

Abstract
Deoxydehydration (DODH) reaction of glycerol (GL) and 1,2-propanediol (1,2-PD), in ionic liquids (ILs), catalyzed by methyltrioxorhenium (MTO) and Re2O7, was studied in detail. To better understand the ability of ILs to improve the catalytic performance of the rhenium catalyst, several experiments, employing eight different cations and two different anions, were carried out. Among the anions, bis(trifluoromethylsulfonyl)imide (TFSI) appears to be more appropriate than PF6–, for its relatively lower volatility of the resulting IL. Regarding the choice of the most appropriate cation, the presence of a single aromatic ring seems to be a necessary requirement for a satisfying and convenient reactivity. With the aim to extend the recyclability of the catalyst, experiments involving the readdition of polyol to the terminal reaction mixture were carried out. Worthy of interest is the fact that the presence of the IL prevents the inertization process of the catalyst, allowing us to obtain the alkene also after a readdition of fresh polyol.
1. Introduction
Biomass, especially which is available as a byproduct, could be used in the future both as renewable chemicals and as an alternative clean energy source.1 However, biomass-derived resources are relatively oxygen-rich, at least compared with the traditional fossil resources. This drawback can be remedied by saving the carbon skeleton of the bioresource and concurrently reducing the oxygen content.2
Among the available biomass, glycerol (GL) is probably the most representative polyol and the simplest one to employ in obtaining light intermediates with low oxygen content.3 By decreasing the number of oxygens in the GL molecule, different monomers could be obtained: from the simply dehydrated molecules such as acrolein4 and hydroxyacetone5 passing to the deoxygenated propanediols6 or the deoxydehydrated allyl alcohol (AA)7 and up to the fully deoxygenated propene8 and propane.9
In the same trend of many recent studies,10−13 also our group undertook a program focused on the study of the deoxydehydration (DODH) reaction of both GL7,14 and 1,2-propanediol (1,2-PD),8 trying to understand some mechanistic aspects also, using several rhenium catalysts. For example, when methyltrioxorhenium (MTO) is the employed catalyst, a methane release always occurs at the beginning of the reaction, with the concomitant formation of some solvent-assisted rhenium oxo-species. As also observed by Abu-Omar,15 only after a certain delay time, possibly necessary to produce the catalytically active species, AA starts to be formed.
Regarding the current state of the art of the DODH, we believe that it is still too early to consider any application to produce olefins at the industrial scale, and so, further improvements are necessary to obtain greener and more sustainable protocols. For example, it is unfeasible to use rhenium derivatives at relatively high concentrations (1–5 M %),16 at least before a highly efficient catalyst recycling system is available. A viable way could be to replace rhenium with other metals such as vanadium17−19 and molybdenum,20,21 even if the experimental conditions of these catalytic systems are still too harsh. Furthermore, considering the rhenium-catalyzed DODHs, the nature of the solvent, together with the choice of the reductant and the lowering of the metal catalyst amount, represents major hurdles to keep in mind.
Several successful DODH reactions developed in the past were conducted in aromatic solvents22−25 and in the presence of reductants such as phosphines,10,26 molecular H2,22,27 sulfite,28 or alcohols, better if secondary.13−15 From the sustainability point of view, the use of aromatic solvents is not indicated, and phosphines are not considered green reagents, at least when used in stoichiometric amounts, while H2, unfortunately, is not particularly efficient as a reductant (often requiring high pressures) and sulfites produce big amounts of sulfate salt. A good choice could be to use the sugar alcohol itself as both the solvent and reductant, at least when it possesses a melting point which is lower than the DODH working temperature (such as GL14). This choice, in our opinion, could represent a good compromise in terms of efficiency, sustainability, and costs.
However, in all the examples reported above, the recyclability of metal catalysts is not always satisfactory, and further in-depth studies will be necessary to try to industrially apply the entire process. As an alternative to the use of a solvent acting both as a solvent and a reducing agent, a chemically inert, sustainable medium can be proposed, which could make the recyclability of the catalyst easier and smarter. Of course, this solution implies the use of a reducing agent in stoichiometric amounts.
Ionic liquids (ILs) are a class of nonmolecular solvents with relatively low melting points (<373 K), which result from combinations of organic cations (i.e., ammonium or phosphonium) and inorganic or organic anions.29,30 Several ILs exist as liquids at room temperature, which are called room temperature ILs.31 They generally have negligible vapor pressure, high thermal and chemical stability, wide range of the liquid status, and good solvation behavior.32,33 The unique properties of ILs make them an ideal class of substitutes for conventional, often volatile, organic solvents in various applications.34 By suitably modulating the nature of both the cation and anion, it is possible to obtain a very large number of compounds, each one with a unique behavior, as might be requested by the synthetic chemist.35 In the recent past, ILs have found many and diversified applications: they have been considered as promising innovative reaction media in biocatalysis,36,37 in the processes of capture, separation, and reuse of CO2,38,39 as green solvents for extracting and separating metal ions,40 biomolecules, and biopolymers,41,42 and keeping into account their good solvent behavior for transition metal complexes, they have been widely employed in various catalytic organic reactions.43−45
The use of ILs in Re-catalyzed DODH reactions has been scarcely considered in the past: in fact, at the best of our knowledge, only one recent work was published about it.46 Moreover, looking at the hydrodeoxygenation of natural polyols by employing metal catalysts different from Re, scientific interest was recently increased by an interesting report about the production of olefins from sugar alcohols with Ru as the hydrogenation catalyst and tetrabutylphosphonium bromide as the medium of the reaction (but with the concomitant role of the dehydration catalyst).47
2. Materials and Methods
2.1. Materials
Nine ILs were used: 1-methyl-3-octylimidazolium bis(trifluoromethylsulfonyl)imide (MOIm-TFSI), 1-methyl-3-butylimidazolium bis(trifluoromethylsulfonyl)imide (MBIm-TFSI), 1-methyl-3-butylimidazolium hexafluorophosphate (MBIm-PF6), 1,3-diethoxyimidazolium bis(trifluoromethylsulfonyl)imide (EEIm-TFSI), N,N-dimethyl-N-butyl-N-benzyl ammonium bis(trifluoromethylsulfonyl)imide (MMBBAm-TFSI), N,N-dimethyl-N-octyl-N-benzyl ammonium bis(trifluoromethylsulfonyl)imide (MMOBAm-TFSI), N,N,N-triethyl-N-benzyl ammonium bis(trifluoromethylsulfonyl)imide (EEEBAm-TFSI), ethyl triphenyl phosphonium bis(trifluoromethylsulfonyl)imide (EPPPPh-TFSI), and 4-butyl-1,1-dimethyl-1-piperazinium bis(trifluoromethylsulfonyl)imide (BMMPz-TFSI).
MOIm-TFSI, MBIm-TFSI, MBIm-PF6, and EEIm-TFSI were purchased from Merck KGaA, while the remaining ILs were synthesized by us (see later). GL (reagent grade; ≥99% of purity by GC) and 1,2-PD (ACS reagent grade; ≥99.5% of purity by GC) were kept under vacuum for 24 h and dried on silica gel before their use. Bis(trifluoromethane)sulfonimide lithium salt, ethyltriphenylphosphonium bromide, N,N-dimethyl-N-butyl amine, benzyl chloride, N,N-dimethyl-N-octyl amine, N,N,N-triethylamine, N-butyl-N,N-dichloroethyl amine, 1,2-dibromopropane (1,2-DBP), 3-trimethylsilyl-2,2′,3,3′-tetradeutero propionic acid sodium salt, AA, MTO, Br2, and Re2O7 oxide were purchased from Merck KGaA and used without any manipulation.
2.2. Instruments
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance DRX-300 spectrometer (7.05 T) equipped with a high-resolution multinuclear probe operating in the range of 30–300 MHz. Spectra were recorded in an NMR tube (5 mm) filled with deuterated chloroform solutions (i.e., for the detection of 1,2-DBP) or with aqueous solutions containing, respectively, the starting reaction mixture or the AA aqueous trap contents. Only for the aqueous solutions, to lock the NMR signal and to obtain reliable quantitative data, inside the NMR tube, a closed coaxial capillary tube filled with 30 mM D2O solution of TSP was inserted. Free induction decays were acquired at 22 °C using, for 1H, a pulse sequence (made by Bruker; zgcppr) that suppresses the water signal at 4.71 ppm. The spectral width was −1 to 12 ppm (3894.081 Hz), while a 90° excitation pulse, together with a 1 s relaxation delay, was used to collect 32 scans. Similarly, 13C spectra were acquired using again a suitable proton decoupling excitation pulse (zgdc) with 3 s of delay and a spectral width of 250 ppm (from −10 to +240 ppm).
The gas chromatography–mass spectrometry (GC–MS) equipment includes a Thermo Scientific Focus series gas chromatograph coupled to an ISQ mass-selective detector equipped with a split/splitless injection system (injections made in the split mode) and a low-polar HP-5 MS (cross-linked 5% phenyl methyl siloxane) capillary 30 m in length, 0.25 mm in diameter, and with 0.25 μm film thickness, using high-purity helium as the carrier gas at a constant pressure of 30 kPa. For the headspace analysis, the sample was thermostated at 45 °C for 5 min, after which 200 μL of aerial top layer solution was injected into the GC apparatus using a gas-tight syringe at a 5:1 split ratio. The electron impact (EI) ion source was held at 250 °C, with a filament bias of 70 eV, transfer line at 250 °C, mass EI range 33–350 a, injector temperature of 250 °C, initial temperature of the analyses of 38 °C (1 min) and then 10 °C min–1 up to 250 °C (kept for 1 min), for a total acquisition time of 26 min.
GC analyses were performed using a Hewlett–Packard 6890 Series gas chromatography system equipped with a flame-ionization detector, using a 30 m HP-5 capillary column (cross-linked 5% Ph Me siloxane, 0.32 mm in diameter, 0.25 μm film thickness) with the injection port kept at 250 °C (carrier gas: high-purity helium with a pressure at the head of the column of 35 kPa). For the water solution, 0.5 μL was injected, while for headspace analysis, the sample was thermostated at 45 °C for 5 min, after which 200 μL of aerial top layer solution was injected using a gas-tight syringe at a 5:1 split ratio. The temperature programs were as follows: start at 60 °C with a hold time of 2 min, first increased at a rate of 5 °C min–1 to 130 °C (holding time 0 min) and then increased at a rate of 20 °C min–1 to 230 °C with a holding time of 2 min.
The UV–vis spectra were recorded using a Jenway 6505 system. Spectra were recorded in a 3 mL high-precision quartz cell (made of Suprasil quartz; Hellma), and the wavelength range was 200–600 nm.
Infrared spectra were measured on a Varian Scimitar 1000 FT-IR spectrometer with a cooled DTGS detector and Varian Resolutions 4.0 software. The samples were prepared on KBr pellets (1 mg in 100 mg of KBr).
The Raman spectra were acquired using an XploRA PLUS Raman spectrometer by HORIBA, at a temperature of T = 300 K within the 125–3200 cm–1 range. The excitation wavelengths were 532 and 638 nm, the spectral resolution was no worse than 1 cm–1, and the laser power was 1 mW or less (0.1 mW), at least when the heating is quite consistent. In fact, since some oxidation states of Re (i.e., Re(VI)) might possess weak adsorption bands in the visible region of the laser, we diluted them with KBr by preparing a tablet to minimize sample heating.
2.3. Syntheses of ILs
2.3.1. Synthesis of EPPPPh-TFSI
Ethyltriphenylphosphonium bromide was dissolved (37.125 g; 0.10 mol) in a minimum amount of water under stirring, and this solution was added to a saturated aqueous solution of lithium bis(trifluoromethane)sulfonimide (31.58 g; 0.11 mol). During the addition, the mixture turned milky due to the poor solubility of the onium salt having the bis(trifluoromethane)sulfonimide as the counterion, which tends to phase-separate as a second liquid phase. At the end of the addition, the mixture was kept at room temperature under stirring for 1–2 h, and then, the aqueous solution was extracted with petroleum ether. The aqueous phase was extracted three times with the same solvent; then, the collected organic phases were washed with water. The final organic phase was dried over sodium sulfate and filtered, and the solvent was removed under rotary evaporation. The final liquid was further dried under vacuum at 60 °C for a couple of days.
2.3.2. Synthesis of MMBBAm-TFSI, MMOBAm-TFSI, and MMBBAm-TFSI
In an oven-dried 500 mL flask, equipped with a stir bar, 15.84 g (0.157 mol) of freshly distilled N,N-dimethyl-N-butyl amine and 100 mL of ethyl acetate were introduced (alternatively, instead of N,N-dimethyl-N-butyl amine, 25 g of freshly distilled N,N-dimethyl-N-octyl amine or 15.85 g of freshly distilled N,N,N-triethylamine was added). To the clear and colorless solution, 19.80 g (18 mL) of benzyl chloride was added at once. The mixture was left at room temperature under magnetic stirring. After few seconds, the mixture turned milky. Upon standing, the formation of a second liquid phase was observed. After 5 h, the second liquid phase started to solidify. At this point, 50 mL of CH3CN was added, and the mixture was heated to obtain a clear, colorless solution. After cooling at room temperature, a white crystalline solid was formed, which was collected by filtration on a Buchner filter and washed several times with ethyl ether. A yield of 98% was obtained. This solid was used without further purification for the ion-exchange reaction with the TFSI anion, which was conducted essentially as described above for EPPPPh-TFSI.
2.3.3. Synthesis of BMMPz-TFSI
Into a 1 L flask, equipped with a reflux condenser, 23.4 g of N-butyl-N,N-dichloroethyl amine HCl, 400 mL of CH3CN, 36.6 mL of a 33 wt % solution of dimethylamine in MeOH, and 25.0 g of dry potassium carbonate were added. The mixture was refluxed for 24 h and then left to cool at room temperature. Upon cooling, a white solid precipitated, which was redissolved by adding CH2Cl2. The inorganic solids present in the reaction mixture were collected by filtration on a filter paper. The filtered solution was concentrated using a rotary evaporator, until a solid material appeared. The latter was again redissolved by heating and then left to crystallize. The white solid was filtered and washed with diethyl ether and then recrystallized twice from acetone. A white crystalline solid was obtained (71% yield), which was the desired piperazinium compound having chloride as the counterion. Introduction of the TFSI counterion was carried out by ion-exchange reaction, which was conducted essentially as described for EPPPPh-TFSI.
2.4. DODH Reaction Procedure
In a 10 mL round glass vessel equipped with a stir bar and a rubber septum, 1.5 g of the IL and 0.06 mmol of MTO (15 mg) or 0.03 mmol of Re2O7 (14.5 mg) were initially added; subsequently, after the complete dissolution of the catalyst, 5.4 mmol (0.5 g) of GL or 5.9 mmol of 1,2-PD (0.45 g) was added to the mixture. The reactants were heated to the desired temperature (145–150 °C) in a thermostatic oil bath and continuously purged with air or hydrogen (≈1 bubble per second) for 22 h. The vessel was connected, through a three-valve way, to two cold traps (25 mL, CHCl3 and water, both at around 0 °C) by a steel double-pointed needle equipped with a sparger; then, the gas flux coming from the DODH reaction with GL has been conveyed to the aqueous trap, while the gas flux coming from the DODH reaction with 1,2-PD, has been conveyed to the CHCl3 trap. Quantitative data for DODH reaction with GL were obtained by sampling 600 μL of the aqueous trap and transferring them to an NMR tube with a coaxial sealed capillary placed inside, previously filled with a 30 mM TSP D2O solution; the concentration of AA was obtained by the use of the normalized area of the NMR signal at 6.0 ppm (two proton) and compared with a calibration straight, built daily, using four known concentrations of AA. Before proceeding to the quantitative analytical step for DODH reaction with 1,2-PD, it was necessary to remove the excess of Br2 by a washing treatment with an aqueous sodium metabisulfite solution: in a 25 mL separatory funnel, 5 mL of 0.5 M sodium metabisulfite was shaken threefold together with the CHCl3 trap content, and the resulting CHCl3 fraction was then dried by adding anhydrous MgSO4. Quantitation of 1,2-DBP was obtained, by sampling 100 μL of CHCl3 trap which was then diluted with fresh CHCl3 and analyzed by both GC-FID and GC–MS. Quantitative data were obtained by GC using a calibration straight built with known titer solutions containing 1,2-DBP, while qualitative confirmations were carried out by GC–MS and 1H NMR.
3. Results
Re catalysts were chosen according to literature data.48 Taking into consideration the DODH reactions carried out in alcoholic solvents or conducted in the absence of any solvents (i.e., with the polyol acting simultaneously as the solvent and as the reducing agent), the higher oxidation state of Re, matching with relatively high catalytic activities, was selected. MTO (the most used DODH catalyst) was chosen, while for comparison purposes, we selected another, very common, oxo-Re(VII) catalyst, that is, Re2O7.
Regarding the use of ILs as dispersing medium, we first considered the choice of an appropriate counterion. We focused on two ILs with the same cation (MBIm) and different anions (TFSI and PF6) (see Chart 1). A comparison between these two ILs in the DODH reaction showed that with both reagents (i.e., GL and 1,2-PD), in the presence of MTO or Re2O7 and under H2 or air, substantial conversions appeared only when the anion was TFSI, while the MBIm-PF6, probably due to its higher volatility, was found in appreciable amounts inside the cold trap, thus making it difficult to manage the reaction. We also checked the final reaction mixture on the integrity of the used IL, but, at least for the TFSI derivative (for reference regarding the thermal stability of ILs containing the TFSI anion, please see refs (49) and (50)), we did not notice any evidence that may suggest an active degradative process (e.g., gravimetrically, the reaction mixture does not show any significant weight loss, and furthermore, the pH of the aqueous extracts of the final reaction mixture did not change, compared with the same of the initial one). These data prompted us to have the TFSI anion as the only choice without any further attempts with the other one.
Chart 1. ILs used in this study; only cations have been reported here, while as the counterion, they contain the bis(trifluoromethane)sulfonimide anion (TFSI), and only for MBIm, also the hexafluorophosphate derivative (MBIm-PF6) has been considered.

The standard reaction was conducted by heating GL (or 1,2-PD) in the IL at 140 °C for 22 h (or as specified) under air or H2 flux at ambient pressure in the presence of 1 mol % Re catalyst (MTO or Re2O7) (Figure 1 and Table 1); the gas flux conveyed the formed alkene in a cold aqueous trap (AA) (Figures S1–S3) or inside a CHCl3 trap containing bromine that acted as a double bond-derivatizing agent (to the dibromoderivative of propene) for a more accurate quantitative analysis (GC and GC–MS, Figures S4–S7). Our reaction apparatus was designed to remove the gaseous products (i.e., allyl alcohol and propene) instantaneously by air or H2 flux and separate them from the Re catalyst, thus avoiding the potential chance of catalytic oxidation of the alkenes which, especially for the reaction under H2, seems to be a very unlikely hypothesis of reactivity.
Figure 1.
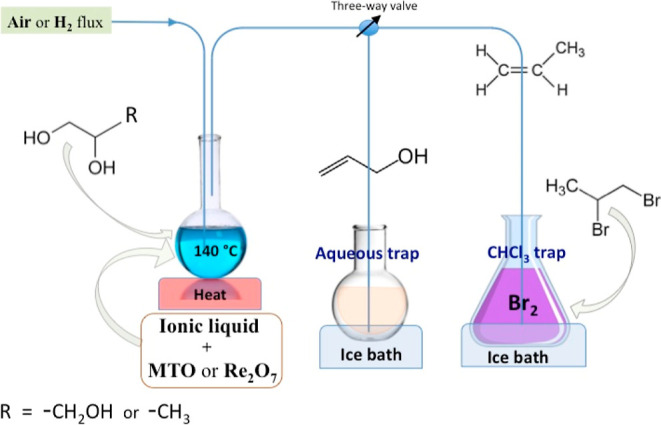
Schematic representation of the DODH reactions. The apparatus is sealed, just allowing a gas flux of about 1 bubble per second (air or H2), which helps to remove continuously the alkene and transfer it inside the appropriate ice cold trap. Temperature of the oil bath is 140 °C.
Table 1. Alkene Yields Obtained in the DODH Reaction between GL (or 1,2-PD) and MTO (or Re2O7) as a Metal Catalyst, Conducted at 140 °C and in the Absence of Any External Reducing Agentab.
| GL |
1,2-PD |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | IL | MTO |
Re2O7 |
MTO |
Re2O7 |
||||
| air | H2 | air | H2 | air | H2 | air | H2 | ||
| 1 | MMBBAm-TFSI | 17.5 (<5) | 24 (<5) | 2 (15) | 23 (<5) | 6 (0) | 16 (0) 10c | 8 (0) | 10.5 (0) |
| 2 | MMOBAm-TFSI | 20.5 (<5) | 31 (<5) | 18 (7) | 11 (5) | 90 | 110 | 11.50 | 150 |
| 3 | EEEBAm-TFSI | 18 (<5) | 25 (<5) | 10 (8) | 21 (<5) | 5 (0) | 12 (0) | 5 (0) | 13.5 (0) |
| 4 | EPPPPh-TFSI | 9 (12) | 1 (20) | 25 (<5) | 14 (12) | 13 (0) | 6 (0) | 12 (0) | 4 (0) |
| 5 | BMMPz-TFSI | - (70) | - (45) | - (55) | - (72) | 0.7 (0) | 4 (0) | - (0) | - (0) |
| 6 | EEIm-TFSId | 7 (15) | 12 (12) | 5 (19) | 20 (9)– | 5 (0) | 10 (0) | 5 (0) | 7 (0) |
| 7 | MOIm-TFSI | 12 (<5) | 22 (<5) | 12 (<5) | 14 (<5) | 7 (0) | 20 (0) | 2 (0) | 18 (0) |
| 8 | MBIm-TFSI | 8 (15) | 10 (18) | 16 (<5) | 23 (<5) | 8 (0) | 10 (0) | 12 (0) | 13.5 (0) |
| 9 | MBIm-PF6e | - | - | - | - | - (0) | - (0) | - (0) | - (0) |
In each experiment, a different IL was used as reaction media. In round brackets, the residual diol amounts (mol %) are reported. For detailed reaction conditions, see the Materials and Methods section.
Reaction conditions: GL (5.4 mmol) or 1,2-PD (5.9 mmol), Re (0.06 mmol; 1% catalyst/substrate), IL (1.5 g), 140 °C, room pressure, gas flux (air or H2 at about 2–3 bubble/sec), 22 h. Data obtained by NMR and GC–MS.
DODH reaction conducted with 5.9 mmol of 1,2-PD, with 1.5 g of MMBBAm-TFSI, in H2 current, and using, as a catalyst, the solid residue recovered from an exhaust analogous reaction mixture catalyzed by MTO; the catalyst recovery step was carried out by an ethyl acetate extraction procedure followed by filtration of the solid residue.
The reaction occurred, even if a partial degradation of ILs was observed, pointed out by the detection of ethanol inside the final reaction mixture.
Product yields, in all the experiments, were very scarce, and furthermore, the IL decomposed and probably partially evaporated as we recovered discrete amounts of it, inside the trapping solutions.
EPPPPh-TFSI is the only IL which is a solid at room temperature and liquefies at 110 °C. GL and MTO dissolved well in all tested solvents even when they are in a heterogeneous state at room temperature: in fact, the initial reaction mixtures were generally heterogeneous at the beginning (room temperature), while they became clear when the temperature was increased to the value chosen for the DODH reaction (140 °C). The reaction products (AA or 1,2-DBP) were quantified by NMR and GC–MS at several fixed time intervals by withdrawing an aliquot of the trap mixture.
Product yields are shown in Table 1; we would point out that the maximum nominal product yield was 50% since the other 50% of the reagent was necessary for the reduction reaction. Furthermore, in the absence of the catalyst and using the IL, both glycols were recovered unchanged.
Other than the air as gaseous flux, as well as the reaction environment, we used H2 because we had past evidence that although it does not act as a reductant, it equally contributes to increase the alkene yield, and furthermore, it helps to reduce the amounts of byproducts, such as acrolein,14 derived by an alternative and parallel pathway.51
The first observation to make is to consider the relative reactivity of the two polyols and the catalytic activities of the two catalysts. Basically, GL was converted to the corresponding alkene more efficiently than 1,2-PD, while the two catalysts seem to possess comparable catalytic activities. Hydrogen often increases the product yields, with some exceptions, above all, related to the nature of the IL. For example, the only IL with a phosphorus unit (EPPPPh-TFSI; entry 4 in Table 1) showed product yields under aerobic conditions higher than those in H2.
Among the selected ILs, BMMPz-TFSI, that is, the only one without π electrons, seems to almost completely inhibit the DODH (entry 5 in Table 1), at least under aerobic conditions (also under H2, the alkene was absent or produced in limited amounts). Another IL, which resulted as not suitable for this reaction, was EEIm-TFSI (entry 6 in Table 1). It was selected because of the presence of a couple of alkoxy groups that, in our opinion, could have a positive effect on the formation of Re diolate. Unfortunately, we did not observe a net increase in product yield, and furthermore, we observed partial chemical instability of the IL since ethanol was always found inside the cold trap.
The remaining ILs (three ammonium—MMOBAm, MMBBAm, and EEEBAm—and two imidazole—MOIm and MBIm derivatives; entries 1–3, 7, and 8 in Table 1) showed almost quantitative conversions of polyols with satisfying alkene yields. Taking into account reagents and alkene products, a lack of mass balance is apparent. With 1,2-PD, the possible reason is the formation of a couple of acetals that subtracts part of the reagent, transferring it inside the cold trap.8 For GL, the explanation is less immediate since we did not detect other reaction products inside the trap, and the free GL found inside the reactor was often negligible. A polymerization process, which is always present because of the formation of the two potential oxidation products of GL when it acts as a reducing agent, that is, hydroxyacetone and/or glyceraldehyde, can subtract part of the reagent, thus decreasing the reactivity and making the recovery more difficult.
The peculiarity of the DODH conducted in a nonreducing medium is that there is a need to clarify who plays the role of the reductant. We have already reminded that H2 is not suitable to act as a reducing agent, at least under ambient pressure, so we had to understand what could be the best reductant to transform 100% of the polyol to alkene. Selecting MOIm-TFSI as the reference IL, we tested a series of reducing agents under different experimental conditions, taking into consideration the sustainability, above all the other green aspects (Table 2).
Table 2. AA Yields (mol %) and Reaction Conditions of DODH Experiments Performed in the Presence of a Series of Reducing Agents and Employing GL as the Substrate, MTO as the Catalyst, and MOIm-TFSI as the IL.
| reducing agent | 2,4-dimethyl-3-pentanol | 2,4-dimethyl-3-pentanol | sodium metabisulfite | PPh3 | PPh3 | PPh3 | PPh3 |
|---|---|---|---|---|---|---|---|
| amount (mmol) | 7.2 | 21.6 | 10.5 | 7.6 | 7.6 | 7.6 | 7.6 |
| GL (mmol) | 5.4 | 2 | 5.4 | 5.4 | 5.4 | 5.4 | 5.4 |
| MTO (mmol) | 0.06 | 0.02 | 0.06 | 0.06 | 0.06 | 0.06 | 0.06 |
| MOIm-TFSI (g) | 1.5 | 1 | 1.5 | 1.5 | 3 | 3 | 4.5 |
| reaction time(h) | 22 | 22 | 22 | 22 | 22 | 30 | 30 |
| AA yields (mol % of AA) | 32 | 56 | 43.5 | 63 | 80 | 92 |
Following the criteria of chemical reactivity and also taking a look at the relevant literature, it clearly appeared that triphenylphosphine (PPh3) was a functional option, although not particularly green, also because it is hard to accept the production of a stoichiometric amount of triphenylphosphine oxide (Ph3P=O). Nevertheless, we tested the DODH of GL with PPh3, using MTO as the catalyst and different amounts of MOIm-TFSI as dispersing medium and extending the reaction times. As the amount of IL increased, the produced AA increased to a very good value of 92 mol %. Also, the reaction time resulted to be an important parameter, and at 140 °C, probably, the reaction required a longer reaction time (30 rather than 22 h). Therefore, for a reductant such as PPh3, the IL must be used in a higher amount to obtain an almost quantitative product yield.
Other reducing agents tested were 2,4-dimethyl-3-pentanol and sodium sulfite. Although the salt showed several drawbacks, such as the scarce reproducibility of yields, due probably to the heterogeneous state of the reaction mixture, 2,4-dimethyl-3-pentanol gave good product yields, but only with adding an amount of reducing agent that is above the requested stoichiometry.
The reuse and the recovery of the catalyst are key operations particularly important for any potential industrial application. Currently, high amounts of metal catalyst are still employed in the DODH, and we believe that the presence of the IL should facilitate these steps. To this purpose, we added to the reaction mixture further aliquots of the reagent just after the consumption of the polyol (Table 3).
Table 3. Yields of DODH Reaction Catalyzed with Both Catalysts (MTO and Re2O7) and Both Polyols (GL—Odd Entries—and 1,2-PD—Even Entries), Performed with Four Different Reagent configurations. Run 1 is Relative to the Standard DODH Reactions (See the Footnote at the Bottom of the Table); in Runs 2 and 3, a Refilling of 5.4 mmol of GL (or 5.9 mmol of 1,2-PD) Has Been Carried Out (See the Materials and Methods Section for Details)a.
| product
yields (mol %) |
|||||
|---|---|---|---|---|---|
| entries | polyol | MTO | Re2O7 | ||
| 1 | polyol | run 1 (standard experimental conditions) | GL | 27 | 19 |
| 2 | 1,2-PD | 25 | 22 | ||
| 3 | run 2 (after a reload of 6 mmol of polyol) | GL | <5 | <5 | |
| 4 | 1,2-PD | <5 | <5 | ||
| 5 | polyol + IL | run 1 (standard experimental conditions) | GL | 22 | 14 |
| 6 | 1,2-PD | 20 | 18 | ||
| 7 | run 2 (after a reload of 6 mmol of polyol) | GL | 14 | 7 | |
| 8 | 1,2-PD | 14 | 10 | ||
| 9 | run 3 (after a second reload of 6 mmol of polyol) | GL | 8 | <5 | |
| 10 | 1,2-PD | 10 | 7 | ||
| 11 | polyol + PPh3 | run 1 (standard experimental conditions) | GL | 90 | 91 |
| 12 | 1,2-PD | 88 | 82 | ||
| 13 | run 2 (after a reload of 6 mmol of polyol) | GL | 15 | 25 | |
| 14 | 1,2-PD | 18 | 10 | ||
| 15 | polyol + PPh3 + IL | run 1(standard experimental conditions) | GL | 92 | 93 |
| 16 | 1,2-PD | 88 | 89 | ||
| 17 | run 2(after a reload of 6 mmol of polyol) | GL | 40 | 45 | |
| 18 | 1,2-PD | 45 | 47 | ||
Reaction conditions: GL (5.4 mmol) or 1,2-PD (5.9 mmol), MTO or Re2O7 (0.06 mmol; 1% catalyst/substrate), MOIm-TFSI: 1.5 g (entries 5–10) or 4.5 g (entries 15–18), under H2 current (about 2–3 bubble/sec), 140 °C, room pressure, reaction time: 22 h (entries 1–10) or 30 h (entries 11–18), PPh3 (when present) 7.6 mmol. Data obtained by NMR and GC–MS.
This readdition of the reagent was made for two consecutive times (run 2 and run 3), always waiting until GL (entries 7 and 9) or 1,2-PD (entries 8 and 10) disappeared (Table 3). Therefore, we measured the AA and propene yields, noting that at least in the presence of the IL and without any reducing agent inside the reaction mixture (polyol + IL; entries 5–10), yields were only slightly lower than that found in run 1 reactions. The same experiments were carried out also without ILs (only polyol; entries 1 and 2): for both polyols, AA and propene were formed in negligible amounts only even after the first readdition (entries 3 and 4). On the other hand, when the readditions of the reagent were carried out in systems containing an external reductant such as PPh3 (entries 11 and 12), the alkene yield was noticeably lower if the IL was absent (polyol + PPh3; entries 13 and 14) and definitely higher in the presence of the IL (polyol + PPh3 + IL; entries 15–18).
From the mechanistic point of view, the delay time and the formation of both methane and methanol from MTO-catalyzed DODH reaction, which is a direct consequence of its transformation, are of great interest. We already described the origin of methane elsewhere14 as the respective demethylated Re oxide started to catalyze the DODH reaction. In this study, we also noted that methanol was equally formed, always before the catalytic production of olefin starts. The formation of methanol was observed with both polyols, and quantitatively, it did not exceed 10 mol % of the total amount of MTO. Also, its hypothetical formation from the degradation of the polyol was discarded because, in both cases, namely, in a model reaction conducted in the absence of any catalyst and in a model reaction conducted with Re2O7, methanol was never detected.
In DODH reactions involving both GL and 1,2-PD, we observed delay times comparable with that noted in the alcoholic solvent or in neat polyol.7,14,52 The typical sigmoid trends were always recorded, with a more or less extended delay time, probably necessary to make available an active form of the catalyst (see Supporting Information, Figures S8–S13).
4. Discussion
DODH reactions on GL and 1,2-PD were performed at 140 °C in ILs as reaction media. Considering the tested ILs, the alkene yields, and their selectivities, we are able to make some intriguing considerations. Both the physical and chemical stabilities of ILs are crucial since the DODH reaction is commonly performed at temperature ≥140 °C. For example, the ethoxy derivative (EEIm) tends to lose ethanol, and consequently, any other ILs susceptible to chemical changes at such temperatures may not be employed. The same concept can be applied to ILs with appreciable volatility (MBIm-PF6),53 at least at the relatively high temperatures such as those used herein, since the reaction products must then be recovered by distillation or entrapment in inert cold media, and a further purification step would be counterproductive to the economy of the reaction.
Taking a look at the data reported in Table 1, it seems apparent that the most appropriate ILs in terms of alkene yields, in the DODH reaction, are the benzyl and imidazoyl derivatives. On the other hand, neither the piperazine nor the triphenylphosphine derivatives seem to activate the metal-catalyzed DODH reaction. We may comment that the presence of a single aromatic ring seems to facilitate the reactivity, even if it is hard to propose here a deeper explanation of such a behavior. Further experiments, possibly with a wider range of ILs, both with and without one or more aromatic rings, will be necessary.
As previously stated, one priority regarding the Re-catalyzed DODH reaction is to improve the performance of the catalyst.54 This is achievable through two conceptually different strategies: to support or to recover and to reuse the metal catalyst. Regarding the heterogenization techniques, many attempts have been made on supporting Re.55,56 However, the issue has not yet been completely resolved; in fact, under the typical experimental conditions, the metal leaching can be significant, and in addition, the distinction between the reaction that occurred in homogeneous or heterogeneous conditions could be troublesome.55 A second strategy involves the recovery and reuse of the catalyst, possibly avoiding tedious and costly operations, and/or trying to increase the olefin to catalyst ratio. We preferred, for the moment, to study the reaction in the homogeneous mode, trying to improve the recovery and reusing steps. We have carried out appropriate experiments by refilling the batch with starting polyol, always after its total exhaustion. In Table 3, data regarding this modus operandi are reported, and you can see that the presence of ILs clearly contribute to preserve the catalyst in its active form since it continues to catalyze the production of alkene even after two sequential refills of the polyol (run 2 and run 3; red text). In contrast, exhaust catalysts coming from reactions conducted in the absence of any IL were completely inactive, leading to only marginal yield values of alkenes (run 2; black text).
We can tentatively explain this behavior by considering the polymerization reaction that could potentially occur at 140 °C when the oxidized forms of the polyols (dihydroxyacetone + glyceraldehyde for GL and hydroxyacetone + lactaldehyde for 1,2-PD) are present. We just remind that these trioses, at high temperatures and in the presence of metal derivatives, can be further converted into pyruvaldehyde, followed by lactic acid formation that can easily be transformed to the well-known polylactic acid.57 This side reaction can contribute to modify the catalyst surface, making it unsuitable for further catalytic turnovers.
To support our explanation, we replicated the same experiments by introducing a reducing agent such as triphenylphosphine, which maximized the alkene yields and minimized the formation of easily polymerizable derivatives such as carbonyl compounds coming from the oxidized polyols. In fact, in terms of alkene yields, here, we obtained a satisfying recycle of the catalyst in both cases, namely, in the absence and in the presence of ILs (runs 2 in green text and run 2 in blue text, Table 3).
Some mechanistic aspects of the DODH reaction have been tentatively elucidated by Raman spectroscopy. We recorded spectra of Re compounds both alone and treated with a low-boiling-point secondary alcohol such as 2-propanol, at 140 °C, in a stainless-steel closed vessel at times of 0, 15, 45, and 120 min (Figures 2 and 3). The choice to use 2-propanol rather than the typical reducing agents previously employed in the DODH reactions was made since we thought that, at the end of thermal treatment, we obtained a sample containing only the Re compound to be analyzed spectroscopically; in fact, both 2-propanol and its oxidized counterpart such as acetone can be easily removable using a rotavapor at the end of treatment.
Figure 2.

Series of Raman spectra of MTO treated at 140 °C with 2-propanol: from the top, 0 min (dark blue), 15 min (green), 45 min (red), and 120 min (light blue).
Figure 3.

Series of Raman spectra of Re2O7 treated at 140 °C with 2-propanol: from the top, 0 min (dark blue), 15 min (green), 45 min (red), and 120 min (light blue) (we omitted the part of spectra from 2000 to 3000 cm–1 since no signals appeared).
By comparing the spectra of the untreated and treated samples, it is easy to note the changes that occurred with heating in the presence of 2-propanol. Taking into consideration MTO, the disappearance of more intense signals ascribable to the methyl group (2893 and 2987 cm–1, respectively, the symmetric and asymmetric stretching) and a bathochromic shift of about 29 cm–1 of the symmetric stretching of the ReO3 group (from 989 to 960 cm–1) clearly demonstrate its loss after only 45 min of reaction time. These data are quite superimposable with our previously reported measurements by NMR,14 and again, this explains well the presence of delay times >30 min, observed in our experiments. On the other hand, the Raman shifts relative to the treated Re2O7 samples are less evident, where only some large bands appeared (around 750 and 1575 cm–1; this is probably attributable to amorphous carbon dispersed inside the residual metal catalyst58), while the other Raman signals, assigned to the Re–O bonds, remained unchanged.
To understand if other oxidation states of Re are present during the evolution of the reaction, we recorded the Raman shift of ReO2, ReO3, and ReO4– (Figure S14). As also confirmed by some literature references,59−62 the signals are not very different, and therefore, it is difficult to say if some of them were present after the thermal treatment of MTO and Re2O7. We believe that the presence of ReO3 could be equally conceivable since the red color, typical of Re(VI), clearly appears after the delay time. In favor of the ReO3 presence, we can mention the noticeable heat developed during the 532 laser irradiation of both Re catalysts. In this regard, we also tried to record the spectra using the 638 nm wavelength rather than the 532 nm wavelength, but, as shown in Figure S15, the signal positions are identical, with only the intensity noticeably lower. This side effect is exactly the same as that observed when we recorded the Raman spectra of neat ReO3, where a satisfying Raman pattern could be recorded only with a very diluted sample (in the KBr tablet) and employing a very low laser power.
Regarding the formation of both methane and methanol, we can tentatively propose a thermal homolytic fragmentation of the Re–CH3 with a subsequent competition for the methyl radical between a hydrogen abstraction assisted by the alcohol and an oxygen transfer, leading to the alcoholate and then to methanol.
5. Conclusions
It is significantly true that Re is an expensive and relatively rare metal and that it is advisable to discover new catalytic systems to deoxygenate the polyols, but at present, the traditional Re(VII) compounds are still the best choice. This study outlines several improvements in the reaction conditions adding ILs as novel innovative solvents for DODH reaction. When the IL, stable at moderate temperature, was a benzyl or imidazolyl derivative like in MOIm, MBIm, MMOBAm, MMBBAm, and EEEBAm, good yields of alkene derivatives were observed, and it was possible to reuse the Re catalyst, seeing only a slight decrease in product yield, affirming the possibility of catalyst recycling, a crucial point for any potential industrial use. The preserved activity of the Re catalyst is probably due to the involvement of the IL with the resulting suppression of the polymerization of byproducts (e.g., hydroxyacetone, lactic acid, and glyceraldehyde). Raman spectra confirmed the transformation of the MTO during the early times of the reaction and the suitable presence of Re(VI) during the active steps of the DODH.
It will be necessary to elucidate the potential role of the ILs with aromatic rings in the reaction mechanism of the DODH, by designing suitable experiments with several families of ILs. The present study opens a new route toward the sustainable deoxygenation of polyols with the help of specific ILs.
Acknowledgments
The authors are grateful for the financial support to the ITQSA of Abruzzo Region, Italy. We thank Silvia Bruni of University of Milan for the helpful discussion with us and for the valuable advices on execution and interpretation of Raman spectra.
Glossary
Abbreviations
- DODH
deoxydehydration
- GL
glycerol
- 1,2-PD
1,2-propanediol
- IL
ionic liquid
- MTO
methyltrioxorhenium
- 1,2-DBP
1,2-dibromopropane
- TSP
3-trimethylsilyl-2,2′,3,3′-tetradeutero propionic acid sodium salt
- GC–MS
gas chromatography–mass spectrometry
- TFSI
bis(trifluoromethylsulfonyl)imide
- PF6
hexafluorophosphate
- MOIm
1-methyl-3-octylimidazolium
- MBIm
1-methyl-3-butylimidazolium
- EEIm
1,3-diethoxyimidazolium
- MMBBAm
N,N-dimethyl-N-butyl-N-benzyl ammonium
- MMOBAm
N,N-dimethyl-N-octyl-N-benzyl ammonium
- EEEBAm
N,N,N-triethyl-N-benzyl ammonium
- EPPPPh
ethyl triphenyl phosphonium
- BMMPz
4-butyl-1,1-dimethyl-1-piperazinium
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01803.
GC–MS chromatogram and mass spectrum of allyl alcohol; NMR spectrum of allyl alcohol; calibration straight of allyl alcohol; GC–MS chromatogram of dibromopropane; mass spectrum of dibromopropane; 1H NMR spectrum of dibromopropane; calibration straight of dibromopropane; time course of DODH reaction involving GL and MTO in the presence of MOIm-TFSI; time course of DODH reaction involving GL and Re2O7 in the presence of MOIm-TFSI; time course of DODH reaction involving GL and MTO in the presence of MMOBAm-TFSI; time course of DODH reaction involving GL and Re2O7 in the presence of MMOBAm-TFSI; time course of DODH reaction involving GL and MTO in the presence of MBIm-TFSI; time course of DODH reaction involving GL and MTO in the presence of MBIm-TFSI; Raman spectra of NH4ReO4, ReO2, and ReO3; and Raman spectra of MTO recorded with 532 and 638 nm laser wavelength (PDF)
Author Contributions
A.M. and G.S. contributed equally. N.d.A and L.T. conceived the study. N.d.A. and P.D.P wrote the manuscript. A.M. and G.S. carried out DODH experiments. R.G. synthetized the ILs. L.T., A.M., and G.S. recorded NMR spectra and carried out GC–MS analyses. V.C. recorded the Raman spectra. All authors have given approval to the final version of the manuscript.
Research project by ITQSA “Consorzio per l’Innovazione Tecnologica—Qualità e Sicurezza degli Alimenti” (ITQSA): Utilization of agroindustrial byproducts. Project no. DM 61318.
The authors declare no competing financial interest.
Supplementary Material
References
- Garedew M.; Lin F.; Song B.; DeWinter T. M.; Jackson J. E.; Saffron C. M.; Lam C. H.; Anastas P. T. Greener routes to biomass waste valorization: Lignin transformation through electrocatalysis for renewable chemicals and fuels production. ChemSusChem 2020, 13, 4214–4237. 10.1002/cssc.202000987. [DOI] [PubMed] [Google Scholar]
- Dawes G. J. S.; Scott E. L.; Le Nôtre J.; Sanders J. P. M.; Bitter J. H. Deoxygenation of biobased molecules by decarboxylation and decarbonylation—a review on the role of heterogeneous, homogeneous and bio-catalysis. Green Chem. 2015, 17, 3231–3250. 10.1039/c5gc00023h. [DOI] [Google Scholar]
- Salvi B. L.; Panwar N. L. Biodiesel resources and production technologies—A review. Renewable Sustainable Energy Rev. 2012, 16, 3680–3689. 10.1016/j.rser.2012.03.050. [DOI] [Google Scholar]
- Possato L. G.; Chaves T. F.; Cassinelli W. H.; Pulcinelli S. H.; Santilli C. V.; Martins L. The multiple benefits of glycerol conversion to acrolein and acrylic acid catalyzed by vanadium oxides supported on micro-mesoporous MFI zeolites. Catal. Today 2017, 289, 20–28. 10.1016/j.cattod.2016.08.005. [DOI] [Google Scholar]
- Mazarío J.; Concepción P.; Ventura M.; Domine M. E. Continuous catalytic process for the selective dehydration of glycerol over Cu-based mixed oxide. J. Catal. 2020, 385, 160–175. 10.1016/j.jcat.2020.03.010. [DOI] [Google Scholar]
- El Doukkali M.; Iriondo A.; Gandarias I. Enhanced catalytic upgrading of glycerol into high value-added H2 and propanediols: Recent developments and future perspectives. Mol. Catal. 2020, 490, 110928. 10.1016/j.mcat.2020.110928. [DOI] [Google Scholar]
- Lupacchini M.; Mascitti A.; Canale V.; Tonucci L.; Colacino E.; Passacantando M.; Marrone A.; d’Alessandro N. Deoxydehydration of glycerol in presence of rhenium compounds: reactivity and mechanistic aspects. Catal. Sci. Technol. 2019, 9, 3036–3046. 10.1039/c8cy02478b. [DOI] [Google Scholar]
- Scioli G.; Tonucci L.; Di Profio P.; Proto A.; Cucciniello R.; d’Alessandro N.; d’Alessandro N. New green route to obtain (bio)-propene through 1,2-propanediol deoxydehydration. Sustainable Chem. Pharm. 2020, 17, 100273. 10.1016/j.scp.2020.100273. [DOI] [Google Scholar]
- Taher D.; Thibault M. E.; Di Mondo D.; Jennings M.; Schlaf M. Acid-, water- and high-temperature-stable ruthenium complexes for the total catalytic deoxygenation of glycerol to propane. Chem.—Eur. J. 2009, 15, 10132–10143. 10.1002/chem.200901427. [DOI] [PubMed] [Google Scholar]
- Morris D. S.; van Rees K.; Curcio M.; Cokoja M.; Kühn F. E.; Duarte F.; Love J. B. Deoxydehydration of vicinal diols and polyols catalyzed by pyridinium perrhenate salts. Catal. Sci. Technol. 2017, 7, 5644–5649. 10.1039/c7cy01728f. [DOI] [Google Scholar]
- Dethlefsen J. R.; Fristrup P. In situ spectroscopic investigation of the rhenium-catalyzed deoxydehydration of vicinal diols. ChemCatChem 2015, 7, 1184–1196. 10.1002/cctc.201403012. [DOI] [Google Scholar]
- Shiramizu M.; Toste F. D. Deoxygenation of biomass-derived feedstocks: oxorhenium-catalyzed deoxydehydration of sugars and sugar alcohols. Angew. Chem., Int. Ed. 2012, 124, 8206–8210. 10.1002/ange.201203877. [DOI] [PubMed] [Google Scholar]
- Arceo E.; Ellman J. A.; Bergman R. G. Rhenium-catalyzed didehydroxylation of vicinal diols to alkenes using a simple alcohol as a reducing agent. J. Am. Chem. Soc. 2010, 132, 11408–11409. 10.1021/ja103436v. [DOI] [PubMed] [Google Scholar]
- Canale V.; Tonucci L.; Bressan M.; d’Alessandro N. Deoxydehydration of glycerol to allyl alcohol catalyzed by rhenium derivatives. Catal. Sci. Technol. 2014, 4, 3697–3704. 10.1039/c4cy00631c. [DOI] [Google Scholar]
- Liu S.; Senocak A.; Smeltz J. L.; Yang L.; Wegenhart B.; Yi J.; Kenttämaa H. I.; Ison E. A.; Abu-Omar M. M. Mechanism of MTO-catalyzed deoxydehydration of diols to alkenes using sacrificial alcohols. Organometallics 2013, 32, 3210–3219. 10.1021/om400127z. [DOI] [Google Scholar]
- Tshibalonza N. N.; Monbaliu J.-C. M. The deoxydehydration (DODH) reaction: a versatile technology for accessing olefins from bio-based polyols. Green Chem. 2020, 22, 4801–4848. 10.1039/d0gc00689k. [DOI] [Google Scholar]
- Chapman G. Jr.; Nicholas K. M. Vanadium-catalyzed deoxydehydration of glycols. Chem. Commun. 2013, 49, 8199–8201. 10.1039/c3cc44656e. [DOI] [PubMed] [Google Scholar]
- Petersen A. R.; Nielsen L. B.; Dethlefsen J. R.; Fristrup P. Vanadium-catalyzed deoxydehydration of glycerol without an external reductant. ChemCatChem 2018, 10, 769–778. 10.1002/cctc.201701049. [DOI] [Google Scholar]
- Kwok K. M.; Choong C. K. S.; Ong D. S. W.; Ng J. C. Q.; Gwie C. G.; Chen L.; Borgna A. Hydrogen-Free Gas-Phase Deoxydehydration of 2,3-Butanediol to Butene on Silica-Supported Vanadium Catalysts. ChemCatChem 2017, 9, 2443–2447. 10.1002/cctc.201700301. [DOI] [Google Scholar]
- Tran R.; Kilyanek S. M. Deoxydehydration of polyols catalyzed by a molybdenum dioxo-complex supported by a dianionic ONO pincer ligand. Dalton Trans. 2019, 48, 16304–16311. 10.1039/c9dt03759d. [DOI] [PubMed] [Google Scholar]
- Beckerle K.; Sauer A.; Spaniol T. P.; Okuda J. Bis(phenolato)molybdenum complexes as catalyst precursors for the deoxydehydration of biomass-derived polyols. Polyhedron 2016, 116, 105–110. 10.1016/j.poly.2016.03.053. [DOI] [Google Scholar]
- Davis J.; Srivastava R. S. Oxorhenium-catalyzed deoxydehydration of glycols and epoxides. Tetrahedron Lett. 2014, 55, 4178–4180. 10.1016/j.tetlet.2014.05.044. [DOI] [Google Scholar]
- Denning A. L.; Dang H.; Liu Z.; Nicholas K. M.; Jentoft F. C. Deoxydehydration of glycols catalyzed by carbon-supported perrhenate. ChemCatChem 2013, 5, 3567–3570. 10.1002/cctc.201300545. [DOI] [Google Scholar]
- Cook G. K.; Andrews M. A. Toward nonoxidative routes to oxygenated organics: stereospecific deoxydehydration of diols and polyols to alkenes and allylic alcohols catalyzed by the metal oxo complex (C5Me5)ReO3. J. Am. Chem. Soc. 1996, 118, 9448–9449. 10.1021/ja9620604. [DOI] [Google Scholar]
- Raju S.; Moret M.-E.; Klein Gebbink R. J. M. Rhenium-catalyzed dehydration and deoxydehydration of alcohols and polyols: opportunities for the formation of olefins from biomass. ACS Catal. 2015, 5, 281–300. 10.1021/cs501511x. [DOI] [Google Scholar]
- Raju S.; Jastrzebski J. T. B. H.; Lutz M.; Klein Gebbink R. J. M. Catalytic deoxydehydration of diols to olefins by using a bulky cyclopentadiene-based trioxorhenium catalyst. ChemSusChem 2013, 6, 1673–1680. 10.1002/cssc.201300364. [DOI] [PubMed] [Google Scholar]
- Vkuturi S.; Chapman G.; Ahmad I.; Nicholas K. M. Rhenium-catalyzed deoxydehydration of glycols by sulfite. Inorg. Chem. 2010, 49, 4744–4746. 10.1021/ic100467p. [DOI] [PubMed] [Google Scholar]
- Ahmad I.; Chapman G.; Nicholas K. M. Sulfite-driven, oxorhenium-catalyzed deoxydehydration of glycols. Organometallics 2011, 30, 2810–2818. 10.1021/om2001662. [DOI] [Google Scholar]
- Forsyth S. A.; Pringle J. M.; MacFarlane D. R. Ionic Liquids-An Overview. Aust. J. Chem. 2004, 57, 113–119. 10.1071/CH03231. [DOI] [Google Scholar]
- Singh S. K.; Savoy A. W. Ionic liquids synthesis and applications: An overview. J. Mol. Liq. 2020, 297, 112038. 10.1016/j.molliq.2019.112038. [DOI] [Google Scholar]
- Marsh K. N.; Deev A.; Wu A. C.-T.; Tran E.; Klamt A. Room temperature ionic liquids as replacements for conventional solvents - A review. Korean J. Chem. Eng. 2002, 19, 357–362. 10.1007/BF02697140. [DOI] [Google Scholar]
- Zhang S.; Sun N.; He X.; Lu X.; Zhang X. Physical properties of ionic liquids: database and evaluation. J. Phys. Chem. Ref. Data 2006, 35, 1475. 10.1063/1.2204959. [DOI] [Google Scholar]
- Pádua A. A. H.; Costa Gomes M. F.; Canongia Lopes J. N. A. Molecular solutes in ionic liquids: A structural perspective. Acc. Chem. Res. 2007, 40, 1087–1096. 10.1021/ar700050q. [DOI] [PubMed] [Google Scholar]
- Earle M. J.; Seddon K. R. Ionic liquids. Green solvents for the future. Pure Appl. Chem. 2000, 72, 1391–1398. 10.1351/pac200072071391. [DOI] [Google Scholar]
- Tokuda H.; Hayamizu K.; Ishii K.; Susan M. A. B. H.; Watanabe M. Physicochemical properties and structures of room temperature ionic liquids. 1. Variation of anionic species. J. Phys. Chem. B 2004, 108, 16593–16600. 10.1021/jp047480r. [DOI] [Google Scholar]
- Xu P.; Liang S.; Zong M.-H.; Lou W.-Y. Ionic liquids for regulating biocatalytic process: Achievements and perspectives. Biotechnol. Adv. 2021, 51, 107702. 10.1016/j.biotechadv.2021.107702. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Lau R. M.; Sorgedrager M. J.; van Rantwijk F.; Seddon K. R. Biocatalysis in ionic liquids. Green Chem. 2002, 4, 147–151. 10.1039/B110008B. [DOI] [Google Scholar]
- Yan X.; Anguille S.; Bendahan M.; Moulin P. Ionic liquids combined with membrane separation processes: A review. Sep. Purif. Technol. 2019, 222, 230–253. 10.1016/j.seppur.2019.03.103. [DOI] [Google Scholar]
- Lian S.; Song C.; Liu Q.; Duan E.; Ren H.; Kitamura Y. Recent advances in ionic liquids-based hybrid processes for CO2 capture and utilization. J. Environ. Sci. 2021, 99, 281–295. 10.1016/j.jes.2020.06.034. [DOI] [PubMed] [Google Scholar]
- Stojanovic A.; Keppler B. K. Ionic liquids as extracting agents for heavy metals. Sep. Sci. Technol. 2012, 47, 189–203. 10.1080/01496395.2011.620587. [DOI] [Google Scholar]
- Zhu S.; Wu Y.; Chen Q.; Yu Z.; Wang C.; Jin S.; Ding Y.; Wu G. Dissolution of cellulose with ionic liquids and its application: a mini-review. Green Chem. 2006, 8, 325–327. 10.1039/B601395C. [DOI] [Google Scholar]
- Han X.; Armstrong D. W. Ionic liquids in separations. Acc. Chem. Res. 2007, 40, 1079–1086. 10.1021/ar700044y. [DOI] [PubMed] [Google Scholar]
- Welton T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- Sheldon R. Catalytic reactions in ionic liquids. Chem. Commun. 2001, 2399–2407. 10.1039/B107270F. [DOI] [PubMed] [Google Scholar]
- Vekariya R. L. A review of ionic liquids: Applications towards catalytic organic transformations. J. Mol. Liq. 2017, 227, 44–60. 10.1016/j.molliq.2016.11.123. [DOI] [Google Scholar]
- Shin N.; Kwon S.; Moon S.; Hong C. H.; Kim Y. G. Ionic liquid-mediated deoxydehydration reactions: Green synthetic process for bio-based adipic acid. Tetrahedron 2017, 73, 4758–4765. 10.1016/j.tet.2017.06.053. [DOI] [Google Scholar]
- Stalpaert M.; Janssens K.; Marquez C.; Henrion M.; Bugaev A. L.; Soldatov A. V.; De Vos D. Olefins from biobased sugar alcohols via selective, Ru-mediated reaction in catalytic phosphonium ionic liquids. ACS Catal. 2020, 10, 9401–9409. 10.1021/acscatal.0c02188. [DOI] [Google Scholar]
- Wozniak B.; Li Y.; Tin S.; de Vries J. G. Rhenium-catalyzed deoxydehydration of renewable triols derived from sugars. Green Chem. 2018, 20, 4433. 10.1039/c8gc02387e. [DOI] [Google Scholar]
- Huang Y.; Chen Z.; Crosthwaite J. M.; N.V.K. Aki S.; Brennecke J. F. Thermal stability of ionic liquids in nitrogen and air environments. J. Chem. Thermodyn. 2021, 161, 106560. 10.1016/j.jct.2021.106560. [DOI] [Google Scholar]
- Zhang Z.; Yang L.; Luo S.; Tian M.; Tachibana K.; Kamijima K. Ionic liquids based on aliphatic tetraalkylammonium dications and TFSI anion as potential electrolytes. J. Power Sources 2007, 167, 217–222. 10.1016/j.jpowsour.2007.01.033. [DOI] [Google Scholar]
- Yi J.; Liu S.; Abu-Omar M. M. Rhenium-catalyzed transfer hydrogenation and deoxygenation of biomass-derived polyols to small and useful organics. ChemSusChem 2012, 5, 1401–1404. 10.1002/cssc.201200138. [DOI] [PubMed] [Google Scholar]
- Li J.; Lutz M.; Gebbink R. J. M. K. N-Donor Ligand Supported “ReO2+”: A pre-catalyst for the deoxydehydration of diols and polyols. Catalysts 2020, 10, 754. 10.3390/catal10070754. [DOI] [Google Scholar]
- Valderrama J. O.; Forero L. A.; Rojas R. E. Critical properties and normal boiling temperature of ionic liquids. Update and a new consistency test. Ind. Eng. Chem. Res. 2012, 51, 7838–7844. 10.1021/ie202934g. [DOI] [Google Scholar]
- Jang J. H.; Sohn H.; Camacho-Bunquin J.; Yang D.; Park C. Y.; Delferro M.; Abu-Omar M. M. Deoxydehydration of biomass-derived polyols with a reusable unsupported rhenium nanoparticles catalyst. ACS Sustainable Chem. Eng. 2019, 7, 11438–11447. 10.1021/acssuschemeng.9b01253. [DOI] [Google Scholar]
- Sharkey B. E.; Jentoft F. C. Fundamental insights into deactivation by leaching during rhenium-catalyzed deoxydehydration. ACS Catal. 2019, 9, 11317–11328. 10.1021/acscatal.9b02806. [DOI] [Google Scholar]
- Sandbrink L.; Klindtworth E.; Islam H.-U.; Beale A. M.; Palkovits R. ReOx/TiO2: A Recyclable Solid Catalyst for Deoxydehydration. ACS Catal. 2016, 6, 677–680. 10.1021/acscatal.5b01936. [DOI] [Google Scholar]
- Luo Q.; Lu T.; Xu J.; Yang X.; Zhou L. Conversion of dihydroxyacetone to methyl pyruvate catalyzed by hybrid molecular sieves at low temperature: A strategy for the green utilization of glycerol. Catal. Lett. 2020, 150, 1641–1649. 10.1007/s10562-019-03064-3. [DOI] [Google Scholar]
- Tomasini E. P.; Halac E. B.; Reinoso M.; Di Liscia E. J.; Maier M. S. Micro-Raman spectroscopy of carbon-based black pigments. J. Raman Spectrosc. 2012, 43, 1671–1675. 10.1002/jrs.4159. [DOI] [Google Scholar]
- Parker S. F.; Herman H. The vibrational spectrum of methyltrioxorhenium(VII). Spectrochim. Acta, Part A 2000, 56, 1123–1129. 10.1016/S1386-1425(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Purans J.; Kuzmin A.; Cazzanelli E.; Mariotto G. Disorder-induced Raman scattering in rhenium trioxide (ReO3). J. Phys. Condens. Matter 2007, 19, 226206. 10.1088/0953-8984/19/22/226206. [DOI] [Google Scholar]
- Hofmann B. J.; Harms R. G.; Schwaminger S. P.; Reich R. M.; Kühn F. E. Reactivity of Re2O7 in aromatic solvents – Cleavage of a β-O-4 lignin model substrate by Lewis-acidic rhenium oxide nanoparticles. J. Catal. 2019, 373, 190–200. 10.1016/j.jcat.2019.03.042. [DOI] [Google Scholar]
- Gurzęda B.; Buchwald T.; Krawczyk P. Thermal exfoliation of electrochemically synthesized graphite intercalation compound with perrhenic acid. J. Solid State Electrochem. 2020, 24, 1363–1370. 10.1007/s10008-020-04642-x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.