
Fig. 3 |. The conserved organization of the classical AAA+ domain.
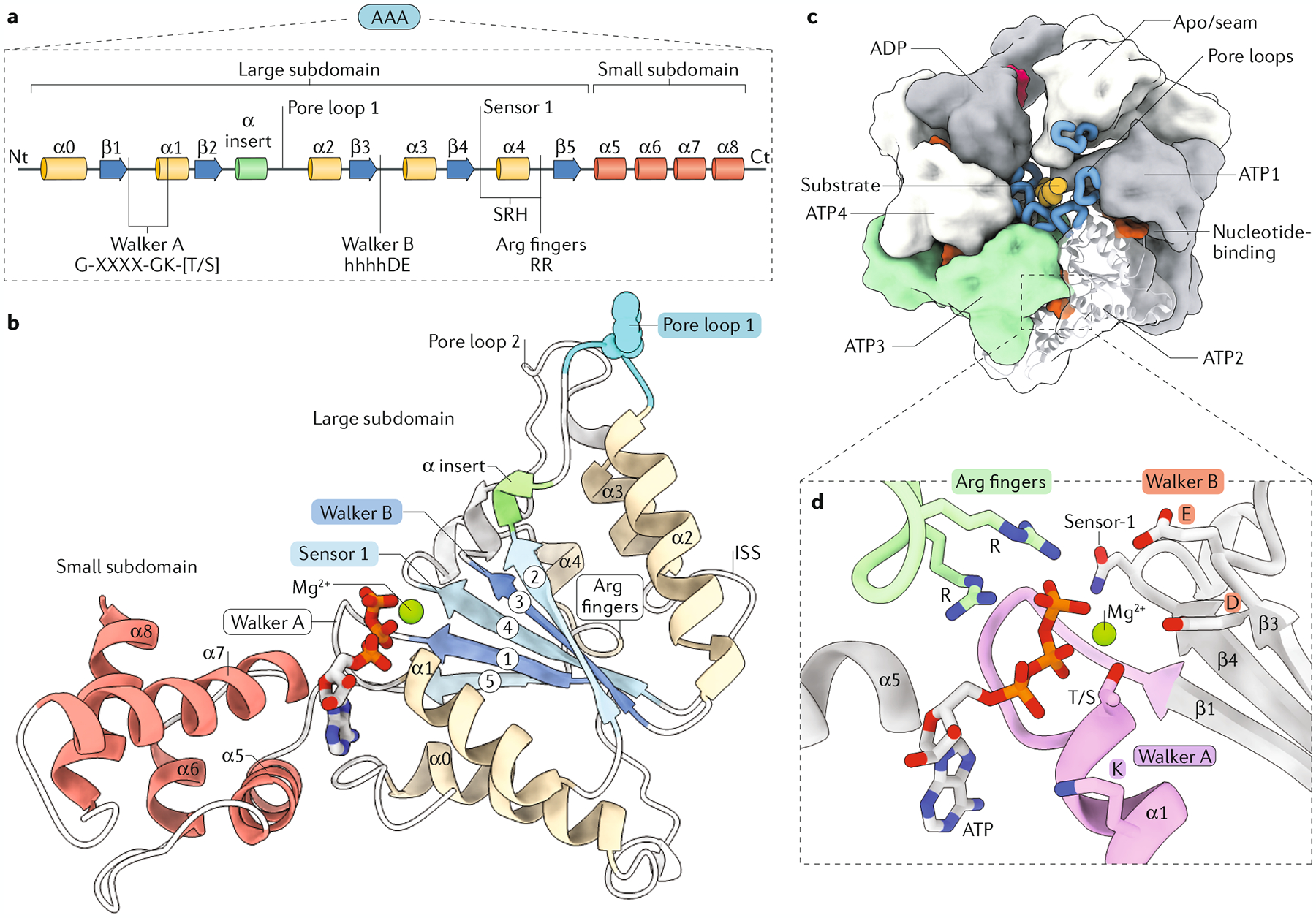
a | Linear diagram of the conserved secondary structure elements of the ATPase associated with diverse cellular activities (AAA+) domain, highlighting the position of the motifs required for activity: Walker A (G-XXXX-GK-[T/S] sequence motif, where X can be any amino acid), pore loop 1, Walker B (comprising conserved aspartic acid (D) and glutamate (E) preceded by several hydrophobic amino acids — hhhhDE), sensor 1 and the arginine (Arg) fingers (RR) found in the second region of homology (SRH). b | Representative atomic model (pdb:6AZ0) of the AAA+ domain labelled as in part a. Inter-subunit signalling (ISS) motif functions in inter-subunit coupling and coordination of the ATPase cycle in some AAA+ proteins (FIG. 7). c | Overview of the hexameric, ring-like organization characteristic of AAA+ proteins, with the pore loops (blue) interacting with the substrate (orange), which is threaded through the centre of the complex. The nucleotide-binding pocket localizes to the inter-subunit interface and is enlarged in part d. The nucleotide state for each subunit is highlighted (bound to ATP (ATP1–4), bound to ADP, nucleotide-free (apo)). d | The ATP-bound nucleotide-binding pocket with the conserved motifs and residues involved in ATP binding and hydrolysis in cis, with the Arg fingers from the adjacent subunit shown in green. Ct, carboxy terminus; Nt, amino terminus.