

Abstract
Phylogenetic analysis has been widely used to describe, display, and infer the evolutionary patterns of viruses. The unprecedented accumulation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomes has provided valuable materials for the real-time study of SARS-CoV-2 evolution. However, the large number of SARS-CoV-2 genome sequences also poses great challenges for data analysis. Several methods for subsampling these large data sets have been introduced. However, current methods mainly focus on the spatiotemporal distribution of genomes without considering their genetic diversity, which might lead to post-subsampling bias. In this study, a subsampling method named covSampler was developed for the subsampling of SARS-CoV-2 genomes with consideration of both their spatiotemporal distribution and their genetic diversity. First, covSampler clusters all genomes according to their spatiotemporal distribution and genetic variation into groups that we call divergent pathways. Then, based on these divergent pathways, two kinds of subsampling strategies, representative subsampling and comprehensive subsampling, were provided with adjustable parameters to meet different users’ requirements. Our performance and validation tests indicate that covSampler is efficient and stable, with an abundance of options for user customization. Overall, our work has developed an easy-to-use tool and a webserver (https://www.covsampler.net) for the subsampling of SARS-CoV-2 genome sequences.
Keywords: SARS-CoV-2, phylogenetics, subsampling
Introduction
As of 9 July 2022, coronavirus disease 2019 (COVID-19) has caused more than 550 million confirmed cases and more than 6 million deaths globally (https://covid19.who.int/). The etiologic agent of COVID-19 is severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). To monitor the prevalence of this virus globally, SARS-CoV-2 genomes have been extensively sequenced worldwide. As of July 2022, millions of SARS-CoV-2 genomes have been submitted to public databases such as the National Center for Biotechnology Information (NCBI) GenBank and the Global Initiative on Sharing All influenza Data (GISAID) (Elbe and Buckland‐Merrett 2017; Shu and McCauley 2017; Khare et al. 2021). This large set of continuously sequenced genome data provides unprecedented materials for the real-time study of viral evolution. Several nomenclature systems for SARS-CoV-2 variants have been introduced to classify and track these variants, such as Nextstrain clades (Hadfield et al. 2018) and Phylogenetic Assignment of Named Global Outbreak (Pango) lineages (Rambaut et al. 2020).
Phylogenetic analysis has been frequently used by researchers to describe, display, and infer the evolutionary pattern of SARS-CoV-2 (Fauver et al. 2020; Gonzalez-Reiche et al. 2020; Alpert et al. 2021; Washington et al. 2021; Zeller et al. 2021). However, the large amount of SARS-CoV-2 sequence data poses great challenges for phylogenetic analysis (Frost et al. 2015; McBroome et al. 2021; Morel et al. 2021; Hodcroft et al. 2021a). For example, the alignment of a data set with millions of sequences, let alone the reconstruction of a phylogenetic tree with so many tips, will call for a large amount of computational resources (Hodcroft et al. 2021a). To date, the obstacles presented by this large amount of viral data have been solved mainly by subsampling the data set (Ladner et al. 2020; Jackson et al. 2021; Lemieux et al. 2021; Planas et al. 2021; Wu et al. 2021; Yaglom et al. 2021; Hodcroft et al. 2021b). However, phylogenetic tree reconstruction is highly dependent on the input data, and subsampled data sets obtained with different strategies might produce different phylogenetic trees and thus inconsistent results. Moreover, the geographic bias of the ongoing accumulation of SARS-CoV-2 sequencing data caused by disparities in resources for genomic sequencing worldwide (Fig. 1A), the different geographic distributions of variants (Fig. 1B), and genetic diversity among sequences (Fig. 1C) are factors to be considered during subsampling. Therefore, reasonable and effective subsampling methods for SARS-CoV-2 genomes are urgently needed.
Figure 1.

Overview of 4,947,593 SARS-CoV-2 genomes collected from 23 December 2019 to 24 June 2022 in NCBI GenBank. (A) Spatiotemporal distribution of SARS-CoV-2 genomes. The y-axis is log-transformed. (B) Geographic distribution of SARS-CoV-2 variants. (C) Accumulation and diversity of mutations in SARS-CoV-2 genomes. Blue dots indicate the mean number of mutations in genomes. Shaded regions indicate the SD from the mean. These mutations are relative to the reference genome, Wuhan-Hu1/2019 (GenBank accession: NC_045512) (Wu et al. 2020).
Recently, several tools have been developed to facilitate subsampling from the large viral data set. Nextstrain (Hadfield et al. 2018) developed a hierarchical subsampling method that divides sequences into groups by geographic and temporal distribution. Another program, named genome-sampler (Bolyen et al. 2020), supports the sampling of collections of viral genomes across multiple variables, including time of genome isolation, location of genome isolation, and viral diversity. In addition, Nybbler is another tool for subsampling SARS-CoV-2 genomes (https://github.com/nodrogluap/nybbler). However, these methods mainly focus on the spatiotemporal distribution of genomes or consider this distribution separately from genetic characteristics, which might lead to post-subsampling bias. In addition, these methods either support the adjustment of only a few parameters or require large amounts of computational resources and time, which restrict their wide application.
Here, we developed a subsampling method, named covSampler, based on the spatiotemporal distribution and genetic variation of SARS-CoV-2 genomes and provided a web application of covSampler (https://www.covsampler.net) for subsampling the SARS-CoV-2 genomes deposited in NCBI GenBank. covSampler includes three main steps (Fig. 2A): (1) Determination of sites of spreading mutations of SARS-CoV-2 and subsequent construction of haplotype sequences. (2) Construction of divergent pathways. The divergent pathways are defined as groups containing genomes with close spatiotemporal distribution and genetic similarity. Each divergent pathway reflects the local dynamic transmission and evolution of viruses over a period of time. (3) Subsampling based on the divergent pathways. Two types of subsampling strategies, representative subsampling and comprehensive subsampling, are included in the method. With representative subsampling, a full data set with millions of genomes is subsampled to a data set containing variable numbers of genomes with a geographic distribution and genetic variation similar to the genomes in the original data set. In contrast, with comprehensive subsampling, a data set with the uniform geographic distribution across continents and relatively higher genetic diversity compared with representative subsampling is provided. Our performance and validation tests indicate that covSampler is efficient and stable, with an abundance of options for user customization. Overall, our work provides an easy-to-use tool for subsampling the SARS-CoV-2 genomes.
Figure 2.

The covSampler workflow. (A) Schematic diagram showing the major steps of covSampler. The workflow is managed using Snakemake (Mölder et al., 2021). (B) Determination of sites of spreading mutations of SARS-CoV-2. Genome sites of nonsynonymous mutations that increased in frequency per week for 4 consecutive weeks on at least one continent are defined as sites of spreading mutations. (C) Construction of divergent pathways. Small circles indicate individual viral genomes. For each pair of genomes, a link between them is constructed if they are close in space and time and genetically similar. After the construction of all links, all viruses are clustered into multiple networks, the network-connected components are interpreted as individual clusters, which are defined as divergent pathways. (D) Representative and comprehensive subsampling based on the divergent pathways. Genomes in the original data set are grouped by their continent, divergent pathway, month, and haplotype. The desired number of subsamples is then assigned according to the selected strategy.
Materials and methods
Data collection and processing
Nextclade (Aksamentov et al. 2021) is used for sequence alignment, mutation calling, Nextstrain clade assignment, and quality control for SARS-CoV-2 genomes. The reference genome is set as the default in Nextclade (Aksamentov et al. 2021), which is Wuhan-Hu-1/2019 (GenBank accession: NC_045512) (Wu et al. 2020). Genomes less than 27,000 nucleotides in length or with Nextclade-assessed errors or warnings will be removed. In addition, we provide a web application of covSampler using sequence data from NCBI GenBank. These data are updated regularly. Since covSampler relies on the geographic and temporal information of genomes, genomes without one of the following information will not be included in the covSampler web application: (1) collection time specifying year, month, and date; (2) continent; (3) country; and (4) administrative division.
In this study, we downloaded all SARS-CoV-2 genomes collected from 23 December 2019 to 24 June 2022 and their related metadata in NCBI GenBank (accessed 2 July 2022) to test the performance of covSampler. The filtering criterion for these genomes was the same as that in the covSampler web application mentioned earlier. After filtering, 4,947,593 genomes were eligible for downstream analysis.
Determination of sites of spreading mutations of SARS-CoV-2
Genome sites of nonsynonymous mutations that increased in frequency per week for 4 consecutive weeks on at least one continent are defined as sites of spreading mutations (Fig. 2B). To avoid selecting an excessive number of sites of spreading mutations, we set a minimum threshold for the absolute number of nonsynonymous mutations in each of four consecutive increasing weeks. The minimum threshold is 1/50,000 of the total number of genomes in the original data set. After identifying these sites of spreading mutations, we construct the haplotype sequence of each genome (Fig. 2A). The haplotype sequence of each genome is a short pseudosequence composed of sites of spreading mutations of the genome.
Construction of divergent pathways
Divergent pathways are defined as groups containing genomes with close spatiotemporal distribution and genetic similarity. Each divergent pathway reflects the local dynamic transmission and evolution of the virus over a period of time. First, we construct links between pairs of viral genomes by determining their relationship according to three aspects: geographic distribution, temporal distribution, and genetic similarity. Pairs of genomes collected from the same geographic administrative division, such as a province or state, are defined as close in space. Pairs of genomes collected less than or equal to 14 days apart are defined as close in time. Pairs of genomes with a Hamming distance less than or equal to 1 between their haplotype sequences are defined as genetically similar. Among all pairs of genomes, we construct links between pairs of genomes that are close in space and time and genetically similar. Then, we cluster all genomes into multiple networks based on the links between genomes, and the network connected components are interpreted as individual clusters, which are defined as divergent pathways (Fig. 2C). After the construction of the divergent pathways, genomes in divergent pathways containing fewer than three genomes are removed due to likely problematic sequencing or assembly.
Representative and comprehensive subsampling based on the divergent pathways
After constructing the divergent pathways, the subsampling procedure is performed based on these divergent pathways. First, genomes in the original data set are hierarchically divided into subsampling units, which are groups containing genomes with or within the same continent, divergent pathway, month, and haplotype (Figs 2D and S1). This procedure is performed as follows: (1) All genomes in the original data set are divided by continent. (2) Genomes in each continent are divided into divergent pathways in the continent. (3) Genomes in each divergent pathway are divided by month. (4) Genomes in each month within divergent pathways are divided by haplotype. After these procedures, all genomes are divided into multiple mutually exclusive ‘continent–divergent pathway–month–haplotype’ groups, which we defined as subsampling units. Genomes within the same continent are defined as those belonging to the same continent branch. The same logic applies to the definitions of divergent pathway branch, month branch, and haplotype branch.
Subsequently, we assign the desired total number of subsamples to the subsampling units along branches (Figs 2D and S1). In principle, we assign the desired number of subsamples from basal branches to their descended branches. We first assign the desired total number of subsamples to the descended branches of the original data set, which are the continent branches. Then, for each continent branch, we assign the desired number of subsamples of the continent branch to its descended branches, which are the divergent pathway branches. The assignment procedures are the same from divergent pathway branches to month branches and from month branches to haplotype branches. Finally, each subsampling unit is assigned a desired number of subsamples, and we take the subsamples from these subsampling units according to the assigned number.
During the assignment, representative subsampling and comprehensive subsampling are performed using different strategies. Before introducing the different strategies used by these two subsamplings, we would like to introduce an assignment approach that is widely used below, which is ‘assign in turn’. When the desired number of subsamples is assigned in turn from one basal branch to branches descending from this basal branch, these descended branches are sorted according to different criteria mentioned below. The sorted descended branches are then labeled from 1 to N, where N is the number of these descended branches. And the desired number of subsampled genomes of these descended branches is calculated as follows:
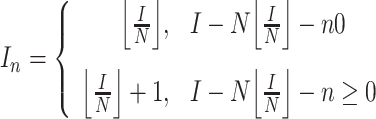 |
(1) |
where In is the desired number of subsamples of the descended branch n when assigning in turn, I is the desired number of subsamples of the basal branch, and the descended branch label from 1 to N represents the assignment priority from high to low, and the descended branches with higher priority will be preferentially assigned with the desired number of subsamples.
For representative subsampling, the assignment strategy is as follows (Fig. 2D): (1) At the continent branch level, the proportion of the desired number of subsamples assigned to each continent branch is the same as the proportion of the number of genomes in the continent branch. (2) At the divergent pathway branch level, the desired number of subsamples assigned to each divergent pathway branch is proportional to the number of genomes in the divergent pathway branch. The ratio is calculated to ensure that the desired number of subsamples assigned to each continent branch is the same as the desired number of subsamples assigned to all divergent pathway branches descending from the continent branch. (3) At the month branch level, the desired number of subsamples of month branches descending from the same divergent pathway branch is assigned in turn. The month branches descending from the same divergent pathway branch are sorted chronologically, with later months having higher priority in the assignment. (4) At the haplotype branch level, the desired number of subsamples of haplotype branches descending from the same month branch is assigned in turn. The haplotype branches descending from the same month branch are sorted according to the number of times the haplotypes are present in genomes belonging to the month branch, with haplotypes present in more genomes belonging to the month branch having higher priority in the assignment.
For comprehensive subsampling, the assignment strategy is as follows (Fig. 2D): (1) At the continent branch level, the desired number of subsamples of each continent branch is assigned in turn. The continent branches are sorted by the number of genomes they contain, with continent branches containing more genomes having higher priority in the assignment. (2) At the divergent pathway branch level, the desired number of subsamples of divergent pathway branches descending from the same continent branch is assigned in turn. The divergent pathway branches descending from the same continent branch are sorted by the number of genomes they contain, with divergent pathway branches containing more genomes having higher priority in the assignment. (3) At the month branch level, the assignment process is the same as that used in the representative subsampling strategy. (4) At the haplotype branch level, the desired number of subsamples of haplotype branches descending from the same month branch is assigned in turn. The haplotype branches descending from the same month branch are sorted by the number of mutations the haplotypes have, with haplotypes with more mutations having higher priority in the assignment.
Analysis of comprehensively and representatively subsampled genomes
We extracted comprehensively and representatively subsampled genomes with sample sizes of 500, 5,000, and 10,000 from 23 December 2019 to 24 June 2022 worldwide for subsample analysis. First, we compared the spatiotemporal and variant distributions of global genomes and subsampled genomes. Then, the diversity and consistency of mutations of global genomes and subsampled genomes were evaluated using Pearson correlation, which was calculated through the scipy.stats.pearsonr function (scipy v1.6.2). Finally, principal coordinate analysis (PCoA) of Hamming distance between haplotype sequences of subsampled genomes and analysis of the Pango lineage (with version as of 2 July 2022) coverage of the subsampled genomes were performed to evaluate the genetic differences between the genome sets generated by the two subsampling strategies. In the PCoA, the Hamming distance matrix between all comprehensively and representatively subsampled genomes with the same subsample size was constructed. Then, the PCoA assigned each virus a location in a two-dimensional space based on the distance matrix. For the Pango lineage coverage analysis, Pango lineages with genome counts less than or equal to 0.01 per cent of all genomes in the global data set were discarded. In the phylogenetic visualization section of Pango lineage coverage analysis, the phylogenetic tree summarizing the evolutionary relationships among different SARS-CoV-2 Pango lineages was downloaded from CoVizu (https://filogeneti.ca/CoVizu/) (Ferreira et al. 2021) and manipulated with ggtree (Yu et al. 2017).
Webserver construction
The covSampler webserver was constructed based on the Vue.js (https://vuejs.org/) and Node.js (https://nodejs.org/en/) frameworks. Data were stored in MongoDB (https://www.mongodb.com/). The phylogenetic tree in the webserver summarizing the evolutionary relationships among different SARS-CoV-2 Pango lineages was downloaded from CoVizu (https://filogeneti.ca/CoVizu/) (Ferreira et al. 2021) and manipulated with ggtree (Yu et al. 2017). Visualization of the phylogenetic tree was implemented with phylotree.js (Shank, Weaver, and Pond 2018). Visualization of bar graphs was implemented with ECharts (https://echarts.apache.org/en/index.html) (Li et al. 2018). The related scripts were written in Python and R and are available in our GitHub repository (https://github.com/wuaipinglab/covSampler).
Results
Performance of global comprehensive and representative subsampling
To test the performance of covSampler, 500, 5,000, and 10,000 subsampled genomes from 23 December 2019 to 24 June 2022 worldwide with comprehensive and representative characteristics were extracted for examination. Four aspects, namely, the spatiotemporal distribution, variant distribution, mutation frequencies, and genetic diversity, of these subsampled genomes were evaluated.
As shown in Fig. 3A, comprehensively subsampled genomes were evenly distributed across all continents, while the distribution of the representatively subsampled genomes was similar to that in the global genome data set. In the early period of pandemic, the temporal distribution of comprehensively subsampled genomes globally was different from that of representatively subsampled genomes and global genomes (Fig. 3B). We looked at the temporal distribution of these comprehensive subsampled genomes and global genomes across continents and noticed that the number of comprehensively subsampled genomes changed with the number of global genomes for each continent (Fig. S2). Due to the relatively large number of genomes in the early period of the pandemic on continents other than Europe and North America and the consistent number of comprehensively subsampled genomes in all continents, the number of comprehensively subsampled genomes in the early period of the pandemic was relatively large. Furthermore, the temporal distribution of representatively subsampled genomes was similar to that of the global genomes. Differences in the proportion of variants of concern (VOC) in each strategy could be detected (Fig. 3C). The proportions of Alpha, Delta, and Omicron variants in the comprehensively subsampled genomes were lower than their proportions in the original data set of global genomes, whereas variants with relatively small proportions globally but relatively high proportions in Africa and South America, such as Beta and Gamma, were present at a higher proportion in the comprehensively subsampled genomes than in the global genomes. In contrast, the distribution of VOC in the representatively subsampled genomes was similar to that in the global genome data set.
Figure 3.

Performance of global comprehensive and representative subsampling by covSampler. (A–C) Spatiotemporal and variant distribution of global SARS-CoV-2 genomes and 500, 5,000, and 10,000 comprehensively and representatively subsampled genomes. Bars represent the mean of subsampled genomes with different sample sizes. Error bars represent the SD of subsampled genomes with different sample sizes. (D) Pearson correlation of mutation frequency at each genome site between global genomes and comprehensively subsampled genomes. (E) Pearson correlation of mutation frequency at each genome site between global genomes and representatively subsampled genomes. (F) PCoA of Hamming distance between haplotype sequences of 5,000 comprehensively subsampled genomes and 5,000 representatively subsampled genomes. (G,H) Pango lineage coverage of 5,000 comprehensively subsampled genomes and 5,000 representatively subsampled genomes.
To evaluate the diversity and consistency of mutation frequency between the subsampled genomes and the global genome data set, we calculated the Pearson correlation of the mutation frequency at each genome site between subsampled genomes and global genomes. For comprehensively subsampled genomes, most genome sites with a high mutation frequency in global genomes showed a decreasing trend in mutation frequency in the subsampled genomes (Fig. 3D). For representatively subsampled genomes, the mutation frequency of each genome site in subsampled genomes was similar to its mutation frequency in the global genome data set (Fig. 3E).
In addition, the genetic diversity between comprehensively and representatively subsampled genomes was examined by calculating the Hamming distance between all haplotype sequences of comprehensively subsampled genomes and those of representatively subsampled genomes. Figure 3F displays the PCoA based on the Hamming distance matrix of 5,000 comprehensively subsampled genomes and 5,000 representatively subsampled genomes. In the two-dimensional space of PCoA, each point represents a viral genome. The differences between haplotypes of genomes are proportional to the distance in the two-dimensional space between the points, and the closeness of the points in the two-dimensional space is related to the genetic diversity of the sequences. The distribution of representatively subsampled genomes was more compact, while the distribution of comprehensively subsampled genomes was more scattered and covered areas that were not covered by the representatively subsampled genomes. The coverage of the Pango lineage, which represents a variant in the widely used SARS-CoV-2 Pango nomenclature system (Rambaut et al. 2020), was also compared between comprehensively subsampled genomes and representatively subsampled genomes. Figure 3G and H displays the coverage of the Pango lineage of 5,000 representatively subsampled genomes and 5,000 comprehensively subsampled genomes. Of all 277 Pango lineages with genome counts greater than 0.01 per cent of all genomes in the global data set, 134 were present in both subsampled genome sets, 68 were present only among the comprehensively subsampled genomes, and 31 were present only among the representatively subsampled genomes (Fig. 3G). We plotted these 277 Pango lineages in the phylogenetic tree (Fig. 3H). The Pango lineages that were present in both subsample sets covered most areas of the phylogenetic tree. However, Pango lineages that were present only in comprehensively subsampled genomes covered more area than Pango lineages that were present only in representatively subsampled genomes. We also found that for all comparisons of subsampled genomes of the same sizes, the comprehensively subsampled genomes had a wider distribution than the representatively subsampled genomes (Fig. S3). Thus, it could be concluded that both subsampling strategies worked well for subsampling, with comprehensive subsampling tending to yield higher coverage of the original data set and representative subsampling tending to yield the pivotal skeleton of the original data set.
Webserver interface and features
An online webserver (https://www.covsampler.net) was constructed to allow users to perform SARS-CoV-2 subsampling with covSampler (Fig. 4). The sequence data used in the web application are sourced from NCBI GenBank. The location, date range, variant, distribution characteristic, and desired number of subsampled genomes can be customized by users. After 3–5 min of calculation, the GenBank accessions of the subsampled genomes can be downloaded. At the same time, an interactive phylogenetic tree constructed from corresponding Pango lineages and the spatiotemporal distribution of these subsampled genomes are presented.
Figure 4.

covSampler web application interface. The user selects the location, date range, variants, subsampling characteristics, and desired subsample size, and then the GenBank accessions of subsampled genomes can be downloaded after calculation. The phylogenetic tree constructed from corresponding Pango lineages and the spatiotemporal distribution of these subsampled genomes are also presented.
Discussion
To date, numerous mutations have arisen in the SARS-CoV-2 genome. However, most of these mutations are random mutations and have little effect on the properties of the virus. These mutations are usually present at low frequency in the viral population (Martin et al. 2021; Sun et al. 2022). Some adaptive mutations may change the inherent transmissibility, immune escape ability, and other properties of the virus and have a growth advantage in population. We therefore target potential adaptive mutations by successive increases in frequency and define them as the spreading mutations. Subsequently, the haplotype sequence of each genome is constructed from the sites of spreading mutations. By combining the genetic characteristics contained in these haplotype sequences and the spatiotemporal distribution of genomes, we determined the relationships between each pair of genomes in the original data set. In brief, pairs of genomes with close spatiotemporal distribution and genetic similarity were considered to share a recent common ancestor. By connecting each pair of genomes with close spatiotemporal distribution and genetic similarity, we clustered all genomes into multiple networks and defined each network as a divergent pathway. The divergent pathways are clusters of similar genomes that are close in time and space, and each divergent pathway reflects the evolution of viruses over time. Therefore, subsampling along the divergent pathway with time and genetic variation can effectively capture the evolutionary trajectory of the viruses, and the subsamples can be used to represent the entirety of the large-scale genome data set for phylogenetic analysis.
Comprehensively subsampled genomes and representatively subsampled genomes can be extracted from the same data set by using different criteria. Comprehensively subsampled genomes are evenly distributed across each continent. We extract only a small number of genomes from each divergent pathway to ensure that the subsampled genomes come from as many divergent pathways as possible. Within each divergent pathway, genomes with more genetic variation in each month will be extracted with higher priority because these genomes may reflect a more detailed evolution process in this divergent pathway. Consequently, comprehensively subsampled genomes have a uniform geographic distribution across continents and relatively high genetic diversity. In contrast, the geographic distribution and genetic variation of representatively subsampled genomes are similar and proportional to those of the original data set. The number of subsampled genomes extracted in each divergent pathway is proportional to the number of genomes contained in that divergent pathway. Then, within each divergent pathway, genomes with the most common haplotypes in each month are preferentially extracted, which allows the subsampled genomes to reflect the dynamic evolution of viruses over time. As a result, representatively subsampled genomes can present key genetic variation contained in the original data with a reliable distribution.
Different subsampling strategies are suitable for different application scenarios. Comprehensive subsampling can provide a rich genetic background for exploring the distribution and evolution of specific viruses. In addition, more detailed information will be contained in these subsamples, which is helpful when exploring emerging variants or mutations. On the other hand, representative subsampling better reflects the actual and pivotal phylogenetic relationships and structure of the large-scale genome data set. Thus, researchers can use representatively subsampled genomes as a representative of the original data set for exploring the molecular evolution and epidemiology of viruses.
In summary, covSampler is a publicly available method for subsampling SARS-CoV-2 genomes based on geographic distribution, temporal distribution, and genetic variation. In addition, customized subsamples can be obtained under different requirements by covSampler. We have also developed a user-friendly webserver that provides an online application of covSampler to better serve the scientific community.
Supplementary Material
Acknowledgements
We gratefully acknowledge NCBI GenBank and all the authors, originating and submitting laboratories of the SARS-CoV-2 sequences for sharing their work in open databases. We would like to thank the technical support from Beijing Cloudna Technology Co., Ltd.
Contributor Information
Yexiao Cheng, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China; School of Life Science and Technology, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District Nanjing 211198, China.
Chengyang Ji, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Na Han, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Jiaying Li, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Lin Xu, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China; School of Life Science and Technology, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District Nanjing 211198, China.
Ziyi Chen, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Rong Yang, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Hang-Yu Zhou, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Aiping Wu, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, 9 Dongdan Santiao, Dongcheng District, Beijing 100730, China; Suzhou Institute of Systems Medicine, 100 Chongwen Road, Suzhou Industrial Park, Suzhou 215123, China.
Data availability
All sequence data used in this study are available at NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/). The phylogenetic tree summarizing the evolutionary relationships among different SARS-CoV-2 lineages is downloaded from CoVizu (https://filogeneti.ca/CoVizu/) (Ferreira et al. 2021). The source code of covSampler is released at GitHub under an MIT license (https://github.com/wuaipinglab/covSampler).
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
We appreciate the support we received from National key research and development program (2021YFC2301300); the CAMS Innovation Fund for Medical Sciences (2021-I2M-1-061); the National Natural Science Foundation of China (92169106); the special research fund for central universities, Peking Union Medical College (2021-PT180-001); China postdoctoral science foundation grants (2019M660548 and 2020T130007ZX); Suzhou science and technology development plan (szs2020311); and the Youthful Teacher Project of Peking Union Medical College (3332019114). Funding for open access charge: CAMS Innovation Fund for Medical Sciences.
Conflict of interest:
None declared.
Author contributions
A.W., Y.C., and H.-Y.Z. contributed to the design of this study and the manuscript draft. Y.C. and H.-Y.Z. contributed to the design of the method and webserver. C.J., N.H., and Z.C. contributed to data analysis and workflow optimization. J.L. contributed to data analysis. L.X. contributed to the design of webserver. R.Y. contributed to material preparation.
References
- Aksamentov I. et al. (2021) ‘Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes’, Journal of Open Source Software, 6: 3773. [Google Scholar]
- Alpert T. et al. (2021) ‘Early Introductions and Transmission of SARS-CoV-2 Variant B. 1.1. 7 in the United States’, Cell, 184: 2595–2604. e2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E. et al. (2020) ‘Reproducibly Sampling SARS-CoV-2 Genomes across Time, Geography, and Viral Diversity’, F1000Research, 9: 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., and Buckland‐Merrett G. (2017) ‘Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health’, Global Challenges, 1: 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J. R. et al. (2020) ‘Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States’, Cell, 181: 990–996. e995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R.-C. et al. (2021) ‘CoVizu: Rapid Analysis and Visualization of the Global Diversity of SARS-CoV-2 Genomes’, Virus Evolution, 7: veab092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost S. D. et al. (2015) ‘Eight Challenges in Phylodynamic Inference’, Epidemics, 10: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche A. S. et al. (2020) ‘Introductions and Early Spread of SARS-CoV-2 in the New York City Area’, Science, 369: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodcroft E. B. et al. (2021a) ‘Want to Track Pandemic Variants Faster? Fix the Bioinformatics Bottleneck’, Nature, 591: 30–3. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020’, Nature, 595: 707–12. [DOI] [PubMed] [Google Scholar]
- Jackson B. et al. (2021) ‘Generation and Transmission of Interlineage Recombinants in the SARS-CoV-2 Pandemic’, Cell, 184: e5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S. et al. (2021) ‘GISAID’s Role in Pandemic Response’, China CDC Weekly, 3: 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner J. T. et al. (2020) ‘An Early Pandemic Analysis of SARS-CoV-2 Population Structure and Dynamics in Arizona’, MBio, 11: e02107–02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux J. E. et al. (2021) ‘Phylogenetic Analysis of SARS-CoV-2 in Boston Highlights the Impact of Superspreading Events’, Science, 371: eabe3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2018) ‘ECharts: A Declarative Framework for Rapid Construction of Web-based Visualization’, Visual Informatics, 2: 136–46. [Google Scholar]
- Martin D. P. et al. (2021) ‘The Emergence and Ongoing Convergent Evolution of the SARS-CoV-2 N501Y Lineages’, Cell, 184: e5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBroome J. et al. (2021) ‘A Daily-updated Database and Tools for Comprehensive SARS-CoV-2 Mutation-annotated Trees’, Molecular Biology and Evolution, 38: 5819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölder F. et al. (2021) ‘Sustainable Data Analysis with Snakemake F1000Research’, F1000Research, 10: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel B. et al. (2021) ‘Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult’, Molecular Biology and Evolution, 38: 1777–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D. et al. (2021) ‘Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization’, Nature, 596: 276–80. [DOI] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank S. D., Weaver S., and Pond S. L. K. (2018) ‘Phylotree. Js-a JavaScript Library for Application Development and Interactive Data Visualization in Phylogenetics’, BMC Bioinformatics, 19: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., and McCauley J. (2017) ‘GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality’, Eurosurveillance, 22: 30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q. et al. (2022) ‘VarEPS: An Evaluation and Prewarning System of Known and Virtual Variations of SARS-CoV-2 Genomes’, Nucleic Acids Research, 50: D888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington N. L. et al. (2021) ‘Emergence and Rapid Transmission of SARS-CoV-2 B. 1.1. 7 In the United States’, Cell, 184: 2587–2594. e2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A. et al. (2021) ‘One Year of SARS-CoV-2 Evolution’, Cell Host & Microbe, 29: 503–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaglom H. D. et al. (2021) ‘Applying Genomic Epidemiology to Characterize a COVID-19 Outbreak in a Developmentally Disabled Adult Group Home Setting, Arizona’, Frontiers in Public Health, 9: 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2017) ‘GGtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
- Zeller M. et al. (2021) ‘Emergence of an Early SARS-CoV-2 Epidemic in the United States’, Cell, 184: e4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data used in this study are available at NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/). The phylogenetic tree summarizing the evolutionary relationships among different SARS-CoV-2 lineages is downloaded from CoVizu (https://filogeneti.ca/CoVizu/) (Ferreira et al. 2021). The source code of covSampler is released at GitHub under an MIT license (https://github.com/wuaipinglab/covSampler).