

Summary
Genetic heterogeneity, reduced penetrance, and variable expressivity, the latter including asymmetric body axis plane presentations, have all been described in families with congenital limb malformations (CLMs). Interfamilial and intrafamilial heterogeneity highlight the complexity of the underlying genetic pathogenesis of these developmental anomalies. Family-based genomics by exome sequencing (ES) and rare variant analyses combined with whole-genome array-based comparative genomic hybridization were implemented to investigate 18 families with limb birth defects. Eleven of 18 (61%) families revealed explanatory variants, including 7 single-nucleotide variant alleles and 3 copy number variants (CNVs), at previously reported “disease trait associated loci”: BHLHA9, GLI3, HOXD cluster, HOXD13, NPR2, and WNT10B. Breakpoint junction analyses for all three CNV alleles revealed mutational signatures consistent with microhomology-mediated break-induced replication, a mechanism facilitated by Alu/Alu-mediated rearrangement. Homozygous duplication of BHLHA9 was observed in one Turkish kindred and represents a novel contributory genetic mechanism to Gollop-Wolfgang Complex (MIM: 228250), where triplication of the locus has been reported in one family from Japan (i.e., 4n = 2n + 2n versus 4n = 3n + 1n allelic configurations). Genes acting on limb patterning are sensitive to a gene dosage effect and are often associated with an allelic series. We extend an allele-specific gene dosage model to potentially assist, in an adjuvant way, interpretations of interconnections among an allelic series, clinical severity, and reduced penetrance of the BHLHA9-related CLM spectrum.
Keywords: allelic series, Alu/Alu-mediated rearrangement, birth defect, clinical genomics, congenital limb malformation, developmental genomics, exome sequencing analysis, gene dosage effect, limb development, SV mutagenesis
Graphical abstract
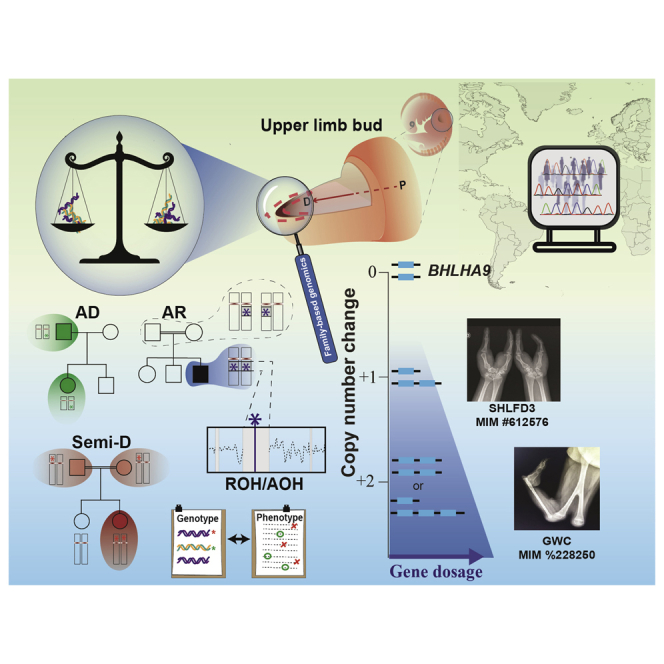
We implemented family-based genomics and rare variant analyses to explore the developmental genomics of congenital limb malformations. The results indicate that both allelic series and gene dosage effects contribute to the observed clinical variability. The allele-specific gene dosage model (AsGD) explains the BHLHA9-related clinical spectrum including the Gollop Wolfgang Complex (MIM 228250).
Duan et al. implemented family-based genomics and rare variant analyses in 18 families with CLMs. The results show that allelic series and gene dosage effects contribute to the clinical variability . The allele-specific gene dosage model (AsGD) explains observations of the BHLHA9-related clinical spectrum including the Gollop-Wolfgang complex (MIM: 228250).
Introduction
Vertebrate limb development is a sophisticated, patterned process with the limbs formed in the early embryonic stages because of underlying mesodermal-ectodermal cellular interactions. Orchestrated by complex “fine-tuned” gene dosage expression, tissue/cell gradients of developmental factors or signal molecules are formed through inductive interactions between mesoderm/ectoderm tissues and oscillation of several signal transduction pathways.1, 2, 3 3D axes (anterior/posterior [A/P], dorsal/ventral [D/V], and proximal/distal [P/D]) are interconnected to coordinate homeostatic cell population/tissue/organismal development of the apical ectodermal ridge (AER), zone of polarizing activity (ZPA), and limb bud development (Figure 1). Disruption of gene action or gene regulation for genes executing tissue proliferation, differentiation, cell migration, or stratification for each axial patterning plane can cause congenital limb malformation (CLM).
Figure 1.

A schematic of embryonic upper limb development
An illustrative drawing describes a human embryo at Carnegie stage 12 (top left; approximately 4 weeks after conception), when the limb bud forms as a ventrolateral bulge and begins to emerge outwardly. Also shown is a magnified illustration depicting the 3D patterning of the upper limb bud and a dissected view of the anatomical position between ectoderm and mesoderm. The P/D, A/P, and D/V axes are labeled with different colors. The AER is labeled at the brown epithelial region, ZPA is shown as a gradient-based yellow zone. Bottom right: illustration of a mutual genetic antagonism between the short isoform of GLI3 (GLI3R) and HAND2 that is required to establish a “pre-patterning” of the A/P axis prior to formation of the SHH-ZPA gradient. Top right: P/D elongation is regulated by many signaling elements, including FGF8, TP63, and BHLHA9.
CLM represents a broad spectrum of prenatal developmental defects, including limb reduction, appendicular skeletal anomalies, and/or abnormalities in the number, length, anatomical positioning, and anatomy of the digits.4 CLMs affect approximately 1 in 500 newborns, with occurrence as a deformation, disruption, or isolated developmental event (i.e., malformation), or it can be a manifestation of an endophenotype of a rare Mendelian disease trait.5,6 In addition to extensive interfamilial and intrafamilial clinical and genetic heterogeneity, incomplete penetrance and variable expressivity have been frequently highlighted in CLM studies; these genetic features are among some of the inherent challenges of concluding molecular diagnosis(es) for children and families manifesting CLM and for genetic diagnoses from unphased genomic information.
During the past decade, elucidation of the molecular etiology of CLM has been facilitated by advances in high-throughput family-based genomics, rare variant allele studies, computational tools, and population variation studies, driving the molecular diagnostic “solved rate” of a random CLM-related cohort to be reported with a range of 10%–35% in different populations.7, 8, 9, 10, 11 To date, more than 150 “disease trait genes or loci” have been attributed to CLM in affected individuals and families. A diverse mutational landscape comprised of single-nucleotide variant (SNV) alleles, small insertion or deletion (indel) events, simple sequence repeat expansions, and locus copy number variant (CNV) alleles involving coding or non-coding regulatory regions has been elucidated.8,12,13
Despite the continuing efforts of genome-wide screening and novel molecular etiological insights provided for CLM, many candidate genes/loci remain uncharacterized or under-characterized, or allelic series at a locus are not explored. Some clinically well-described CLMs with numerous case reports published through decades of research still lack a clear underlying contributing genetic variant explanation: examples include Gollop-Wolfgang complex (GWC; MIM: 228250) and fibular aplasia, tibial campomelia, and oligosyndactyly (FATCO) syndrome (MIM: 246570). Recurrent CLM-associated CNVs and other variant types are often poorly elucidated and mechanistically enigmatic from a mutational mechanism standpoint, whether as de novo allelic variants associated with a sporadic congenital anomaly or contributing as a recently arising mutation in clan ancestors.14 Contributions to a biallelic variant state are not always genetically clear as to when homozygosity results from identity by state (IBS; identical alleles/genomic segments do not share a common ancestor) versus identity by descent (IBD; identical alleles/genomic segments inherited from a common ancestor without intervening recombination). In this study, we implemented family-based genomics and rare variant analyses as a molecular “entry point” to decipher the biology, mutational features, and mutational mechanisms underlying the genetic and developmental basis of CLM.
Methods and materials
Sample preparation
Prior to enrollment for genomic studies, the phenotype of each affected individual was initially investigated by local clinical geneticists and re-evaluated by physicians, including pediatricians, pediatric neurologists, and medical geneticists, in our research genomics group. To reduce the clinical heterogeneity, the recruited cases were primarily those with phenotypic findings involving the appendicular skeleton, focusing on limb anomalies as their major structural morphogenesis defects. Peripheral blood from each registered individual and associated family members was collected locally in EDTA tubes and shipped to our lab for genomic DNA extraction using standard protocols.
Exome sequencing analysis and rare variant SNV prioritization
Family-based exome sequencing (ES) of all subjects was implemented at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) under previously described protocols through the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) initiative.15, 16, 17 In brief, pre-capture library preparation was performed using KAPA HyperPrep Kits, followed by the HGSC-designed core capture protocol (52 Mb, Roche NimbleGen).18 Paired-end sequencing with multiplex pools was executed using an Illumina Hiseq2000 or NovaSeq6000, generating an average depth of coverage of ∼100×. Subsequently, post-sequencing data were mapped to the haploid human genome reference sequence assembly (hg19) together with variant calling, post-processing, annotation, and quality control under a standardized HGSC-Mercury pipeline as implemented previously and deployed in the cloud.19
An unbiased, stepwise computational analysis workflow was employed for SNV filtering, parsing, and prioritization.20 Rare variants were extracted comprehensively based on their minor allele frequency (MAF), calculated collectively from an internal BCM-HGSC exome variant database consisting of personal genomes from ∼13,000 individuals and a variety of public genomic databases, including the Genome Aggregation Database (gnomAD), 1000 Genomes Project (TGP), and the Atherosclerosis Risk in Communities (ARIC) study database. Filtered variants were annotated and prioritized for further study based on MAFs (<0.5% as threshold) and pathogenicity prediction algorithms, including combined annotation-dependent depletion (CADD; >20 as deleteriousness)21 and the rare exome variant ensemble learner (REVEL; >0.5 as likely damaging)22 for missense variants. Then each annotated variant was further parsed and prioritized via a comprehensive data-mining process based on additional gene- and variant-level information from publicly available databases, including Online Mendelian Inheritance in Man (OMIM; http://www.omim.org), PubMed, the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Variant validation and segregation analysis
All candidate SNVs and indels were orthogonally validated by Sanger dideoxy sequencing of the target region amplified using conventional PCR (HotStar Taq, QIAGEN). Familial segregation analysis was examined in accordance with Mendelian expectations on all identified candidate variants alleles, with potential penetrance evaluated by physical examination and radiographs.23
CNV identification and analysis
Large CNVs (>3-exon deletion/duplication) were initially called in silico from exome data via several read-depth-based algorithms, including exome-hidden Markov model (XHMM) and HMZDelFinder for smaller homozygous deletion.24,25 Using a family-based genomics approach and rare CNV filtering, potential pathogenic CNVs were detected (families HOU1397, HOU3358, and HOU3586).
The genomic architecture of each potential pathogenic CNV was experimentally investigated in affected individuals and selected family members by customized high-definition array-based comparative genomic hybridization (HD-aCGH), using arrays designed by implementing the eArray system (https://earray.chem.agilent.com/suredesign/) and array platforms manufactured by Agilent (AMAID: 086724). Microarray protocols, including DNA digestion, probe labeling, gender-matched hybridization, and post-washing with minor modifications, were performed as described previously.26 Agilent SureScan and Feature Extraction software was utilized to achieve the image-to-digital transition, with further data analysis and visualization on the Agilent Genomic Workbench.
Genes mapping within genomic intervals potentially susceptible to genomic instability or Alu/Alu-mediated rearrangement (AAMR) were investigated by AluAluCNVpredictor for a relative risk genomic instability index or score, implementing a recently described informatics tool.27,28 Approximate genomic intervals of CNVs were determined at single W-C base-pair resolution via breakpoint junction analysis using Sanger dideoxynucleotide sequencing.
Absence of heterozygosity (AOH) and co-efficient of consanguinity analysis
Genomic data evidence for potential consanguinity was investigated quantitatively based on AOH, which can serve as a surrogate measure of runs of homozygosity (ROHs), to profile potential IBD genomic intervals and a coefficient of consanguinity (i.e., the fraction of the assayable human genome haploid reference), as described previously.29,30 For an individual genome, ROH genomic regional analyses often provide additional evidence for variant prioritization, serving as an adjuvant tool to probe the genetic and allelic genomic architecture and autozygous intervals (IBD). In this study, AOH data visualization and analysis for individual personal genomes were achieved through BafCalculator, an in-house-developed bioinformatics method for B allele frequency computation from unphased ES-variant data.31
Study approval
Eighteen families with clinical diagnoses of CLM were recruited into the BHCMG under an institutional review board (IRB; H-29697)-approved study.15 All participating subjects signed informed consent regarding data sharing and publication of medical information and photographs.
Results
Characteristics of study subjects and family-based genomic analyses
From 2012–2020, 18 families with rare congenital defects involving limbs or the appendicular skeleton were enrolled into the BHCMG, now the BCM-Genomics Research to Elucidate the Genetics of Rare (BCM-GREGoR) disease consortium, with a mandate to “solve the unsolved.” Using family-based genomics and rare variant analyses, 7 SNV and 3 CNV alleles were identified as plausible molecular diagnostic etiologies for 11 of the 18 studied families, interpreted by American College of Medical Genetics and Genomics (ACMG)-based variant classification criteria (Table 1 and Table 2).32,33 Most gene products involve the P/D and A/P axial formation of the limbs.
Table 1.
Summary of identified SNVs and CNVs in the cohort
| Family | Country of origin | Consanguinity | Gene/locus | Variant type | Variant (GRCh37\hg19) | Zygosity | Allele count (gnomAD) | CADD score (Phred-scale) | Conservation, (phyloP20way_mammalian) | ACMG variant classification criteria |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Evidence category/section | Classification | |||||||||||
| SNV | HOU2084 | Turkey | no | GLI3 | nonsense | NM_000168: | Htz | 0 Htz−0 Hmz | N/A | 1.048 | PVS1, PM2, PP1, PP3 | pathogenic |
| c.1673C>A (p.Ser558∗) | ||||||||||||
| HOU3022 | Turkey | no | HOXD13 | missense | NM_000523: | Htz | 0 Htz−0 Hmz | 28.8 | 0.977 | PM2, PP2, PP3 | uncertain significance | |
| c.623A>T (p.Asp208Val) | ||||||||||||
| HOU3212 | Turkey | yes (1O cousins) | NPR2 | missense | NM_003995: c.1673T>C (p.Ile558Thr) | Hmz | 2 Htz−0 Hmz | 25.5 | 0.997 | PS1, PM2, PP1, PP3 | likely pathogenic | |
| HOU2130 | Turkey | yes (1O cousins) | NPR2 | missense | NM_003995: c.277C>A (p.Pro93Thr) | Hmz | 0 Htz−0 Hmz | 24.4 | 1.026 | PM2, PP1, PP2, PP3, PP4 | likely pathogenic | |
| HOU1409 | Turkey | yes (1O cousins) | NPR2 | nonsense | NM_003995: c.1087C>T (p.Arg363∗) | Hmz | 1 Htz–0 Hmz | N/A | N/A | PVS1, PM2, PP1, PP3, PP4 | pathogenic | |
| HOU1410 | Turkey | yes (1O cousins) | NPR2 | missense | NM_003995: c.2720C>T (p.Thr907Met) | Hmz | 0 Htz−0 Hmz | 32 | 1.048 | PS1, PM2, PP1, PP3 | likely pathogenic | |
| HOU2346 | Turkey | yes (1O cousins) | WNT10B | frameshift | NM_003394: c.741delC (p.Cys247∗) | Hmz | 0 Htz−0 Hmz | N/A | N/A | PVS1, PM2, PP1, PP3, PP4 | pathogenic | |
| HOU3360 | ||||||||||||
| CNV | HOU1397 | Turkey | no | 2q31.1 | deletion | chr2:171,524,396–178,694,337; 7.16 mb | Htz | 1A, 2A, 3C, 5A | pathogenic | |||
| HOU3586 (F35a) | Brazil | no | 17p13.3 | duplication | chr17:1,124,394–1,186,190, 61.8 kb | Htz | ` | 1A, 2A, 3A, 4H, 4L, 5D | pathogenic | |||
| HOU3358 | Syria | yes (1O cousins) | 17p13.3 | duplication | chr17:1,164,471–1,239,336, 74.9 kb | Hmz | 1A, 2A, 3A, 4L, 5D | pathogenic | ||||
Hmz, homozygous; Htz, heterozygous; N/A, not available; CADD, combined annotation-dependent depletion.
This family was published previously as family 35 (F35) by da Rocha et al.53
Table 2.
Summary of clinical information in 18 unrelated families with phenotypic findings on limbs and appendicular skeletons
| Family | Individual | Dz, OMIM | Phenotype of upper limbs |
Phenotype of lower limbs |
Other phenotypes and information | ||||
|---|---|---|---|---|---|---|---|---|---|
| Left | Right | Left | Right | ||||||
| A/P | HOU2084 | BAB5343 | polydactyly preaxial type 4; MIM: 174700 | syndactyly between third and fourth digits, postaxial polydactyly | bilateral preaxial polydactyly of halluces, syndactyly involving second and third toes | Solved | |||
| HOU3022 | BAB8289 | synpolydactyly type 1; MIM: 186000 | mesoaxial polydactyly and syndactyly on the fourth digit | postaxial polydactyly on fifth toe, syndactyly involving fourth toe | |||||
| P/D | HOU3212 | BAB8908 | acromesomelic dysplasia, Maroteaux type (AMDM); MIM: 602875 | bilateral macrodactyly of halluces with brachydactyly of toes | |||||
| HOU2130 | BAB5498 | AMDM; MIM: 602875 | brachydactyly; short, broad metacarpals/metatarsals and phalanges; restricted elbow extension | pectus excavatum, micromelia with predominant rhizomelia | |||||
| BAB5499 | short, bowed forearm; platyspondyly; thoracic kyphosis; and congenital hip dislocation | ||||||||
| HOU1410 | BAB3611 | AMDM; MIM: 602875 | Brachydactyly; short, broad metacarpals/metatarsals and phalanges; hyperextensibility of metacarpals; restricted elbow extension | short and thick humeri, platyspondyly | |||||
| BAB3612 | Brachydactyly; short, broad metacarpals/metatarsals and phalanges; tubular bone shortness | short and thick humeri, platyspondyly, flattened acetabulum and coxa vara | |||||||
| HOU1409 | BAB3606 | AMDM; MIM: 602875 | Brachydactyly; short, broad metacarpals/metatarsals and phalanges | short and thick humeri, platyspondyly | |||||
| BAB3607 | |||||||||
| HOU2346 | BAB6262 | split-hand/foot malformation (SHFM) type 6; MIM: 225300 | syndactyly involving third and fourth digit | bilateral split foot and syndactyly | |||||
| BAB8610 | joint contracture on second digit | ||||||||
| HOU3360 | BAB9281 | bilateral split hand, oligodactyly, and syndactyly | bilateral split foot and syndactyly | ||||||
| BAB9286 | |||||||||
| BAB9316 | |||||||||
| HOU1397 | BAB4812 | SHFM5; MIM: 606708 | bilateral split hand, oligodactyly, and syndactyly | bilateral syndactyly involving third and fourth toes | see Supplemental Note | ||||
| HOU3586 (F35a) | BAB9661 (F35-1) | SHFM with long bone deficiency type 3; MIM: 612576 | bilateral split hand | bilateral tibial aplasia | |||||
| BAB9662 (F35-2) | bilateral split hand, triphalangeal thumbs | bilateral tibial aplasia | |||||||
| HOU3358 | BAB9273 | GWC; MIM: 228250 | bilateral femur bifurcation, absent of tibia and monodactyly | ||||||
| N/A | HOU1558 | BAB4015 | N/A | preaxial polydactyly (seven digits) with duplicated hallux and second toe | sacral dimple, age-appropriate neuromotor development, secundum type atrial septal defect, normal brain MRI, abdominal ultrasound and eye examination; no consanguinity between parents, and our child is the first child of the couple | unsolved | |||
| HOU1780 | BAB4381 | N/A | anteriorly placed fifth toe | 5-day-old male infant born at 33rd gestational week with a birth weight of 1,800 g; atresia of anus and duodenum, hypospadias, high and narrow palate, atypical hair color; karyotype and cranial ultrasound unremarkable; died because of multiple medical comorbidities; parents were first-degree cousins | |||||
| HOU1841 | BAB4486 | fibular aplasia, tibial campomelia, and oligosyndactyly (FATCO) syndrome; MIM: 246570 | unilateral (right) fibular aplasia, tibial bowing and hypoplasia (campomelia), rudimentary right foot with oligosyndactyly | parents are unrelated, he has one healthy sister | |||||
| HOU2133 | BAB5510 | oligosyndactyly with micromelia on second and third digits | oligodactyly with absence of fifth ray | bilateral fibular aplasia, tibial campomelia, and oligosyndactyly | case report was previously published by Sezer et al.99 | ||||
| HOU2245 | BAB5902 | N/A | bilateral camptodactyly with left hand oligodactyly, single palmar crease | brachydactyly on fourth and fifth toes | short statute, facial dysmorphism, narrow thorax, patent ductus arteriosus, ID, and splenomegaly | ||||
| HOU4419 | BAB11857 | N/A | bilateral upper limb deformity with absence of thumbs | normal weight, height, and head circumference; epilepsy; no consanguinity between parents | |||||
| HOU4083 | BAB11205 | N/A | syndactyly between second and third digits | bilateral oligodactyly, short and bowing femora | see Supplemental Note | candidate (NLK: c.982C>T [p.Gly328∗]) | |||
Dz, disease; A/P, anterior/posterior; P/D, proximal-distal.
This family was published previously as family 35 (F35) by da Rocha et al.53
Intrafamilial phenotypic variability was commonly observed in the studied families and associated with reduced penetrance and variable expressivity (Table 2). Sometimes, variability in presentation manifests as unilateral or asymmetric anatomical patterns (e.g., left/right, upper/lower limbs) for an individual, with malformation observed specifically in an asymmetric pattern on only the left/or right side or upper/lower limbs.
Novel variant identification involving genes contributing to A/P axis patterning
Polydactyly preaxial type IV (PPD4; MIM: 174700), alternatively known as polysyndactyly, represents manifestations characterized by a bilateral polydactyly presenting preaxially in the thumb/hallux, with variable syndactyly seen in other digits (fingers/toes).34,35 In our cohort, PPD4 was observed in multiple members of a Turkish family (HOU2084) as an apparent autosomal dominant (AD) trait (Figures S1A–S1B). ES analyses of three affected individuals revealed a novel heterozygous nonsense variant in the Glioma-associated oncogene (GLI)-Kruppel family member 3 gene, GLI3 (NM_000168:c.1673C>A [p.Ser558∗]). Segregation studies fulfilled Mendelian expectations for a dominant allele with complete penetrance (Figure S1A). This variant allele is predicted by conceptual translation to result in a premature termination codon (PTC) within the N-terminal zinc-finger domain (ZFD) of GLI3; this PTC is predicted to trigger nonsense-mediated decay (NMD), generating a loss-of-function (LoF) variant allele. This interpretation is further supported here by the evidence of a genotype-phenotype observation in the GLI3-nonsense variant carriers in family HOU2084, showing only the essential clinical features of PDD4 but no additional findings (Figure S1B).
During limb A/P axial development, a mutual genetic antagonism between GLI3 and heart- and neural crest derivative-expressed protein 2 (HAND2) is required to establish a “pre-patterning” of the A/P axis prior to ZPA formation of the Sonic hedgehog (SHH) gradient (Figure S1C). In contrast to GLI3, HAND2 is expressed in a cell-restricted manner in the posterior mesenchymal region and activated by a direct upstream regulator, Homeobox D13 (HOXD13) (Figure S1C).
Synpolydactyly (SPD1; MIM: 186000) is another “phenotypic combination” with a typical clinical presentation characterized by mesoaxial polydactyly and syndactyly, particularly involving fingers 3/4 and toes 4/5.36,37 Trio-ES rare variant analyses of a sporadic proband, BAB8289, who manifested a typical SPD1 phenotype, found a maternally inherited and novel heterozygous missense mutation of HOXD13 (NM_000523:c.623A>T [p.Asp208Val]) (Figures S1D and S1E). This variant allele is absent in gnomAD and BCM-GREGoR internal databases comprising exome data from ∼13,000 individuals. The mother of the proband seems non-penetrant because variant reads are not supportive of mosaicism (variant read to total read ratio as 82/158). This finding in BAB8289 showed that a dominant cause of the disease could be identified in a family from an admixed population with a high coefficient of consanguinity.
Multiple mutation types for pathogenic variant alleles of HOXD13 segregate with SPD1.38 Aggregated evidence implicates LoF variants of the HOXD13 functional domain driving the typical features of SPD1 and anticipated haploinsufficiency as the most plausible mechanism for the molecular pathogenesis of SPD1.39,40 The p.Asp208Val substitution altered an evolutionarily conserved residue outside of, but adjacent to, the homeodomain of HOXD13 (Figure S1D); this variant allele mapped 12 amino acids from a previously reported pathogenic missense variant, p.Gly220Val. As measured by dosage changes, the latter variant allele has been shown experimentally to jeopardize protein stability through aberrant cytosolic accumulation.41
Biallelic disruption of NPR2 dysregulating longitudinal skeletal growth
Natriuretic peptide receptor B/guanylyl cyclase B, encoded by NPR2, is one of the homodimeric transmembrane hormonal receptors regulating longitudinal skeletal growth during the process of endochondral ossification. Biallelic LoF variants in NPR2 are known to cause autosomal recessive (AR) acromesomelic dysplasia, Maroteaux type (AMDM; MIM: 602875), an endochondral bone growth disorder characterized by significant short stature, predominantly affecting the forearms and hands.42,43 Here we identified two novel and two known pathogenic variants of NPR2 from four unrelated consanguineous Turkish families (HOU2130, HOU1409, HOU3212, and HOU1410) (Figure 2A). Short stature and acromesomelia, characterized by short and bowed forearms and symmetrically short and broad metacarpals and phalanges with fingernail hypoplasia, are observed in most of the affected individuals (Figures 2B and S2; Table 1B).
Figure 2.

Biallelic disruption of NPR2 dysregulating longitudinal skeletal growth
(A) Four pedigrees show the segregation of disease-causing variant alleles in each family with AMDM. From left to right: HOU2130 (c.277C>A [p.Pro93Thr]), HOU1409 (c.1087C>T [p.Arg363∗]), HOU1410 (c.2720C>T [p.Thr907Met]), and HOU3212 (c.1673T>C [p.Ile558Thr]).
(B) Clinical images and radiographs from selected individuals with NPR2-related skeletal dysplasia. The top panel shows brachydactyly, nail hypoplasia, and symmetrically short and broad metacarpals and phalanges. From left to right: BAB5498, BAB5499, BAB3606, BAB3607, and BAB3611. The left bottom panel shows similar findings in the feet. From left to right: BAB5499, BAB3607, and BAB3611. The right bottom panel shows a distinct feature in BAB8908, with macrodactyly of halluces with long and wide metatarsals and phalanges of the great toes as well as brachydactyly of other toes because of short metatarsals and phalanges.
(C) A linear protein structure of the transmembrane receptor NPR2, with four identified variant alleles marked in correlated positions (missense variants are labeled as a black line, and a purple line denotes a nonsense variant). Different functional domains and extracellular/cytoplasmic territories of NPR2 are highlighted in different colors.
(D) AOH studies on chromosome 9, visualized by B allele frequency data from personal genomes of two affected individuals in family HOU2130 and proband from family HOU3212. The top two panels for family HOU2130 describe a 2.1 Mb (for BAB5498) and 2 Mb (for BAB5499) interval of AOH genomic interval haplotype block (gray shade) surrounding the causative variant of NPR2 (red vertical line), marked with thick gray rectangles. The bottom panel for HOU3212 denotes a 5.2 Mb AOH interval encompassing the phenotype-associated NPR2 variant allele identified in individual BAB8908.
(E) An illustrative drawing showing the NPR2 homodimer and process of CNP-induced cGMP production. Shown are four identified variant alleles located in different functional domains, and each side of transmembrane territories.
In family HOU2130, a novel homozygous missense variant (NPR2: c.277C>A [p.Pro93Thr]) was identified through ES analysis of two affected siblings (BAB5498 and BAB5499) with AMDM. This missense variant results in alteration of a conserved residue (p.Pro93Thr) in the C-type natriuretic peptide-binding domain (CNP) of NPR2 and potentially disrupts the CNP binding affinity. AOH studies mapped the variant to overlapping 2-Mb genomic intervals of AOH/ROH in BAB5948 and BAB5949 encompassing NPR2, whereas the total AOH was 295 Mb and 239.5 Mb, respectively, consistent with a clan genomics-derived allele versus a Turkish founder allele (Figures 2A–2D).29
Affected individuals of family HOU1409 and family HOU1410 manifest similar AMDM phenotypes and were directly tested by NPR2-targeted Sanger dideoxy sequencing. A novel homozygous nonsense variant of NPR2 (c.1087C>T [p.Arg363∗]) was found in two affected male siblings (BAB3607 and BAB3608) in family HOU1409 segregating with the AMDM phenotype. This novel stop-gain variant allele resulted in a PTC in exon 4 (total 21 exons) of NPR2 and was predicted to be subject to NMD (Figure 2C). In family HOU1410, we identified a known pathogenic homozygous missense variant allele (NPR2, c.2720C>T [p.Thr907Met]) in two affected siblings (BAB3611 and BAB3612) and their affected paternal aunt (BAB3614). This missense variant altered a conserved residue mapping to the guanylate cyclase domain, potentially jeopardizing the process that produces cytoplasmic cyclic guanosine monophosphate (cGMP) from GTP (Figure 2E).
Proband BAB8908 was born to Turkish kindred (HOU3212) with reported parental consanguinity, and the clinical phenotype was manifested by marked bilateral hallux macrodactyly with brachydactyly of the other toes (Figures 2A and 2B). Trio-ES analysis identified a homozygous missense variant allele (c.1673T>C [p.Ile558Thr]) in the kinase homology domain (KHD) of NPR2 (Figures 2B–2D). Segregation analysis confirmed homozygosity for this variant allele in the proband; homozygosity was also found in an affected sister and affected maternal aunt of the proband. Both parents and the unaffected brother were heterozygous carriers consistent with Mendelian expectations (Figure 2A). The same allele has been reported previously in two affected siblings with short-limbed short stature in association with brachycephaly and a dysplastic middle phalanx of the fifth finger.42 AOH studies map the allelic variant to a 5.2 Mb AOH block, whereas the total AOH was 281.6 Mb, consistent with a clan genomics-derived IBD allele versus a Turkish IBS founder allele (Figure 2D).14,29,44
WNT10B dysfunction in limb development and evidence of a potential founder allele
Failure of AER (Figure 1) stratification presumably results in split-hand/foot malformations (SHFM), a subgroup of CLM characterized by maldevelopment of central rays of the distal extremities. SHFM type 6 (SHFM6; MIM: 225300) is a non-syndromic AR trait associated with biallelic frameshift, nonsense, or missense variants of WNT10B.45,46 Studies in mouse embryonic limb bud development reveal Wnt10b as an endogenous ectodermal Wnt ligand specifically expressed in the AER at embryonic day 11.5 (E11.5), whereas its regulatory mechanism remains largely unclear. In this cohort, we identified a novel indel variant that leads to a nonsense allele of WNT10B (c.741delC [p.Cys247∗]); autozygosity of this novel frameshift allele causes a similar SHFM6 phenotype in two unrelated consanguineous Turkish families in our cohort because of IBD (Figures 3A and 3C).
Figure 3.

A novel homozygous NMD-escaping frameshift variant of WNT10B driving the onset of SHFM6 in two unrelated families from Turkey
(A) Pedigree structure with allelic information, showing segregation of the novel indel variant that leads to a nonsense allele (WNT10B: c.741delC [p.Cys247∗]) in family HOU2346. Proband BAB6262 (black arrow) was born from a first-cousin mating. He has two male siblings. One brother, BAB8610, is affected by same SHFM phenotype; the other brother, BAB6263 (shaded green on the pedigree), was diagnosed with cerebral atrophy but no limb malformation.
(B) Clinical images and radiographs showing the hands and feet of two affected individuals (BAB6262 and BAB8610) in association with SHFM6. Note the right-hand 3-4 syndactyly in BAB6262 and hypoplasia with medial deviation of the index finger of the left hand of BAB6263. There is split foot malformation in both, with oligodactyly and syndactyly of lateral toes.
(C) Multigeneration pedigrees of family HOU3360, labeled with allelic information profiling segregation of the same frameshift variant (WNT10B: c.741delC [p.Cys247∗]) and recessive inheritance pattern (i.e., AR trait) in comparison with family HOU2346. ∗BAB9287 is affected by an unknown type of CLM characterized by bilateral hand brachydactyly, clinodactyly, and phalangeal dysplasia. ∗∗BAB9284 has a unilateral polysyndactyly on the second toe of the right foot; its etiology remains unknown).
(D) Clinical photos of the three affected individuals in family HOU3360. From top to bottom: BAB9286, BAB9316, and BAB9281 manifest the SHFM6 phenotype, demonstrating varying degrees of SHFM, oligodactyly, and syndactyly.
(E) AOH genomic intervals mapping to chromosome 12 from ES personal genomes, visualized by B allele frequency computational studies from four affected individuals in families HOU2346 and HOU3360. The top two panels for family HOU2346 describe a 9.6 Mb (for BAB6262) and 21.6 Mb (for BAB8610) interval of AOH genomic interval haplotype block surrounding the causative variant allele of WNT10B (red vertical line), marked with thick gray rectangles. The two bottom panels for HOU3360 denote a shared 19.5 Mb AOH interval encompassing WNT10B variant alleles between individual BAB9281 (proband) and the affected brother of her maternal grandfather, BAB9286.
(F) An illustration from NMDEscPredictor shows the WNT10B cDNA architecture and the position of the frameshift variant located in the NMD escape region (denoted as a green bar).
(G) The top illustration shows a comparison of linear structures or map positions and domains between the WNT10B wild-type (WT) and truncated version (p.Cys247∗, with missing residues denoted by dashed lines). Also shown is a comparison (bottom) of two 3D protein structural models (WNT10B: WT and truncated version) obtained using the AlphaFold platform and visualized on UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). Truncated WNT10B was predicted to lose the entire linker region and CTD, including the amino acids that map to the interface that interacts with the WNT classic receptor (Frizzled protein) and canonical coreceptor LRP5.
In family HOU2346, two male siblings, BAB6262 and BAB8610, manifest an isolated bilateral split-foot malformation characterized by absence of the two central rays. In the upper distal limb, syndactyly is present between the third and fourth digit of the right hand of the proband (BAB6262), whereas his affected brother (BAB8610) has a flexion contracture of his left index finger (Figure 3B). The same WNT10B frameshift variant allele (c.741delC [p.Cys247∗]) was identified in another consanguineous Turkish family in our cohort, HOU3360; homozygosity segregated with SHFM6 phenotypes among three generations (Figure 3C). Similar to the clinical features in family HOU2346, bilateral SHFM with more severe foot manifestations appeared among three affected individuals in family HOU3360 (proband BAB9281 and two siblings of her maternal grandfather, BAB9286 and BAB9316), in association with variable hand abnormalities, including syndactyly, polydactyly, and phalangeal dysplasia.
This frameshift variant allele (WNT10B, c.741delC) results in a PTC in situ (p.Cys247∗) within the last exon; it is predicted by NMDescPredictor to escape NMD surveillance (Figure 3F). Compared with the WNT10B wild-type protein structure, the linker region and C-terminal domain (CTD) of WNT10B that is essential for formation of the Wnt-Frizzled-LRP6 ternary complex, is completely removed in the modeled WNT10B p.Cys247∗ truncation (Figure 3F). These observations suggest the underlying molecular mechanism and that the NMD-escaping frameshift variant allele results in a truncated form of WNT10B that is unable to activate canonical Wnt/β-catenin signaling pathways during AER stratification.
Segregation analysis and ROH/AOH profiling in these two multigeneration families revealed genomic evidence of consanguinity and autozygosity of disease-causing alleles (Figure 3E). In family HOU2346, the WNT10B variant maps within 9.6 Mb and 21.6 Mb genomic intervals of AOH/ROH in BAB6262 and BAB8610, with total AOH/ROH of 251.2 Mb and 217.2 Mb, respectively. In BAB9281 and BAB9286, a similar 19.5 Mb genomic interval of AOH/ROH encompasses WNT10B, which tracked with transmission of the disease-causing allele and provided evidence of intergeneration autozygosity through IBD. This genetic pattern, resulting from a homozygote marrying a recessive carrier, is referred to as a pseudodominant trait.
The youngest child, BAB6263 of family HOU2346, was found to have cerebral atrophy but no limb anomaly (Supplemental Note). Carrier and segregation analysis of BAB6263 identified a homozygous variant in general transcription factor IIIC subunit 1 (GTF3C1: c.4096G>A [p.Glu1366Lys]), and this particular GTF3C1 variant allele has been reported previously by our group, causing cerebellar atrophy in another unrelated Turkish kindred (Figure S3).17 This family illustrates multi-locus pathogenic variation (MPV) within the personal genome of family members rather than the MPV of an individual with different genomic interval autozygosity in different siblings.
Haploinsufficiency of HOXD cluster deletion as a major molecular etiology for SHFM5
Large deletion CNVs encompassing the HOXD cluster have been proposed previously to cause syndromic SHFM5 (MIM: 606708), characterized by SHFM, syndactyly and skeletal hypoplasia in association with ectodermal and craniofacial findings.47, 48, 49
We identified an individual (BAB4812) born to a Turkish family (HOU1397) with no report of parental consanguinity (Supplemental Note). His phenotype resembles a syndromic spectrum of SHFM5: bilateral hand SHFM and variable syndactyly; multiple congenital anomalies, including global developmental delay (GDD) and intellectual disability (ID); hypotonia; and facial dysmorphism (Figures S4A and S4B).
CNV computational analysis from extant ES data of the BAB4812 personal genome via XHMM identified a de novo, ∼7 Mb heterozygous deletion at chromosome 2q31.1. Subsequently, using customized HD-aCGH targeting and interrogating chromosome 2q, a de novo interstitial 7.8 Mb deletion, including 48 genes at chr2q31.1, was identified; this deletion CNV encompassed the entire HOXD gene cluster (HOXD1-3-4-8-9-10-11-12-13) and two adjacent regulatory topologically associating domains (TADs) (Figures S4A and S4C). Thus, the limb phenotype (SHFM5) of this individual is primarily derived from the haploinsufficiency of the entire HOXD territory.
BHLHA9 CNV and semidominant inheritance underlie GWC
Copy number gains of BHLHA9 in humans have been described in association with the triplosensitivity trait (defined as a phenotype produced by an additional copy of the gene) potentially causing a gain-of-function (GoF) hypermorphic allele of SHFM with long bone deficiency (SHFLD3; MIM: 612576) as an AD trait with less than 50% penetrance and variable phenotypic expression.50, 51, 52 Family HOU3586 was referred from Brazil and has been published previously.53 Three individuals manifest a paternally inherited CLM, including SHFM and bilateral tibial aplasia (Figures S5A–S5C). The whole-genome aCGH of each family member revealed a shared 61.8 kb duplication at chr17p13.3 in three affected and three unaffected individuals. The duplication encompasses BHLHA9 driving the onset of SHFLD3.50, 51, 52
GWC (MIM: 228250) is characterized by unilateral bifid femur and tibial bone absence with monodactyly.54,55 The presence of heterozygous triplication involving BHLHA9 (gene copy number = 4) has been reported only once in a single family associated with GWC in the Japanese population, whereas there remains a lack of additional cases reported.56 Here we found a novel allelic contribution by autozygosity-induced homozygous duplication of chr17p13.3, including the BHLHA9 locus. HOU3358 is a consanguineous family from Syria in which proband BAB9273 was clinically diagnosed as having GWC (Figures 4A and 4B). The proband has two unaffected brothers. The parents also had five pregnancies with fetuses that were prenatally diagnosed by ultrasound with bilateral femur bifurcations.
Figure 4.

Homozygous duplication of BHLHA9 causing GWC, illustrating semidominant inheritance
(A) Pedigree structure of HOU3358 with a customized HD-aCGH interrogating chr17p13.3 for each of the family members. An ∼74.9 kb duplication encompassing the full-length of BHLHA9 and TUSC5 was identified in all family members. The proband (BAB9273) is homozygous for this duplication, and both parents (BAB9274 and BAB9275) and her brother (BAB9276) are unaffected heterozygous carriers.
(B) Clinical photos and radiographs of proband BAB9273, showing bifid femurs, tibial hemimelia, and monodactyly of the lower extremities; the upper extremities are observed as normotypical.
(C) B allele frequency of chromosome 17 for the proband showing a 9.8 Mb interval of AOH surrounding the homozygous BHLHA9 duplication, marked with a thick gray line.
(D) A comparison of log10 reads per thousand base pairs per million reads (RPKM) plots of the duplication region from XHMM raw data. RPKM calculations of the proband (red line) and brother (yellow line) show different amplification levels on the reads mapping to the interrogated region (shaded in pink) in contrast to the gray lines delineated as individual ES samples from an internal database with similar experimental conditions.
(E) An illustration showing the proposed model of BHLHA9 tandem duplication formed by AAMR. A single-strand break that occurred in the 3′ AluSx4+ element (yellow mark) introduced microhomology (marked by a blue bar)-mediated TS into another AluSz+ element located in the 74.9 kb upstream region. A pair of primers was designed to capture the recombinant joint of the tandem duplicated allele by producing an amplicon size of ∼2.5 kb encompassing the approximate breakpoint location. A control primer pair was applied on an ∼500 bp unrelated genomic region. The diagram shown below is the 1% agarose electrophoresis of the PCR product; the specific band of ∼2.5 kb is the tandem duplicated allele captured in all members (parents and brother, with the lighter bands denoting heterozygous duplication; while proband with a thick band consistent with homozygous duplication). Sanger sequencing (bottom) of the ∼2.5 kb PCR product of the proband revealed an 8 bp microhomology. Three sequences above the Sanger sequencing trace from top to bottom denote the 3’ AluSx4+ reference (colored in green), the proband “chimeric” sequence, and the 5′ AluSz+ reference.
Initially, CNV analysis using XHMM from family-based exome data detected a potential copy number gain on chr17p13.3 involving BHLHA9 in proband BAB9273 and her unaffected brother, BAB9276 (Figure 4D). Validation studies of each family member via customized chr17p HD-aCGH resolved a 74.9 kb duplication at chr17p13.3 covering the full length of BHLHA9 and tumor suppressor candidate gene 5 (TUCS5) in all four family members (Figure 4A). This duplicated allele was brought into homozygosity in the proband with GWC through IBD, not IBS, resulting in four genomic copies of the gene and genomic duplication interval (Figures 4A and 4C), a gene copy number identical to the heterozygous triplication mentioned above.57
Semidominant inheritance, with a more severe phenotype in the homozygote versus heterozygote, indicates that the individuals who carried heterozygous variant alleles manifest an intermediate phenotype in comparison with the homozygous individual with the same duplication CNV affecting both alleles. Semidominant inheritance has been documented in other human disease traits, such as achondroplasia (ACH; point mutation of FGFR3; MIM: 100800) and Huntington disease (HTT triplet-repat polyglutamine expansion; MIM: 143100), and in gene duplication traits such as that resulting from CMT1A duplication.58,59 Here, homozygous duplication for BHLHA9 results in GWC, a more severe type of limb malformation than SHFLD3; the latter is a rare disease trait caused by heterozygous duplication.
Structural variant (SV) mutagenesis by AAMR
To understand mutagenesis mechanisms underlying the observed CNV events, we applied breakpoint junction analysis on all SV cases, including deletion or duplication CNV alleles. For the de novo HOXD cluster deletion of proband BAB4812 in family HOU1397, analysis of this deleted CNV allele unveiled two flanking Alu elements (AluSx+ and AluSc8−), in the opposite orientation with respect to the haploid reference genome, that generated a complex genomic rearrangement best described as a “deletion-normal/inversion-deletion (DEL-INV-DEL)” sequence involving a two-step template-switching (TS) mechanism (Figures S4D–S4F).60 A 5 bp sequence microhomology and a 6 bp microhomology were found at two breakpoint junctions, respectively, indicating microhomology-mediated break-induced replication (MMBIR) as the likely underlying DNA-replicative repair mutational molecular mechanism (Figures S4D–S4F).
It has been established that chr17p13 is a “genomic instability hotspot” with high susceptibility for genomic rearrangement that frequently involves Alu/Alu recombination.61 AluAluCNVPredictor revealed that BHLHA9 showed an elevated genomic instability index with a high relative risk score of 0.798 for OMIM genes and 0.782 for RefSeq genes (Figure S5E).27 Breakpoint junction analysis of family HOU3586 showed a chimeric repetitive element fused by an AluSx1 and a few upstream nucleotides adjacent to a long terminal repeat (LTR), potentially derived by MMBIR (Figure S5D). Strikingly, another duplicated allele, including BHLHA9 in family HOU3358, was also demonstrated to be driven by an AAMR mechanism, resulting in a chimeric Alu element resulting from recombination of AluSx4 and AluSz in the same orientation, with an 8 bp microhomology at the breakpoint junction (Figure 4E). Thus, de novo mutagenesis in an antecedent generation of the clan derived this allele by SV mutagenesis via AAMR, with IBD bringing it to homozygosity.
Discussion
Novel variant identification facilitating discovery of an allelic series
Family-based genomics studies and rare variant analyses in human populations throughout the world provide an opportunity to decipher a diverse and complex variant spectrum of disease trait-associated loci potentially contributing to CLM; such allelic diversity would not be sufficiently seen/or easily engineered in model organisms.14,44 Identifying complex variation (such as NMD-escaping variant alleles, gene triplication, etc.) provides new insights into the underlying mutational mechanisms enabling extensive allelic series and genotype-phenotype correlation analysis of a given disease-causing locus.
Allelic series is historically a genetic term referring to different “mutant alleles” of a gene or genomic locus that can generate a range of phenotypic trait manifestations. Each phenotype of this range is ascribed to variant alleles within different regions of the same gene/locus.62 For instance, GLI3 and HOXD13 are two genes regulating limb A/P axial development, and their disruption can cause AD-inherited digit abnormalities, as we showed in the families HOU2084 and HOU3022. However, allelic series in both genes are rather intriguing; extensive variant analysis on genotype-phenotype correlation of GLI3 discovered a unique dichotomous relationship between the typical Greig cephalopolysyndactyly syndrome (GCPS) spectrum (MIM: 175700) and a more severe AD disorder called Pallister-Hall syndrome (PHS; MIM: 146510), depending on the location (i.e., genomic map position) of the causative variant allele.63
In contrast to GLI3, similar observational studies of HOXD13 revealed that its disruption was linked to many different digital malformations, including synpolydactyly (SPD1; MIM: 186000), brachydactyly type D (BDD; MIM: 113200), and brachydactyly type E (BDE; MIM: 113300). Although heterozygous polyalanine expansions, frameshift, and nonsense variant alleles of HOXD13 predominantly associate with SPD1, missense variants have been reported to cause SPD1 or BDD/BDE with no positional differentiation.
“Mirror trait” conditions are special allelic series that are defined as resulting from reciprocal genetic/genomic variants (e.g., duplication versus deletion CNV, LoF versus GoF) of the same gene/loci and can cause the observed phenotypes in individuals to appear at the opposite ends of a phenotypic spectrum (e.g., short stature versus tall stature).57,64 As we addressed previously, biallelic LoF variants of NPR2 were frequently reported, causing AMDM characterized by short-limbed short stature. However, a few of the heterozygous missense variant and in-frame deletion alleles of NPR2 were also reported to cause AD epiphyseal chondrodysplasia, Miura type (ECDM; MIM: 615923), characterized by tall stature, arachnodactyly, and long/broad halluces and long metatarsals because of a GoF (potentially a hypermorphic allele) that results in overactivity of guanylate cyclase (Figure S6).65, 66, 67 Genotype-phenotype correlation of these reciprocal traits highlighted their potential underlying etiologies related to abnormal CNP-induced cGMP production. In contrast to AMDM, ECDM-causing alleles were seldomly described (Figure S6).
WNT signaling perturbation affecting P/D axes of limb development
Wnt ligands associated with canonical and non-canonical signaling cascades are essential for limb and skeletal development, particularly required for AER formation and maintenance during P/D and D/V limb bud axial patterning.68, 69, 70, 71 Perturbation of seven genes (DVL1, DVL2, DVL3, FZD2, NXN, ROR2, and WNT5A) acting on non-canonical WNT/planar cell polarity (PCP) signaling pathways are known causes of Robinow syndrome (RS), a congenital skeletal dysplasia with mesomelic limb shortening, distinct craniofacial findings enabling a recognizable pattern of human malformation, and other characteristics.72, 73, 74 In association with clinical and locus heterogeneity, limb shortening has been described as a common endophenotype in RS, and mesomelia (disproportionately short middle portions of the limb) is the most often observed consistent associated clinical finding in individuals with RS.74,75
Although biallelic variants of WNT10B have been described to cause SHFM6 because of LoF alleles, heterozygous missense variant alleles of WNT10B, like WNT10A, can result in tooth agenesis, selective type 8 (STHAG8; MIM: 617073) with no distinct mutational location or nature.76,77 Homozygous mutations in WLS, encoding the Wnt ligand secretion mediator essential for secretion of all Wnt proteins, cause human syndromic structural defects with bilateral split foot and syndactyly.78 For human WNT signaling, we also reported limb findings of bilateral foot oligodactyly and left-hand syndactyly in the individual with a de novo, likely damaging variant allele of Nemo-like kinase (NLK), an inhibitor of the WNT/β-catenin signaling pathway (Figure S7; Supplemental Note).79,80 These clinical observations may not be readily synthesized nor mutational bases discovered and assessed in most standard model organism approaches; thus, the pioneering organism Homo sapiens must be utilized for the allelic series.
CNV of BHLHA9, evidence of extension of an allele-specific gene dosage effect model
Gene dosage is a measurement of the number of copies of a gene in a species genome, whose alteration can simply be made by a CNV encompassing the given locus or gene-transcriptional processes, such as TAD disruptions, that may affect gene action or expression potentially related to a position effect. Rare CNVs are prominently associated with disease in humans and defined the entire field of genomic disorders.81, 82, 83, 84
Disease-causing CNVs are often large (hundreds of kb to Mb sizes), can include more than 1 gene, and are often significantly enriched for genes for transcription factors and/or signaling molecules contributing to developmental processes. Such genes may be subject to a gene dosage effect and may be dosage sensitive, and this aspect may serve as the major determinant of its pathogenicity resulting in haploinsufficient and triplosensitive traits and even mirror traits.57 CNV studies of dosage-sensitive genes have contributed significantly to the progress of disease gene discovery and genomic instability/genome integrity mutagenesis; i.e., SV mutagenesis of the human genome.
BHLHA9 regulates apoptosis during mammalian autopod skeletogenesis and patterning.85,86 In the Bhlha9 knockout mouse, interdigital programmed apoptosis is jeopardized, resulting in various severities of syndactyly of the fingers and hindlimbs/forelimbs.85,86 Abnormal expression patterns of Trp63 and other AER morphogens have been observed in Bhlha9 knockout mice.86 Functional depletion of BHLHA9 has also been observed in individuals with biallelic missense variants involving the DNA-binding domain of BHLHA9, causing mesoaxial synostotic syndactyly with phalangeal reduction (MSSD; MIM: 609432).87 Complex camptosynpolydactyly (CCSPD; MIM: 6097539), characterized by grossly malformed hands with digits arising from the dorsum of the hand and syn-polydactyly, has also been reported in one Indian family.88 The BHLHA9 variant allele we describe here was homozygous, with the position of amino acid change mapping adjacent to the residue substitution observed in association with MSSD.
BHLHA9 is essential for interdigital apoptosis in central limb mesenchyme cells, and biallelic LoF variants ablate this regulatory ability. We hypothesized that copy number gains involving BHLHA9 might enhance its regulatory potential; thus, the apoptosis activity may be overly or ectopically increased in central limb mesenchymal cells during limb development, causing SHFM with or without long bone deficiency. These phenomena are somewhat analogous to a “mirror trait”;64 this, together with its strict spatiotemporal expression pattern, suggested BHLHA9 as a dosage-sensitive gene. Further support for that contention comes from disrupted TAD findings in association with duplication CNV.89 Therefore, individually subtle differences of perturbations in gene expression caused by CNVs of BHLHA9 may explain the phenotypic variability frequently described in the literature.90, 91, 92 Evidence shows that such dominant disease-causing CNVs with less than 50% penetrance, despite having a large effect size, could arise ancestrally and form as “founder pathogenic alleles” frequently transmitted in the Japanese population.56 Mirror traits at the Smith-Magenis syndrome (MIM: 182290)/Potocki-Lupski syndrome (MIM: 610883) locus and the schizophrenia and autism chr1q21.1 loci associated with microcephaly/macrocephaly are mirror trait loci for loci manifesting gene dosage effects.57,64,93
Triplication and quadruplication involving this locus have also been described in different families associated with SHFLD3 and GWC phenotypes in different populations, with the common observation that the copy number gain value (i.e., the number of gene copies gained) enhances the likelihood of penetrance and severity of the phenotype.56,88 We observed that homozygous duplication of BHLHA9 can cause GWC, in which SHFLD3 can be seen as an intermediate phenotype. Here we propose that BHLHA9-associated limb malformations serve as a disease model with the association of penetrance and phenotype severity determined by its magnitude of effect on gene function (Figure 5).44,94, 95, 96 The resultant potential distortions of biological homeostasis could parsimoniously explain how such a “gene dosage expression model” significantly contributes to limb anomalies and other developmental birth defects.44,57
Figure 5.

Allele-specific gene dosage model for perturbation of BHLHA9 action
This illustration uses BHLHA9 as a “molecular entry point” describing the potential genotype-phenotype correlation, penetrance, and expressivity. These observations can be parsimoniously explained by the perturbation of gene dosage underlying different genomic events, e.g., duplication versus triplication or quadruplication of the locus. Copy number gain of BHLHA9 can abnormally enhance apoptosis during limb P/D development, causing the SHFLD phenotype. For individuals, an increased gene copy number increases the gene dosage, reinforcing the phenotype severity, likelihood, and penetrance, with homozygosity resulting in the most intense magnitude of effect (GWC). In contrast, biallelic LoF variant of BHLHA9 result in an attenuated gene dosage, driving a distinct MSSD phenotype characterized by insufficient apoptosis activity during AER stratification.
Challenges and prospects
We provided further support for the contention that the etiological elements of developmental anomalies are rather heterogeneous. With current genome-wide methods (ES and aCGH in this study), we were incapable of revealing other types of genomic events, such as non-coding variant alleles (SNVs or CNVs), copy-number-neutral structural variants, and even some types of indels and repeat expansions (e.g., di-, tri-, and tetranucleotide repeats). Other factors known to cause limb defects, such as postzygotic/somatic mutations, mosaicism, fetal deformation, or exposure to environmental mutagens, are challenging to investigate and positively control.
Prospective short-read and long-read whole-genome sequencing (WGS), and RNA sequencing (RNA-seq) will be employed to detect additional causes in unsolved cases. Concerning overall phenotypic heterogeneity, human phenotype ontology (HPO)-based quantitative phenotypic analysis will be adapted in the future to quantify the limb anomaly trait and investigate the allele-specific differences (or to uncover novel allelic series) in similar CLM spectrums as well as to dissect MPV in AR or AD blended traits.74,97 Autozygosity mapping and AOH/ROH analyses from unphased exome data in consanguineous families could assist with additional novel disease gene/locus discovery in consanguineous families.98 These strategies extend the effort to test the hypothesis proposed by Coban-Akdemir et al. that rare, deleterious variants embedded within long-sized AOH/ROH tend to evolve into disease-contributory haplotypes and whether the intrafamilial clinical heterogeneity could be the resultant from distributive AOH/ROH and MPV.30 Interpretation of interconnections among allelic series, clinical severity, and reduced penetrance remains an important and challenging genotype/phenotype interpretive biology issue. Future explorations will compare the allelic-specific gene dosage (AsGD) model proposed in this study with the established compound inheritance gene dosage model (CIGD) model to test this hypothesis and provide evidence as to whether such gene dosage expression models significantly contribute to limb anomalies and other developmental malformations.95,97
Data and code availability
This study did not generate any codes or analyze any public datasets. The identified variants in this paper were submitted to the ClinVar with sequential identifiers SCV002546515–SCV002546524. All exome sequences, consent, and phenotypic data reported in this paper can be requested from controlled-access databases on dbGaP: phs000711.v7.p2.
Acknowledgments
We thank all families for participation in the study and dedicate this study to Prof. Temtamy and Prof. McKusick for their inspiration. This study was supported in part by the National Human Genome Research Institute (NHGRI) and National Heart, Lung, and Blood Institute (NHBLI) (to the Baylor-Hopkins Center for Mendelian Genomics [BHCMG; UM1 HG006542] and Baylor College of Medicine Genomics Research to Elucidate the Genetics of Rare disease [BCM-GREGoR; U01 HG011758]), the National Institute of Neurological Disorders and Stroke (NINDS; R35NS105078 to J.R.L.), the Muscular Dystrophy Association (MDA; 512848 to J.R.L.), and the Spastic Paraplegia Foundation (SPF; to J.R.L.). J.E.P. was supported by NHGRI K08 HG008986. D.P. is supported by the International Rett Syndrome Foundation (IRSF; 3701-1).
Declaration of interests
J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing and genomic testing (ES, WGS, CMA, and aCGH) conducted at Baylor Genetics (BG). J.R.L. serves on the Scientific Advisory Board (SAB) of BG.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xhgg.2022.100132.
Web Resources
1000 Genomes Project (TGP), https://www.internationalgenome.org/
Agilent aCGH for Genomic DNA Analysis with Tecan HS Pro Hybridization Station
https://www.agilent.com/cs/library/usermanuals/public/G4410-90011_CGH_Tecan_6.1.pdf
Agilent Genomic Workbench 7.0,
https://www.agilent.com/library/usermanuals/Public/G3800-90047_ProductOverview.pdf
Agilent SureDesign eArray system, https://earray.chem.agilent.com/suredesign/
AluAluCNVpredictor, http://alualucnvpredictor.research.bcm.edu:3838/
ARIC database, http://www2.cscc.unc.edu/aric/
BaFCalculator Browser, https://github.com/BCM-Lupskilab/BafCalculator
BCM-HGSC Mercury analysis pipeline, https://www.hgsc.bcm.edu/software/mercury.
CADD, https://cadd.gs.washington.edu/
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
ESP Exome Variant Server, https://evs.gs.washington.edu/EVS/
HMZDelFinder Browser, https://github.com/BCM-Lupskilab/HMZDelFinder
Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk
NMDescPredictor, https://nmdprediction.shinyapps.io/nmdescpredictor/
Online Mendelian Inheritance in Man, https://www.omim.org/
PubMed, https://pubmed.ncbi.nlm.nih.gov/
The Genome Aggregation Database (gnomAD), https://gnomad.broadinstitute.org/)
UCSC Genome Browser, https://genome.ucsc.edu/
UCSF Chimera, https://www.cgl.ucsf.edu/chimera/
UniProt, https://www.uniprot.org/
Supplemental information
References
- 1.Zeller R., López-Ríos J., Zuniga A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 2009;10:845–858. doi: 10.1038/nrg2681. [DOI] [PubMed] [Google Scholar]
- 2.Petit F., Sears K.E., Ahituv N. Limb development: a paradigm of gene regulation. Nat. Rev. Genet. 2017;18:245–258. doi: 10.1038/nrg.2016.167. [DOI] [PubMed] [Google Scholar]
- 3.Niswander L. Pattern formation: old models out on a limb. Nat. Rev. Genet. 2003;4:133–143. doi: 10.1038/nrg1001. [DOI] [PubMed] [Google Scholar]
- 4.Biesecker L.G., Aase J.M., Clericuzio C., Gurrieri F., Temple I.K., Toriello H. Elements of Morphology : Standard Terminology for the Hands and Feet. 2009. [DOI] [PMC free article] [PubMed]
- 5.Giele H., Giele C., Bower C., Allison M. The incidence and epidemiology of congenital upper limb anomalies: a total population study. J. Hand Surg. Am. 2001;26:628–634. doi: 10.1053/jhsu.2001.26121. [DOI] [PubMed] [Google Scholar]
- 6.Ekblom A.G., Laurell T., Arner M. Epidemiology of congenital upper limb anomalies in stockholm, Sweden, 1997 to 2007: application of the oberg, manske, and tonkin classification. J. Hand Surg. Am. 2014;39:237–248. doi: 10.1016/j.jhsa.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Jourdain A.S., Petit F., Odou M.F., Balduyck M., Brunelle P., Dufour W., Boussion S., Brischoux-Boucher E., Colson C., Dieux A., et al. Multiplex targeted high-throughput sequencing in a series of 352 patients with congenital limb malformations. Hum. Mutat. 2020;41:222–239. doi: 10.1002/humu.23912. [DOI] [PubMed] [Google Scholar]
- 8.Sun L., Huang Y., Zhao S., Zhao J., Yan Z., Guo Y., Lin M., Zhong W., Yin Y., Chen Z., et al. Deciphering the mutational signature of congenital limb malformations. Mol. Ther. Nucleic Acids. 2021;24:961–970. doi: 10.1016/j.omtn.2021.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elsner J., Mensah M.A., Holtgrewe M., Hertzberg J., Bigoni S., Busche A., Coutelier M., de Silva D.C., Elçioglu N., Filges I., et al. Genome sequencing in families with congenital limb malformations. Hum. Genet. 2021;140:1229–1239. doi: 10.1007/s00439-021-02295-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamoto K., Saitsu H., Nishimura G., Kosaki R., Takayama S., Haga N., Tonoki H., Okumura A., Horii E., Okamoto N., et al. Comprehensive clinical and molecular studies in split-hand/foot malformation: identification of two plausible candidate genes (LRP6 and UBA2) Eur. J. Hum. Genet. 2019;27:1845–1857. doi: 10.1038/s41431-019-0473-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furniss D., Kan S.H., Taylor I.B., Johnson D., Critchley P.S., Giele H.P., Wilkie A.O.M. Genetic screening of 202 individuals with congenital limb malformations and requiring reconstructive surgery. J. Med. Genet. 2009;46:730–735. doi: 10.1136/jmg.2009.066027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flöttmann R., Kragesteen B.K., Geuer S., Socha M., Allou L., Sowińska-Seidler A., Bosquillon de Jarcy L., Wagner J., Jamsheer A., Oehl-Jaschkowitz B., et al. Noncoding copy-number variations are associated with congenital limb malformation. Genet. Med. 2018;20:599–607. doi: 10.1038/gim.2017.154. [DOI] [PubMed] [Google Scholar]
- 13.Allou L., Balzano S., Magg A., Quinodoz M., Royer-Bertrand B., Schöpflin R., Chan W.L., Speck-Martins C.E., Carvalho D.R., Farage L., et al. Non-coding deletions identify Maenli lncRNA as a limb-specific En1 regulator. Nature. 2021;592:93–98. doi: 10.1038/s41586-021-03208-9. [DOI] [PubMed] [Google Scholar]
- 14.Lupski J.R., Belmont J.W., Boerwinkle E., Gibbs R.A. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Posey J.E., O’Donnell-Luria A.H., Chong J.X., Harel T., Jhangiani S.N., Coban Akdemir Z.H., Buyske S., Pehlivan D., Carvalho C.M.B., Baxter S., et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 2019;21:798–812. doi: 10.1038/s41436-018-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pehlivan D., Bayram Y., Gunes N., Coban Akdemir Z., Shukla A., Bierhals T., Tabakci B., Sahin Y., Gezdirici A., Fatih J.M., et al. The genomics of arthrogryposis, a complex trait: candidate genes and further evidence for oligogenic inheritance. Am. J. Hum. Genet. 2019;105:132–150. doi: 10.1016/j.ajhg.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M., et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. 2015;88:499–513. doi: 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bainbridge M.N., Wang M., Wu Y., Newsham I., Muzny D.M., Jefferies J.L., Albert T.J., Burgess D.L., Gibbs R.A. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reid J.G., Carroll A., Veeraraghavan N., Dahdouli M., Sundquist A., English A., Bainbridge M., White S., Salerno W., Buhay C., et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinf. 2014;15:30. doi: 10.1186/1471-2105-15-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eldomery M.K., Coban-Akdemir Z., Harel T., Rosenfeld J.A., Gambin T., Stray-Pedersen A., Küry S., Mercier S., Lessel D., Denecke J., et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. doi: 10.1186/s13073-017-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D., et al. REVEL: an Ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarvik G.P., Browning B.L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 2016;98:1077–1081. doi: 10.1016/j.ajhg.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambin T., Akdemir Z.C., Yuan B., Gu S., Chiang T., Carvalho C.M.B., Shaw C., Jhangiani S., Boone P.M., Eldomery M.K., et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017;45:1633–1648. doi: 10.1093/nar/gkw1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fromer M., Moran J.L., Chambert K., Banks E., Bergen S.E., Ruderfer D.M., Handsaker R.E., McCarroll S.A., O'Donovan M.C., Owen M.J., et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012;91:597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pehlivan D., Hullings M., Carvalho C.M.B., Gonzaga-Jauregui C.G., Loy E., Jackson L.G., Krantz I.D., Deardorff M.A., Lupski J.R. NIPBL rearrangements in Cornelia de Lange syndrome: evidence for replicative mechanism and genotype-phenotype correlation. Genet. Med. 2012;14:313–322. doi: 10.1038/gim.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song X., Beck C.R., Du R., Campbell I.M., Coban-Akdemir Z., Gu S., Breman A.M., Stankiewicz P., Ira G., Shaw C.A., Lupski J.R. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018;28:1228–1242. doi: 10.1101/gr.229401.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan R., Saadi N.W., Grochowski C.M., Bhadila G., Faridoun A., Mitani T., Du H., Fatih J.M., Jhangiani S.N., Akdemir Z.C., et al. A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy. Am. J. Med. Genet. 2021;185:1972–1980. doi: 10.1002/ajmg.a.62192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitani T., Isikay S., Gezdirici A., Gulec E.Y., Punetha J., Fatih J.M., Herman I., Akay G., Du H., Calame D.G., et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 2021;108:1981–2005. doi: 10.1016/j.ajhg.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coban-Akdemir Z, Song X, Ceballos FC, et al. De novo mutation and identity-by-descent drive disease haplotypes, biallelic traits and multilocus pathogenic variation. bioRxiv. Published online July 27, 2022:2020.04.27.064824. 10.1101/2020.04.27.064824. [DOI]
- 31.Karaca E., Posey J.E., Coban Akdemir Z., Pehlivan D., Harel T., Jhangiani S.N., Bayram Y., Song X., Bahrambeigi V., Yuregir O.O., et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 2018;20:1528–1537. doi: 10.1038/gim.2018.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riggs E.R., Andersen E.F., Cherry A.M., Kantarci S., Kearney H., Patel A., Raca G., Ritter D.I., South S.T., Thorland E.C., et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Genet. Med. 2020;22:245–257. doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujioka H., Ariga T., Horiuchi K., Otsu M., Igawa H., Kawashima K., Yamamoto Y., Sugihara T., Sakiyama Y. Molecular analysis of non-syndromic preaxial polydactyly: preaxial polydactyly type-IV and preaxial polydactyly type-I. Clin. Genet. 2005;67:429–433. doi: 10.1111/j.1399-0004.2005.00431.x. [DOI] [PubMed] [Google Scholar]
- 35.Radhakrishna U., Bornholdt D., Scott H.S., Patel U.C., Rossier C., Engel H., Bottani A., Chandal D., Blouin J.L., Solanki J.V., et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; no phenotype prediction from the position of GLI3 mutations. Am. J. Hum. Genet. 1999;65:645–655. doi: 10.1086/302557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salsi V., Vigano M.A., Cocchiarella F., Mantovani R., Zappavigna V. Hoxd13 binds in vivo and regulates the expression of genes acting in key pathways for early limb and skeletal patterning. Dev. Biol. 2008;317:497–507. doi: 10.1016/j.ydbio.2008.02.048. [DOI] [PubMed] [Google Scholar]
- 37.Muragaki Y., Mundlos S., Upton J., Olsen B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science. 1996;272:548–551. doi: 10.1126/science.272.5261.548. [DOI] [PubMed] [Google Scholar]
- 38.Brison N., Debeer P., Tylzanowski P. Joining the Fingers : A HOXD13 Story. 2014. 37-48. [DOI] [PubMed]
- 39.Goodman F., Giovannucci-Uzielli M.L., Hall C., Reardon W., Winter R., Scambler P. Deletions in HOXD13 segregate with an identical, novel foot malformation in two unrelated families. Am. J. Hum. Genet. 1998;63:992–1000. doi: 10.1086/302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurban M., Wajid M., Petukhova L., Shimomura Y., Christiano A.M. A nonsense mutation in the HOXD13 gene underlies synpolydactyly with incomplete penetrance. J. Hum. Genet. 2011;56:701–706. doi: 10.1038/jhg.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fantini S., Vaccari G., Brison N., Debeer P., Tylzanowski P., Zappavigna V. A G220V substitution within the N-terminal transcription regulating domain of HOXD13 causes a variant synpolydactyly phenotype. Hum. Mol. Genet. 2009;18:847–860. doi: 10.1093/hmg/ddn410. [DOI] [PubMed] [Google Scholar]
- 42.Bartels C.F., Bükülmez H., Padayatti P., Rhee D.K., van Ravenswaaij-Arts C., Pauli R.M., Mundlos S., Chitayat D., Shih L.Y., Al-Gazali L.I., et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am. J. Hum. Genet. 2004;75:27–34. doi: 10.1086/422013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plachy L., Dusatkova P., Maratova K., Petruzelkova L., Zemkova D., Elblova L., Kucerova P., Toni L., Kolouskova S., Snajderova M., et al. NPR2 variants are frequent among children with familiar short stature and respond well to growth hormone therapy. J. Clin. Endocrinol. Metab. 2020;105 doi: 10.1210/clinem/dgaa037. dgaa037–e752. [DOI] [PubMed] [Google Scholar]
- 44.Lupski J.R. Clan genomics: from OMIM phenotypic traits to genes and biology. Am. J. Med. Genet. 2021;185:3294–3313. doi: 10.1002/ajmg.a.62434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blattner A., Huber A.R., Röthlisberger B. Homozygous nonsense mutation in WNT10B and sporadic split-hand/foot malformation (SHFM) with autosomal recessive inheritance. Am. J. Med. Genet. 2010;152A:2053–2056. doi: 10.1002/ajmg.a.33504. [DOI] [PubMed] [Google Scholar]
- 46.Kantaputra P.N., Kapoor S., Verma P., Intachai W., Ketudat Cairns J.R. Split hand-foot malformation and a novel WNT10B mutation. Eur. J. Med. Genet. 2018;61:372–375. doi: 10.1016/j.ejmg.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Theisen A., Rosenfeld J.A., Shane K., McBride K.L., Atkin J.F., Gaba C., Hoo J., Kurczynski T.W., Schnur R.E., Coffey L.B., et al. Refinement of the region for split hand/foot malformation 5 on 2q31.1. Mol. Syndromol. 2010;1:262–271. doi: 10.1159/000328405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Del Campo M., Jones M.C., Veraksa A.N., Curry C.J., Jones K.L., Mascarello J.T., Ali-Kahn-Catts Z., Drumheller T., McGinnis W. Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am. J. Hum. Genet. 1999;65:104–110. doi: 10.1086/302467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Svensson A.M., Curry C.J., South S.T., Whitby H., Maxwell T.M., Aston E., Fisher J., Carmack C.E., Scheffer A., Abu-Shamsieh A., Brothman A.R. Detection of a de novo interstitial 2q microdeletion by CGH microarray analysis in a patient with limb malformations, microcephaly and mental retardation. Am. J. Med. Genet. 2007;143:1348–1353. doi: 10.1002/ajmg.a.31775. [DOI] [PubMed] [Google Scholar]
- 50.Petit F., Jourdain A.S., Andrieux J., Baujat G., Baumann C., Beneteau C., David A., Faivre L., Gaillard D., Gilbert-Dussardier B., et al. Split hand/foot malformation with long-bone deficiency and BHLHA9 duplication: report of 13 new families. Clin. Genet. 2014;85:464–469. doi: 10.1111/cge.12219. [DOI] [PubMed] [Google Scholar]
- 51.Paththinige C.S., Sirisena N.D., Escande F., Manouvrier S., Petit F., Dissanayake V.H.W. Split hand/foot malformation with long bone deficiency associated with BHLHA9 gene duplication: a case report and review of literature. BMC Med. Genet. 2019;20:108–117. doi: 10.1186/s12881-019-0839-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klopocki E., Lohan S., Doelken S.C., Stricker S., Ockeloen C.W., Soares Thiele de Aguiar R., Lezirovitz K., Mingroni Netto R.C., Jamsheer A., Shah H., et al. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J. Med. Genet. 2012;49:119–125. doi: 10.1136/jmedgenet-2011-100409. [DOI] [PubMed] [Google Scholar]
- 53.da Rocha L.A., Pires L.V.L., Yamamoto G.L., Magliocco Ceroni J.R., Honjo R.S., de Novaes França Bisneto E., Oliveira L.A.N., Rosenberg C., Krepischi A.C.V., Passos-Bueno M.R., et al. Congenital limb deficiency: genetic investigation of 44 individuals presenting mainly longitudinal defects in isolated or syndromic forms. Clin. Genet. 2021;100:615–623. doi: 10.1111/cge.14041. [DOI] [PubMed] [Google Scholar]
- 54.Wolfgang G.L. Complex congenital anomalies of the lower extremities: femoral bifurcation, tibial hemimelia, and diastasis of the ankle. Case report and review of the literature. J. Bone Joint Surg. Am. 1984;66:453–458. https://journals.lww.com/jbjsjournal/Fulltext/1984/66030/Complex_congenital_anomalies_of_the_lower.21.aspx [PubMed] [Google Scholar]
- 55.Gollop T.R., Lucchesi E., Martins R.M., Nione A.S., Optiz J.M. Familial occurrence of bifid femur and monodactylous ectrodactyly. Am. J. Med. Genet. 1980;7:319–322. doi: 10.1002/ajmg.1320070313. [DOI] [PubMed] [Google Scholar]
- 56.Nagata E., Kano H., Kato F., Yamaguchi R., Nakashima S., Takayama S., Kosaki R., Tonoki H., Mizuno S., Watanabe S., et al. Japanese founder duplications/triplications involving BHLHA9 are associated with split-hand/foot malformation with or without long bone deficiency and Gollop-Wolfgang complex. Orphanet J. Rare Dis. 2014;9:125. doi: 10.1186/s13023-014-0125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lupski J.R. Biology in balance: human diploid genome integrity, gene dosage, and genomic medicine. Trends Genet. 2022;38:554–571. doi: 10.1016/j.tig.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaku D.A., Parry G.J., Malamut R., Lupski J.R., Garcia C.A. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806–1808. doi: 10.1212/WNL.43.9.1806. [DOI] [PubMed] [Google Scholar]
- 59.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A., et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 60.Carvalho C.M.B., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gu S., Yuan B., Campbell I.M., Beck C.R., Carvalho C.M.B., Nagamani S.C.S., Erez A., Patel A., Bacino C.A., Shaw C.A., et al. Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum. Mol. Genet. 2015;24:4061–4077. doi: 10.1093/hmg/ddv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. Springer Berlin Heidelberg; 2006. Allelic Series; p. 43. [DOI] [Google Scholar]
- 63.Johnston J.J., Olivos-Glander I., Killoran C., Elson E., Turner J.T., Peters K.F., Abbott M.H., Aughton D.J., Aylsworth A.S., Bamshad M.J., et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and pallister-hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am. J. Hum. Genet. 2005;76:609–622. doi: 10.1086/429346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lupski J.R. Structural variation mutagenesis of the human genome: impact on disease and evolution. Environ. Mol. Mutagen. 2015;56:419–436. doi: 10.1002/em.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hannema S.E., van Duyvenvoorde H.A., Premsler T., Yang R.B., Mueller T.D., Gassner B., Oberwinkler H., Roelfsema F., Santen G.W.E., Prickett T., et al. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J. Clin. Endocrinol. Metab. 2013;98:E1988–E1998. doi: 10.1210/jc.2013-2358. [DOI] [PubMed] [Google Scholar]
- 66.Miura K., Kim O.-H., Lee H.R., Namba N., Michigami T., Yoo W.J., Choi I.H., Ozono K., Cho T.J. Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am. J. Med. Genet. 2014;164A:156–163. doi: 10.1002/ajmg.a.36218. [DOI] [PubMed] [Google Scholar]
- 67.Lauffer P., Miranda-Laferte E., van Duyvenvoorde H.A., van Haeringen A., Werner F., Boudin E., Schmidt H., Mueller T.D., Kuhn M., van der Kaay D.C.M. An activating deletion variant in the submembrane region of natriuretic peptide receptor-B causes tall stature. J. Clin. Endocrinol. Metab. 2020;105 doi: 10.1210/clinem/dgaa190. dgaa190-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Church V.L., Francis-West P. Wnt signalling during limb development. Int. J. Dev. Biol. 2002;46:927–936. doi: 10.1387/ijdb.12455630. [DOI] [PubMed] [Google Scholar]
- 69.Geetha-Loganathan P., Nimmagadda S., Scaal M. Wnt signaling in limb organogenesis. Organogenesis. 2008;4:109–115. doi: 10.4161/org.4.2.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kawakami Y., Capdevila J., Büscher D., Itoh T., Rodríguez Esteban C., Izpisúa Belmonte J.C. WNT signals control FGF-dependent limb initiation and AER induction in the chick embryo. Cell. 2001;104:891–900. doi: 10.1016/S0092-8674(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 71.Witte F., Dokas J., Neuendorf F., Mundlos S., Stricker S. Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr. Patterns. 2009;9:215–223. doi: 10.1016/j.gep.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 72.Zhang C., Mazzeu J.F., Eisfeldt J., Grochowski C.M., White J., Akdemir Z.C., Jhangiani S.N., Muzny D.M., Gibbs R.A., Lindstrand A., et al. Novel pathogenic genomic variants leading to autosomal dominant and recessive Robinow syndrome. Am. J. Med. Genet. 2021;185:3593–3600. doi: 10.1002/ajmg.a.61908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White J.J., Mazzeu J.F., Coban-Akdemir Z., Bayram Y., Bahrambeigi V., Hoischen A., van Bon B.W.M., Gezdirici A., Gulec E.Y., Ramond F., et al. WNT signaling perturbations underlie the genetic heterogeneity of Robinow syndrome. Am. J. Hum. Genet. 2018;102:27–43. doi: 10.1016/j.ajhg.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang C., Jolly A., Shayota B.J., Mazzeu J.F., Du H., Dawood M., Soper P.C., Ramalho de Lima A., Ferreira B.M., Coban-Akdemir Z., et al. Novel pathogenic variants and quantitative phenotypic analyses of Robinow syndrome: WNT signaling perturbation and phenotypic variability. HGG Adv. 2022;3:100074. doi: 10.1016/j.xhgg.2021.100074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abu-Ghname A., Trost J., Davis M.J., Sutton V.R., Zhang C., Guillen D.E., Carvalho C.M.B., Maricevich R.S. Extremity anomalies associated with Robinow syndrome. Am. J. Med. Genet. 2021;185:3584–3592. doi: 10.1002/ajmg.a.61884. [DOI] [PubMed] [Google Scholar]
- 76.Yu P., Yang W., Han D., Wang X., Guo S., Li J., Li F., Zhang X., Wong S.W., Bai B., et al. Mutations in WNT10B are identified in individuals with oligodontia. Am. J. Hum. Genet. 2016;99:195–201. doi: 10.1016/j.ajhg.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Du R., Dinckan N., Song X., Coban-Akdemir Z., Jhangiani S.N., Guven Y., Aktoren O., Kayserili H., Petty L.E., Muzny D.M., et al. Identification of likely pathogenic and known variants in TSPEAR, LAMB3, BCOR, and WNT10A in four Turkish families with tooth agenesis. Hum. Genet. 2018;137:689–703. doi: 10.1007/s00439-018-1907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chai G., Szenker-Ravi E., Chung C., Li Z., Wang L., Khatoo M., Marshall T., Jiang N., Yang X., McEvoy-Venneri J., et al. A human pleiotropic multiorgan condition caused by deficient Wnt secretion. N. Engl. J. Med. 2021;385:1292–1301. doi: 10.1056/NEJMoa2033911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishitani T., Ninomiya-Tsuji J., Matsumoto K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol. Cell Biol. 2003;23:1379–1389. doi: 10.1128/MCB.23.4.1379-1389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sokol S.Y. Wnt signaling through T-cell factor phosphorylation. Cell Res. 2011;21:1002–1012. doi: 10.1038/cr.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lupski J.R., Wise C.A., Kuwano A., Pentao L., Parke J.T., Glaze D.G., Ledbetter D.H., Greenberg F., Patel P.I. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- 82.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 83.Lupski J.R. Genomic disorders ten years on. Genome Med. 2009;1:42–51. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harel T., Lupski J.R. Genomic disorders 20 years on—mechanisms for clinical manifestations. Clin. Genet. 2018;93:439–449. doi: 10.1111/cge.13146. [DOI] [PubMed] [Google Scholar]
- 85.Schatz O., Langer E., Ben-Arie N. Gene dosage of the transcription factor Fingerin (bHLHA9) affects digit development and links syndactyly to ectrodactyly. Hum. Mol. Genet. 2014;23:5394–5401. doi: 10.1093/hmg/ddu257. [DOI] [PubMed] [Google Scholar]
- 86.Kataoka K., Matsushima T., Ito Y., Sato T., Yokoyama S., Asahara H. Bhlha9 regulates apical ectodermal ridge formation during limb development. J. Bone Miner. Metabol. 2018;36:64–72. doi: 10.1007/s00774-017-0820-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malik S., Percin F.E., Bornholdt D., Albrecht B., Percesepe A., Koch M.C., Landi A., Fritz B., Khan R., Mumtaz S., et al. Mutations affecting the BHLHA9 DNA-binding domain cause MSSD, mesoaxial synostotic syndactyly with phalangeal reduction, malik-percin type. Am. J. Hum. Genet. 2014;95:649–659. doi: 10.1016/j.ajhg.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Phadke S.R., Kar A., Bhowmik A.D., Dalal A. Complex Camptosynpolydactyly and Mesoaxial synostotic syndactyly with phalangeal reduction are allelic disorders. Am. J. Med. Genet. 2016;170:1622–1625. doi: 10.1002/ajmg.a.37643. [DOI] [PubMed] [Google Scholar]
- 89.Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bien-Willner G.A., Stankiewicz P., Lupski J.R. SOX9cre1, a cis-acting regulatory element located 1.1 Mb upstream of SOX9, mediates its enhancement through the SHH pathway. Hum. Mol. Genet. 2007;16:1143–1156. doi: 10.1093/hmg/ddm061. [DOI] [PubMed] [Google Scholar]
- 91.Velagaleti G.V.N., Bien-Willner G.A., Northup J.K., Lockhart L.H., Hawkins J.C., Jalal S.M., Withers M., Lupski J.R., Stankiewicz P. Position effects due to chromosome breakpoints that map ∼900 Kb upstream and ∼1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am. J. Hum. Genet. 2005;76:652–662. doi: 10.1086/429252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang F., Seeman P., Liu P., Weterman M.A.J., Gonzaga-Jauregui C., Towne C.F., Batish S.D., De Vriendt E., De Jonghe P., Rautenstrauss B., et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am. J. Hum. Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lupski J.R. Schizophrenia: incriminating genomic evidence. Nature. 2008;455:178–179. doi: 10.1038/455178a. [DOI] [PubMed] [Google Scholar]
- 94.Wu N., Ming X., Xiao J., Wu Z., Chen X., Shinawi M., Shen Y., Yu G., Liu J., Xie H., et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med. 2015;372:341–350. doi: 10.1056/NEJMoa1406829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu J., Wu N., Deciphering Disorders Involving Scoliosis and COmorbidities DISCO study. Takeda K., Chen W., Li W., Du R., Liu S., Zhou Y., Zhang L., et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med. 2019;21:1548–1558. doi: 10.1038/s41436-018-0377-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen W., Lin J., Wang L., Li X., Zhao S., Liu J., Akdemir Z.C., Zhao Y., Du R., Ye Y., et al. TBX6 missense variants expand the mutational spectrum in a non-Mendelian inheritance disease. Hum. Mutat. 2020;41:182–195. doi: 10.1002/humu.23907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herman I., Jolly A., Du H., Dawood M., Abdel-Salam G.M.H., Marafi D., Mitani T., Calame D.G., Coban-Akdemir Z., Fatih J.M., et al. Quantitative dissection of multilocus pathogenic variation in an Egyptian infant with severe neurodevelopmental disorder resulting from multiple molecular diagnoses. Am. J. Med. Genet. 2022;188:735–750. doi: 10.1002/ajmg.a.62565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carr I.M., Bhaskar S., O’Sullivan J., Aldahmesh M.A., Shamseldin H.E., Markham A.F., Bonthron D.T., Black G., Alkuraya F.S. Autozygosity mapping with exome sequence data. Hum. Mutat. 2013;34:50–56. doi: 10.1002/humu.22220. [DOI] [PubMed] [Google Scholar]
- 99.Sezer O., Gebesoglu I., Yuan B., Karaca E., Gokce E., Gunes S. Fibular aplasia, tibial campomelia, and oligosyndactyly: a further patient with a 2-year follow-up. Clin. Dysmorphol. 2014;23:121–126. doi: 10.1097/MCD.0000000000000051. https://journals.lww.com/clindysmorphol/Fulltext/2014/10000/Fibular_aplasia, _tibial_campomelia, _and.2.aspx [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any codes or analyze any public datasets. The identified variants in this paper were submitted to the ClinVar with sequential identifiers SCV002546515–SCV002546524. All exome sequences, consent, and phenotypic data reported in this paper can be requested from controlled-access databases on dbGaP: phs000711.v7.p2.