐rich protein highly expressed in fetal brain and heart. Hum Mol Genet. 1995;4(10):1935‐1944. [DOI] [PubMed] [Google Scholar]
- 11. Fuentes JJ, Pritchard MA, Estivill X. Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics. 1997;44(3):358‐361. [DOI] [PubMed] [Google Scholar]
- 12. Reeves RH, Baxter LL, Richtsmeier JT. Too much of a good thing: mechanisms of gene action in Down syndrome. Trends Genet. 2001;17(2):83‐88. [DOI] [PubMed] [Google Scholar]
- 13. Wu Y, Ly PT, Song W. Aberrant expression of RCAN1 in Alzheimer's pathogenesis: a new molecular mechanism and a novel drug target. Mol Neurobiol. 2014;50(3):1085‐1097. [DOI] [PubMed] [Google Scholar]
- 14. Wu Y, Zhang S, Xu Q, et al. Regulation of global gene expression and cell proliferation by APP. Sci Rep. 2016;6:22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ermak G, Morgan TE, Davies KJ. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem. 2001;276(42):38787‐38794. [DOI] [PubMed] [Google Scholar]
- 16. Sun X, Wu Y, Chen B, et al. Regulator of calcineurin 1 (RCAN1) facilitates neuronal apoptosis through caspase‐3 activation. J Biol Chem. 2011;286(11):9049‐9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu C, Zheng L, Wang H, Ran X, Liu H, Sun X. The RCAN1 inhibits NF‐kappaB and suppresses lymphoma growth in mice. Cell Death Dis. 2015;6:e1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin H, Wang C, Jin G, et al. Regulator of calcineurin 1 gene isoform 4, down‐regulated in hepatocellular carcinoma, prevents proliferation, migration, and invasive activity of cancer cells and metastasis of orthotopic tumors by inhibiting nuclear translocation of NFAT1. Gastroenterology. 2017;153(3):799‐811.e33. [DOI] [PubMed] [Google Scholar]
- 19. Pan XY, You HM, Wang L, et al. Methylation of RCAN1.4 mediated by DNMT1 and DNMT3b enhances hepatic stellate cell activation and liver fibrogenesis through Calcineurin/NFAT3 signaling. Theranostics. 2019;9(15):4308‐4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Y, Yang B, Hu Y, et al. Secretion of Down syndrome critical region 1 isoform 4 in ischemic retinal ganglion cells displays anti‐angiogenic properties via NFATc1‐dependent pathway. Mol Neurobiol. 2017;54(8):6556‐6571. [DOI] [PubMed] [Google Scholar]
- 21. Mendez‐Barbero N, Esteban V, Villahoz S, et al. A major role for RCAN1 in atherosclerosis progression. EMBO Mol Med. 2013;5(12):1901‐1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCarthy KD, de Vellis J. Alpah‐adrenergic receptor modulation of beta‐adrenergic, adenosine and prostaglandin E1 increased adenosine 3′:5′‐cyclic monophosphate levels in primary cultures of glia. J Cyclic Nucleotide Res. 1978;4(1):15‐26. [PubMed] [Google Scholar]
- 23. Liu H, Wu X, Luo J, et al. Pterostilbene attenuates astrocytic inflammation and neuronal oxidative injury after ischemia‐reperfusion by inhibiting NF‐kappaB phosphorylation. Front Immunol. 2019;10:2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun X, He G, Qing H, et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up‐regulating BACE1 gene expression. Proc Natl Acad Sci USA. 2006;103(49):18727‐18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng L, Liu H, Wang P, Song W, Sun X. Regulator of calcineurin 1 gene transcription is regulated by nuclear factor‐kappaB. Curr Alzheimer Res. 2014;11(2):156‐164. [DOI] [PubMed] [Google Scholar]
- 26. Yun Y, Zhang Y, Zhang C, et al. Regulator of calcineurin 1 is a novel RNA‐binding protein to regulate neuronal apoptosis. Mol Psychiatry. 2021;26(4):1361‐1375. [DOI] [PubMed] [Google Scholar]
- 27. Lu M, Zheng L, Han B, et al. REST regulates DYRK1A transcription in a negative feedback loop. J Biol Chem. 2011;286(12):10755‐10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu H, Wang P, Song W, Sun X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone‐mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009;23(10):3383‐3392. [DOI] [PubMed] [Google Scholar]
- 29. Peiris H, Keating DJ. The neuronal and endocrine roles of RCAN1 in health and disease. Clin Exp Pharmacol Physiol. 2018;45(4):377‐383. [DOI] [PubMed] [Google Scholar]
- 30. Cho KO, Kim YS, Cho YJ, Kim SY. Upregulation of DSCR1 (RCAN1 or Adapt78) in the peri‐infarct cortex after experimental stroke. Exp Neurol. 2008;212(1):85‐92. [DOI] [PubMed] [Google Scholar]
- 31. Taklimie FR, Gasterich N, Scheld M, et al. Hypoxia induces astrocyte‐derived lipocalin‐2 in ischemic stroke. Int J Mol Sci. 2019;20(6):1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5(6):437‐448. [DOI] [PubMed] [Google Scholar]
- 33. Engelhardt S, Huang SF, Patkar S, Gassmann M, Ogunshola OO. Differential responses of blood‐brain barrier associated cells to hypoxia and ischemia: a comparative study. Fluids Barriers CNS. 2015;12:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bylicky M, Mueller G, Day R. Mechanisms of endogenous neuroprotective effects of astrocytes in brain injury. Oxid Med Cell Longev. 2018;2018:6501031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harari OA, Liao JK. NF‐kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ridder DA, Schwaninger M. NF‐kappaB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995‐1006. [DOI] [PubMed] [Google Scholar]
- 37. Khandelwal P, Yavagal D, Sacco R. Acute ischemic stroke intervention. J Am Coll Cardiol. 2016;67(22):2631‐2644. [DOI] [PubMed] [Google Scholar]
- 38. Jiang H, Zhang C, Tang Y, et al. The regulator of calcineurin 1 increases adenine nucleotide translocator 1 and leads to mitochondrial dysfunctions. J Neurochem. 2017;140(2):307‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schoch HJ, Fischer S, Marti HH. Hypoxia‐induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125(Pt 11):2549‐2557. [DOI] [PubMed] [Google Scholar]
- 40. Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol. 2012;814:3‐7. [DOI] [PubMed] [Google Scholar]
- 42. Gelderblom M, Weymar A, Bernreuther C, et al. Neutralization of the IL‐17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793‐3802. [DOI] [PubMed] [Google Scholar]
- 43. Parra V, Altamirano F, Hernandez‐Fuentes CP, et al. Down syndrome critical region 1 gene, Rcan1, helps maintain a more fused mitochondrial network. Circ Res. 2018;122(6):e20‐e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zusso M, Lunardi V, Franceschini D, et al. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF‐kB pathway. J Neuroinflammation. 2019;16(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Y, Chen N, Wu C, et al. Galectin‐1 attenuates neurodegeneration in Parkinson's disease model by modulating microglial MAPK/IkappaB/NFkappaB axis through its carbohydrate‐recognition domain. Brain Behav Immun. 2020;83:214‐225. [DOI] [PubMed] [Google Scholar]
- 46. Pradere JP, Hernandez C, Koppe C, Friedman RA, Luedde T, Schwabe RF. Negative regulation of NF‐kappaB p65 activity by serine 536 phosphorylation. Sci Signal. 2016;9(442):ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441(7093):595‐600. [DOI] [PubMed] [Google Scholar]
- 48. Stathatos N, Bourdeau I, Espinosa AV, et al. KiSS‐1/G protein‐coupled receptor 54 metastasis suppressor pathway increases myocyte‐enriched calcineurin interacting protein 1 expression and chronically inhibits calcineurin activity. J Clin Endocrinol Metab. 2005;90(9):5432‐5440. [DOI] [PubMed] [Google Scholar]
- 49. Minami T, Horiuchi K, Miura M, et al. Vascular endothelial growth factor‐ and thrombin‐induced termination factor, Down syndrome critical region‐1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279(48):50537‐50554. [DOI] [PubMed] [Google Scholar]
- 50. Shigetomi E, Saito K, Sano F, Koizumi S. Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int J Mol Sci. 2019;20(4):996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Talbot JD, David G, Barrett EF, Barrett JN. Calcium dependence of damage to mouse motor nerve terminals following oxygen/glucose deprivation. Exp Neurol. 2012;234(1):95‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dolmetsch R, Lewis R, Goodnow C, Healy J. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386(6627):855‐858. [DOI] [PubMed] [Google Scholar]
- 53. Berry CT, May MJ, Freedman BD. STIM‐ and Orai‐mediated calcium entry controls NF‐kappaB activity and function in lymphocytes. Cell Calcium. 2018;74:131‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Quintana A, Griesemer D, Schwarz EC, Hoth M. Calcium‐dependent activation of T‐lymphocytes. Pflugers Arch. 2005;450(1):1‐12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative immunofluorescence images and flow cytometry plots indicated the purity of astrocytes.
Figure S2. Representative immunofluorescence images indicated the infection efficiency of lentiviruses and RCAN1.4 protein expression was confirmed by western blotting.
Figure S3. RCAN1.4 inhibits the protein level of pI κ B α‐Y42 in primary astrocytes under OGD conditions.
Figure S4. RCAN1.4 inhibits the NFAT activity under OGD conditions.
Figure S5. Schematic diagram of the two negative feedback loops of RCAN1.4 regulation.
Data Availability Statement
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiulian Sun (xiulians@gmail.com).