

Using a CRISPR-based suppressor screen, the authors identify Atf7ip and Setdb1 as epigenetic regulators of tumor antigen expression and presentation. Genetic deficiency in these regulators enhances antitumor immunity, suggesting a new immunotherapeutic strategy for overcoming tumor immune evasion.
Abstract
Substantial progress has been made in understanding how tumors escape immune surveillance. However, few measures to counteract tumor immune evasion have been developed. Suppression of tumor antigen expression is a common adaptive mechanism that cancers use to evade detection and destruction by the immune system. Epigenetic modifications play a critical role in various aspects of immune invasion, including the regulation of tumor antigen expression. To identify epigenetic regulators of tumor antigen expression, we established a transplantable syngeneic tumor model of immune escape with silenced antigen expression and used this system as a platform for a CRISPR–Cas9 suppressor screen for genes encoding epigenetic modifiers. We found that disruption of the genes encoding either of the chromatin modifiers activating transcription factor 7–interacting protein (Atf7ip) or its interacting partner SET domain bifurcated histone lysine methyltransferase 1 (Setdb1) in tumor cells restored tumor antigen expression. This resulted in augmented tumor immunogenicity concomitant with elevated endogenous retroviral (ERV) antigens and mRNA intron retention. ERV disinhibition was associated with a robust type I interferon response and increased T-cell infiltration, leading to rejection of cells lacking intact Atf7ip or Setdb1. ATF7IP or SETDB1 expression inversely correlated with antigen processing and presentation pathways, interferon signaling, and T-cell infiltration and cytotoxicity in human cancers. Our results provide a rationale for targeting Atf7ip or Setdb1 in cancer immunotherapy.
Introduction
The immune system plays a key role in preventing carcinogenesis in immunocompetent hosts, a process known as immune surveillance (1). However, cancer cells can escape immune surveillance during the cancer immunoediting process (2). Although considerable knowledge has accumulated on how tumors avoid immune surveillance, the development of effective therapies to overcome tumor immune evasion remains a challenge.
Tumor antigens are processed and presented to CD4+ helper T cells and circulating CD8+ cytotoxic T cells, which leads to activation of adaptive immunity by CD4+ helper T cells or the direct killing of tumor cells by infiltrated CD8+ cytotoxic T cells (3). One common mechanism of tumor cell immune evasion is loss of antigen expression (4). In an autochthonous murine model of sarcomagenesis, chronic T-cell attack on tumor cells can silence the expression of tumor-specific antigens (5). In human lung cancer evolution, promoter hypermethylation of genes encoding neoantigenic mutations is an epigenetic mechanism of immune escape (6). Thus, searching for candidate therapeutic targets that can promote tumor antigen expression and boost antitumor immunity when inhibited may lead to novel strategies for cancer immunotherapy.
Targeting epigenetic modifiers shows potential in enhancing antitumor immune response (7). For example, inhibition of polycomb-repressive complex 2 substituents and histone deacetylases can restore expression of MHC-I antigen processing and presentation machinery and T-cell–mediated antitumor immunity (8, 9). Other studies show that DNA methyltransferase and histone deacetylase inhibitors, or histone demethylase inhibition increase tumor immunogenicity through upregulation of tumor antigen expression, including antigens derived from endogenous retroviruses (ERV; refs. 10–13). These findings suggest that targeting epigenetic regulators offers an attractive avenue for restoring immune surveillance.
Here, we performed a CRISPR–Cas9 suppressor screen in an antigen-silenced immune escaped lung adenocarcinoma tumor model. We identified Atf7ip and its interacting partner Setdb1 as potential therapeutic targets to augment tumor immunogenicity. Functional and mechanistic studies demonstrated that Atf7ip–Setdb1 deficiency stimulated antitumor immunity in multiple tumor models. Thus, our findings provide a rationale for nominating ATF7IP and SETDB1 as potential novel immunotherapeutic targets in patients with cancer.
Materials and Methods
Plasmid construction, lentivirus production, cell lines, and cell culture
Plasmids pLenti-Cas9-Puro (cat. #110837, addgene), lentiCRISPRv2 neo (cat. #98292, addgene), Lenti-LucOS (cat. #22777, addgene), psPAX2 (cat. #12260, addgene), and pMD2.G (cat. #12259, addgene) were directly used. The puromycin resistance of pXPR-047 (cat. #107145, addgene) was replaced with blasticidin resistance to construct the plasmid pXPR-GFP-Blast. Coding sequence for ovalbumin-SIINFEKL (SIN, OVA257–264) was engineered to pMT-025 (cat. #158579, addgene) to construct the plasmid SQ. The puromycin resistance of Lenti ORF clone of Setdb1 (cat. #MR222601L3) and Lenti ORF clone of Atf7ip (cat. #MR211907L3) was replaced with neomycin resistance.
The single-guide RNAs (sgRNAs) synthesized by MilliporeSigma (Supplementary Table S1) of mouse genes were cloned into the pXPR-GFP-Blast vector using Gibson Assembly kit (cat. #E2611L, NEB) according to the manufacturer's instructions and cloned into the lentiCRISPRv2 neo vector using T4 DNA ligase (cat. #M0202S, NEB) according to the manufacturer's instructions. These sgRNAs that can work with Cas9 to edit 25 Atf7ip-interacting partners' gene function were used individually in the second screen to investigate the Atf7ip-interacting partners' function in regulating SQ expression and presentation by either RT-PCR or flow cytometry analysis.
To generate lentivirus, HEK-293T cells were cotransfected with transfer plasmids and packaging plasmids PSPAX2 and PMD2.G using Lipofectamine 3000 (cat. #L3000008, Invitrogen) according to the manufacturer's instructions. Viral particles in the cell culture supernatant were filtered with 0.45-μm filters (cat. #431225, Corning) to remove cellular debris.
HEK-293T cells were purchased from the ATCC and were authenticated by the ATCC using STR profiling (14). The mouse KP cells used were authenticated by PCR genotyping routinely run in the laboratory (14, 15). The mouse KP cell line is very malignant and grows rapidly in B6/J immunocompetent mice, indicating the lack of immunogenicity of tumor cells (15). MC38 cells were kindly received from the Belfer Center of Dana-Farber Cancer Institute in 2018. YUMM1.7 cells were kindly received from Dr. Marcus Bosenberg' laboratory at Yale University in 2020. KP-SQ cells were established by infecting KP cells with filtered SQ lentivirus in the presence of 10 μg/mL polybrene (cat. #TR-1003-G). The medium containing lentivirus and polybrene was replaced by fresh medium after overnight incubation. Infected cells were selected with 5 μg/mL puromycin (cat. #A1113803, ThermoFisher Scientific) after 48 hours and maintained in cell culture media containing 2 μg/mL puromycin once the selection was completed. The single-cell clone (KP-C6) expressing high level of SIN was sorted from KP-SQ cells, expanded in cell culture and injected to B6/J immunocompetent mice orthotopically to undergo tumor immunoediting. Injected KP-C6 cells formed tumors in the lungs of some grafted mice (2 out of 10) after 10 months, consistent with substantially increased immunogenicity. The tumor nodule was harvested to establish the cell line KP-IE1 (first-generation of immune escaped model) as previously described (14). KP-IE1 cells formed tumors only 3 weeks after injection into B6/J mice, consistent with adaptation to immune surveillance against ovalbumin-SIINFEKL. The tumor nodule was again collected to establish the cell line KP-IE2 (second-generation of immune escaped model). KP-LucOS cells were established by infecting KP cells with filtered LucOS lentivirus, the single-cell clone expressing high level of SIN was sorted from KP-LucOS cells, expanded in cell culture, named as C1. C1 cells were orthotopically injected to B6/J mice. The lung tumor nodules developed in the mice were harvested for establishment of the immune escaped cell line IE as previously described (14).
To generate Atf7ip or Setdb1-deficient KP-IE2, KP-LucOS, MC38, and YUMM1.7 cells, Th cells were infected with cas9 and sgAtf7ip or cas9 and sgSetdb1 viral supernatants in the presence of 10 μg/mL polybrene (cat. #TR-1003-G). The medium containing lentivirus and polybrene was replaced by fresh medium after overnight incubation. Infected cells were selected with either 5 μg/mL puromycin (cat. #A1113803, ThermoFisher Scientific) or 5 μg/mL blasticidin (cat. #A1113903, ThermoFisher Scientific) or 600 μg/mL G418 (cat. #10131035, ThermoFisher Scientific) after 48 hours and maintained in cell culture media containing antibiotics once the selection was completed.
Gene disruption was further confirmed by western blot. To generate Atf7ip or Setdb1 overexpressed KP-IE2 and MC38 cells, the cells were infected with Atf7ip or Setdb1 viral supernatants in the presence of 10 μg/mL polybrene (cat. #TR-1003-G). The medium containing lentivirus and polybrene was replaced by fresh medium after overnight incubation. Infected cells were selected with 600 μg/mL G418 (cat. #10131035, ThermoFisher Scientific) after 48 hours and maintained in cell culture media containing antibiotics once the selection was completed. Gene overexpression was further confirmed by western blot.
HEK-293T cells and MC38 cells were cultured in DMEM (cat. #11995–065, Gibco) with 10% FBS (cat. #89510–186, VWR) and 1X antibiotic–antimycotic (cat. #15240–062, Gibco). YUMM1.7 cells were cultured as previously described (16). Mouse lung adenocarcinoma cell lines KP, KP-SQ, KP-C6, KP-IE1, KP-IE2, KP-LucOS, C1 and IE were cultured in RPMI-1640 (cat. # 11875–093, Gibco) with 10% FBS. KP-IE2 was used in both initial epigenetic CRISPR screen and the second screen. All cell lines were cultured in vitro for less than 4 passages before use and all tested as Mycoplasma negative using the Universal Mycoplasma Detection Kit (ATCC 30–1012K).
Quantitative RT-PCR
Total RNA was extracted with RNeasy Plus Mini Kit (cat, #74136, Qiagen), and cDNA was constructed with a High-Capacity cDNA Reverse Transcription Kit (cat. #4374966, Applied Biosystems) using 1 μg total RNA. Quantitative PCR was run in Real-Time PCR System (Applied Biosystems) with the PowerUp SYBR Green Master Mix (cat. #A25742, Thermo Fisher Scientific), primers and cDNA templates, and transcripts were normalized with internal control Actin using ΔΔCt quantification. All the samples were run in triplicates. Gene names and the primers used for ΔΔCt quantification were listed (Supplementary Table S1).
Flow cytometry and cell sorting
Cells in culture were collected and resuspended in PBS with 2% FBS. Cells were stained with Fc receptor blocking antibody (cat. #101319, BioLegend) for 5 minutes at room temperature, then the cells were stained for cell surface markers using fluorochrome-coupled antibodies (1:100 dilution; Supplementary Table S2) on ice for 30 minutes. Cells were washed twice using PBS with 2% FBS. Well stained cells were collected by BD Biosciences LSRFortessa and the data were analyzed with FlowJo software. All the samples were run in triplicates. For cell sorting, well stained cells were re-suspended in culture medium and collected by SY3200 cell sorter and the data were analyzed with FlowJo software. For generating single cell clone, cells in culture were collected and filtered through 40-μmol/L cell strainers (cat. #08–771–1, Thermo Fisher Scientific) to obtain single-cell suspensions. Suspended cells with or without surface marker staining depending on experiment design were sorted by SY3200 cell sorter to distribute single-cell clone to 96-well plate with 100 μL culture medium. Each single-cell clone was expanded for further experiments.
Western blots and antibodies
Cells were lysed in RIPA buffer (cat. #89900, Thermo Fisher Scientific) with protease/phosphatase inhibitor cocktail (cat., #78440, Thermo Fisher Scientific). Protein concentration was determined by BCA assay (cat. #23227, Thermo Fisher Scientific). 30 μg proteins from each sample were loaded in 4% to 12% Bis-Tris gels (Invitrogen), transferred to nitrocellulose membranes, and immunoblotted with primary antibodies (Supplementary Table S2). IRDye 800-labeled goat anti-rabbit IgG (cat. #926–32211, LI-COR) and IRDye 680-labeled goat anti-mouse IgG (cat. #926–68070, LI-COR) were applied as secondary antibodies. The membranes were imaged with an Odyssey platform (LI-COR Biosciences).
Epigenetic CRISPR screen using an immune escaped KP-IE2 lung cancer cell line
Cas9-KP-IE2 lung adenocarcinoma cells were infected at a MOI of 0.2 with lentivirus generated from the epigenetic libraries for at least 1,000-fold coverage (1,000 cells per sgRNA construct) in the presence of 10 μg/mL polybrene in each infection replicate as previously described (15). Lentiviral particles in the supernatant were from the HEK-293T cells cotransfected with pXPR-GFP-sgRNA-blast epigenetic library and packing plasmid psPAX2 and pMD2.G. MOI was determined by the percentage of GFP+ cells after the cells were infected with serial volume of lentivirus for 72 hours (15). Transduced KP-IE2 cells were expanded in vitro for 2 weeks, and then both 15% of SIINFEKL high-expression population and 15% low-expression population were sorted out. Genomic DNAs of these two populations were extracted using the DNeasy Blood and Tissue kit (cat. #69506, Qiagen). sgRNA cassettes were amplified by PCR, and next-generation sequencing (NGS) was performed on an Illumina HiSeq to determine sgRNA abundance as previously described (15).
Data analysis for CRISPR screen
Cutadapt (v1.18) was applied to trim adaptor sequences, and untrimmed reads were discarded. Then the sequences after the 20-base sgRNAs were cut using fastx-toolkit (v0.0.13; http://hannonlab.cshl.edu/fastx_toolkit/index.html), sgRNAs were mapped to the annotation file (0 mismatch), and read count tables were made. The count tables were normalized on the basis of their library size factors using DESeq2 (17). MAGeCK (0.5.8; ref. 18) was applied to normalize the read count tables based on median normalization and fold changes, and significance of changes in the conditions was calculated for genes and sgRNAs (Supplementary Tables S3 and S4). ClusterProfiler R package (v3.6.0) was applied to perform pathway analysis and Gene Set Enrichment Analysis (19). R (v3.1.1) was used to perform all downstream statistical analyses and generate plots (http://www.r-project.org).
Cell proliferation assay
Cells were plated in 96-well plates at a density of 500 cells per well. Cell proliferation was measured by CCK-8 kit (cat. #ALX-850–039-KI02, Enzo Lifesciences) on a FlexStation 3 multi-mode microplate reader using Softmax Pro software (V.5.4.6.005) according to the manufacturer's instructions.
Cell apoptosis assay
Cells were plated in 6-well plates at a density of 100,000 cells per well. After 24 hours, cell apoptosis was measured by Annexin V apoptosis detection kit (cat. #88–8007–72, Thermo Fisher Scientific) and BD Biosciences LSRFortessa according to the manufacturer's instructions. The data were analyzed with FlowJo software.
Animal studies
All animal work was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at NYU School of Medicine. All mice were housed and cared for in specific-pathogen-free facilities. Six-week-old B6/J wild-type (WT) mice were purchased from The Jackson Laboratory. Six-week-old NU/NU Nude mice were purchased from Charles River Laboratories. For the MC38 in vivo model, 1 million cells were resuspended in PBS and subcutaneously inoculated into the flanks of B6/J and NU/NU Nude mice. Tumor size was measured every 3 days using calipers to collect maximal tumor length and width. Tumor volume was calculated with the following formula: (L × W2)/2. For the KP-IE2 and KP-LucOS in vivo models, 1 million cells were injected into each mouse via tail vein. Tumor formation and progression were monitored by MRI as previously described (15). CO2 inhalation was used to euthanize mice when the tumor samples were harvested.
RNA sequencing and data analyses
RNA sequencing (RNA-seq) of MC38 cells with or without Atf7ip deficiency and RNA-seq of MC38 cells with or without Setdb1 deficiency was performed in NYU School of Medicine Genome Technology Core. Total RNA was extracted using RNeasy Plus Mini Kit (cat, #74136, Qiagen) according to the manufacturer's instructions. The RNA library was prepared using TruSeq Stranded Total RNA Library Prep Gold (cat, #20020598) according to the manufacturer's instructions. STAR 2.4.2a (20) was applied to align the RNA-seq samples to the reference mouse genome (mm10) and count the number of reads that map to each gene in the ensembl GRCm38.80 gene model. R (v.3.5.1; http://www.R-project.org/) and the DESeq2 package (v.1.10.0) were used to perform differential gene expression analysis among different sample groups (21). Gene set enrichment analysis was done using GSEA (v.3.0) and gene sets from MSigDB (v.5.0). We used the “preranked” algorithm to analyze gene lists ranked by the negative decadic logarithm of P values multiplied by the value of log2FC obtained from the differential-expression analysis with DESeq2. rMATS version 4.0.2 was used to perform alternative splicing analysis. The exon count tables and differential exon usage was calculated using DEXSeq (v3.10). Isoform predictions and isoform differential expression analysis were analyzed using Cufflinks and Cuffdiff (v 2.2.1; ref. 22). To identify differentially expressed retroelements, paired-end reads were mapped to the mm10 reference genome using bowtie2 (23). Genomic wide coordinates of all ERVs were downloaded from RepeatMasker through the UCSC Table Browser tool (mm10) and BEDTools was used to create count tables from the BAM files. These count tables were then normalized using DESeq2 and differential expression analysis was performed on the ERVs genome wide.
MS identification of Atf7ip interactions
KP-IE2 and MC38 cells were lysed with IP lysis buffer (cat. #87788, Thermo Fisher Scientific). Cell lysates were incubated with rabbit IgG (cat. #2729s, Cell Signaling Technology), anti-Atf7ip (cat. #A300–169A, Bethyl Laboratories), and A-agarose beads (cat. # 20333, Pierce) overnight. The agarose resin with immunoprecipitated proteins was washed with IP lysis buffer 3 times. Samples were then washed 3 times with 100 μmol/L Ammonium Bicarbonate. Samples were reduced with DTT (cat. #43815, MilliporeSigma) at 57°C for 1 hour (2 μL of 0.2 mol/L). Samples were alkylated with Iodoacetamide (cat. #I1149, MilliporeSigma) at RT in the dark for 45 minutes (2 μL of 0.5 mol/L). 200 ng of sequencing grade modified trypsin (cat. #V5111, Promega) was added to each gel sample. Digestion proceeded overnight on a shaker at RT. Beads were removed and peptides extracted. A slurry of R2 20-μm Poros beads (cat. #112906, Thermo Fisher Scientific) in 5% formic acid and 0.2% trifluoroacetic acid (TFA) was added to each sample at a volume equal to that of the ammonium bicarbonate added for digestion. The samples were shaken at 4°C for 3 hours. The beads were loaded onto equilibrated C18 ziptips (Millipore) using a microcentrifuge for 30 seconds at 6,000 rpm. Gel pieces were rinsed three times with 0.1% TFA and each rinse was added to its corresponding ziptip followed by microcentrifugation. The extracted poros beads were further washed with 0.5% acetic acid. Peptides were eluted by the addition of 40% acetonitrile in 0.5% acetic acid followed by the addition of 80% acetonitrile in 0.5% acetic acid. The organic solvent was removed using a SpeedVac concentrator and the sample reconstituted in 0.5% acetic acid. 1/30th of each sample was analyzed individually. LC separation online with MS using the autosampler of an EASY-nLC 1000 (Thermo Scientific). Peptides were gradient eluted from the column directly to a Lumos Mass spectrometer using a 95 minutes gradient (Thermo Scientific). High resolution full MS spectra were acquired with a resolution of 240,000, an AGC target of 1e6, with a maximum ion time of 50 ms, and scan range of 400 to 1,500 m/z. All MS/MS spectra were collected using the following instrument parameters: Ion trap scan rate of Rapid, AGC target of 6e4, maximum ion time of 18 ms, one microscan, 2 m/z isolation window, fixed first mass of 110 m/z, and NCE of 30. MS/MS spectra were searched using a Uniprot Human database plus IgG and supplied sequences using Sequest within Proteome Discoverer (Supplementary Tables S5 and S6).
Chromatin immunoprecipitation sequencing and assay for transposase-accessible chromatin sequencing
Chromatin immunoprecipitation (ChIP) was performed for MC38 cells with or without Atf7ip and Setdb1 using the ChIP-IT High Sensitivity Kit (cat. #53040, Active Motif) according to the manufacturer's instructions. An antibody against Histone H3 (tri methyl K9; cat. #Ab8898, Abcam) was used. ChIP DNA was purified using the same kit according to the manufacturer's instructions and sent to NYU School of Medicine Genome Technology Center for library construction and sequencing. The library was constructed with KAPA HyperPrep Kits (cat. #07962347001, Roche) according to the manufacturer's instructions and was sequenced by Illumina NovaSeq 6000. For assay for transposase-accessible chromatin sequencing (ATAC-seq), freshly harvested KP-C6 cells and KP-IE2 cells were directly sent to NYU School of Medicine Genome Technology Center for library construction and sequencing. The library was constructed with Nextera DNA library Prep Kit (cat. #FC-121–1030, Illumina) according to the manufacturer's instructions and was sequenced by Illumina NovaSeq 6000.
ChIP sequencing, ATAC-seq and Hi-C-seq data analysis
Bowtie2 (v2.2.4; ref. 23) was applied to map all the reads from sequencing to the reference genome and Picard tools (v.1.126; http://broadinstitute.github.io/picard/) were used to remove duplicate reads. Low-quality mapped reads (MQ<20) were discarded from the analysis. BEDTools (v.2.17.0; ref. 24) and the bedGraphToBigWig tool (v.4) were applied to generate read per million (RPM) normalized BigWig files. MACS (v1.4.2; ref. 25) was used to perform peak calling and BEDTools was applied to create peak count tables. DESeq2 (17) was applied to perform differential peak analysis (Supplementary Tables S7 and S8). ChIPseeker (v1.8.0; ref. 26) R package was used for peak annotations and motif discovery was performed with HOMER (v4.10; ref. 27). ngs.plot (v2.47; ref. 27). ChIPseeker were applied for TSS site visualizations and quality controls. clusterProfiler R package (v3.0.0; ref. 19) was used to perform KEGG pathway analysis and Gene Ontology (GO) analysis. To compare the level of similarity among the samples and their replicates, two methods were used: principal-component analysis and Euclidean distance-based sample clustering. Downstream statistical analysis and generating plots were performed in R environment (v3.1.1; https://www.r-project.org/). High-throughput chromatin conformation capture (Hi-C) data were analyzed in the platform of hic-bench (28). First, bwa (29) was used to align them to the mm10 reference genome. Only the read pairs uniquely mapped to the same chromosome were used. The interaction matrix was tabulated in the bin size of 40 kilobases, and normalized using iterative correction method (30). Topologically associated domains (TAD) were called from the interaction matrix and the boundary score, using the “ratio” method defined (28). Each domain called had an “intra-TAD” activity signal associated with it, and it was compared across different sample types.
Profiling of tumor-infiltrating immune cells
Mice were euthanized as indicated in “Animal studies.” Tumors were minced and digested in Hank's Balanced Salt Solution with collagenase D (cat. #11088866001, Roche) and DNase I (cat. #10104159001, Roche) at 37°C for 30 minutes. After digestion, the whole tumors were filtered through 70-μmol/L cell strainers (cat. #08–771–2, Fisher Scientific) to obtain single-cell suspensions. Suspended cells were treated with 1× RBC lysis buffer (cat. #420301, BioLegend) to lyse red blood cells. Live cells were stained with a LIVE/DEAD Fixable Aqua Dead Cell Stain kit (cat. #423102, BioLegend). Well processed cell pellets were resuspended in PBS with 2% FBS for FACS analysis. Cells were stained for cell surface markers using fluorochrome-coupled antibodies (Supplementary Table S2) and then were fixed/permeabilized with fixation/permeabilization kit (cat. #00–5123–43 and #00–5223–56, Invitrogen). Well stained cells were collected by BD Biosciences LSRFortessa and the data were analyzed with FlowJo software. The gating strategy was described previously (31).
IHC
Tumors were fixed in 10% formalin for 24 hours, washed in 70% ethanol for 24 hours, and embedded for histological analysis. Five-micron (5 μm) sections were cut from paraffin blocks. Briefly, all slides underwent deparaffinization and sequential antigen retrieval. Primary antibody against mouse CD3 (cat. #MCA1477T, Bio-Rad) and horse-radish peroxidase–coupled secondaries (cat. #RMR622, Biocare) were used according to the manufacturer's instructions. After whole-slide scanning at ×20 using Hamamatsu NanoZoomer whole-slide scanner, ImageJ was used for the quantifications of all IHC staining.
Sanger sequencing and TIDE analysis
Genomic DNA of tumors or cultured cell was extracted using DNeasy Blood and Tissue Kit (cat. #69504, Qiagen) according to the manufacturer's instructions. 100 ng Genomic DNA template with primers and Phusion polymerase (cat. #M0530, NEB) was used to amplify the editing regions of sgAtf7ip or sgSetdb1 according to the manufacturer's instructions. The amplified products were sent to GENEWIZ for sanger sequencing using the same forward primers. Sanger sequencing files were uploaded to TIDE online tool to check the sgRNA editing, including deletion or insertions (https://tide.nki.nl).
sgAtf7ip_1 editing region:
Forward primer: 5′-GCATAGTGATGGCGGTTACA-3′
Reverse primer: 5′-AGAGAAATGTCATAGCTTCCTTCT-3′
sgAtf7ip_2 editing region:
Forward primer: 5′-CTTAGCAGGTAGCAGTGAGTTT-3′
Reverse primer: 5′-GGATACAACATGCACACAAAGAG-3′
sgSetdb1_1 editing region:
Forward primer: 5′-CCAACCGTTCTTCCCTTTCA-3′
Reverse primer: 5′-TCTCTTCCCTTCCCTAGCTATC-3′
sgSetdb1_2 editing region:
Forward primer: 5′-ACCTTACTGTCCTCTTGACCTT-3′
Reverse primer: 5′-CCAATACGAATAGATGGCACCAG-3′
CpG sites methylation analysis on CMV promoter
Genomic DNA of cultured cells was extracted using DNeasy Blood and Tissue Kit (cat. #69504, Qiagen) according to the manufacturer's instructions. 1 μg Genomic DNA was used with EpiJET Bisulfite Conversion Kit (cat. #K1461, Thermo Fisher Scientific) to generate converted DNA according to the manufacturer's instructions. Converted DNA was amplified with primers and Hot Start DNA Polymerase (cat. #F-555S/L, Thermo Fisher Scientific) according to the manufacturer's instructions. Amplified DNA was sent to GENEWIZ for sanger sequencing using the same forward primer. SnapGene 5.3.2 was used to view cytosine and thymine in CpG site of the converted DNA.
Forward primer: 5′-GGGTACAGTGCAGGGGAAAG-3′
Reverse primer: 5′-CGAATTCGCTAGCTCTAGAATCTT-3′
STRING database analysis
The list of proteins was uploaded to online tool STRING database (https://string-db.org/cgi/input?sessionId=b1uYexAeE5VA&input_page_show_search=on). Biological Process (Gene Ontology), KEGG Pathways, and Reactome Pathways analysis were automatically generated by the online tool.
Copy-number analysis
Genomic DNA of cultured cells (KP, KP-SQ, KP-C6, KP-IE1, and KP-IE2) was extracted using DNeasy Blood and Tissue Kit (cat. #69504, Qiagen) according to the manufacturer's instructions. Quantitative PCR was run in Real-Time PCR System (Applied Biosystems) with the PowerUp SYBR Green Master Mix (cat. #A25742, Thermo Fisher Scientific), primers (forward: 5′-ACGGCCAATTCATTCACTTC-3′, reverse: 5′-ACTCTTTGGTGGTGGACTGG-3′) and 20 ng genomic DNA, and SQ copy number were normalized with internal control Actin using ΔΔCt quantification. SQ copy number in KP-SQ was defined as 2. SQ copy numbers of KP, KP-C6, KP-IE1, and KP-IE2 were further normalized on the basis of the SQ copy number of KP-SQ. All the samples were run in triplicates.
The Cancer Genome Atlas RNA-seq data analysis
Level 3 RNA-seq data from The Cancer Genome Atlas (TCGA) were obtained through the TCGA portal with the ID numbers specified (Supplementary Tables S9 and S10). All the normal samples were excluded from the analysis. Data were sorted on the basis of the level of expression of ATF7IP or SETDB1, and the samples were separated into quarters. The top 25% expression group (high expression; 2,581 patients) was compared with the low 25% expression group (low expression; 2,581 patients) by GSEA as outlined in “RNA sequencing and data analyses.” The gene list for GSEA input was ranked by the value of log2FC, where FC was defined by the ratio of the low-expression group to the high-expression group.
Statistical analysis
GraphPad Prism 7 was used for all statistical analyses. Data were analyzed by the student t test (two tailed). Survival analysis was performed by the Kaplan–Meier method and log rank (Mantel–Cox) test. Error bars represent standard error of the mean (SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Data access
NGS data for the CRISPR screen, ATAC-seq data, RNA-seq data, ChIP-sequencing (ChIP-seq) data, and Hi-C data have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus and are accessible through GEO Series accession number GSE179574 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179574).
Results
Silencing of tumor antigen expression contributes to tumor immune evasion
By mimicking immunoediting in mice, we drove the outgrowth of immune evasive tumor cells from cells that were initially highly immunogenic, establishing an immune escaped cancer cell line, KP-IE2 (Fig. 1A). We next evaluated the characteristics of the KP-IE2 immune escaped tumor model. Prior research indicates that intrinsic MHC downregulation can promote tumor cell immune evasion (32). Although we observed no difference in total cell membrane MHC-I expression, we saw inhibition of SIN presentation in KP-IE1 and KP-IE2 cells (Fig. 1B). We observed no change in PD-L1 expression (Fig. 1B), upregulation of which can also promote tumor immune evasion (33). In addition, SQ was transcriptionally significantly inhibited in the absence of deep deletion (Fig. 1C and D). SQ in this construct is under the control of a CMV promoter, which is well known to be silenced by DNA methylation in vivo. CpG methylation analysis confirmed that the CMV promoter was hypermethylated in KP-IE1 and KP-IE2 cells (Fig. 1E and F). Furthermore, the average SQ reads per million sequencing reads was only 13 in KP-IE2 cells compared with 25 in KP-C6 cells, indicating the SQ region in the chromatin was less accessible in immune escaped cell line (Fig. 1G and H), which also suggested more heterochromatin formed in the SQ genomic region during tumor immune escape. These results suggest that KP-IE2 escaped immune surveillance through silencing of SQ antigen expression via an epigenetic mechanism.
Figure 1.

The characteristics of the immune escaped tumor model. A, Strategy for establishing an immune escaped cancer cell line. B, FACS of H-2Kb–bound SIINFEKL, MHC-I, and PD-L1 (blue represents isotype). C, RT-PCR of SQ transcripts. D, Analysis of genomic SQ copy number. E, Animation of CpG methylation on SQ promoter. F, Quantification of methylated CpG sites on SQ promoter. G, DNA accessibility of SQ region evaluated by ATAC-seq. H, Quantification of DNA accessibility of SQ region. Two-tailed unpaired t tests were used to compare the two arms. Error bars represent mean ± SEM. * P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. NS, not significant.
Atf7ip is an epigenetic regulator controlling tumor antigen expression and presentation
Epigenetic modulators regulate tumor cell antigen expression and presentation (10, 34). To identify novel epigenetic modifiers that mediate tumor antigen expression and presentation, we performed an epigenome-wide CRISPR suppressor screen using the KP-IE2 cells (Fig. 2A). The sgRNA library contained sgRNAs targeting 524 epigenetic genes, described previously (15). KP-IE2 cells were infected with the lentivirus pool, cultured for 2 weeks, and sorted for the top 15% of SIINFEKL-expressing cells (SIINFEKLhigh) and the bottom 15% of cells (SIINFEKLlow; Fig. 2A). By comparing sgRNAs extracted from SIINFEKLhigh cells with those from the SIINFEKLlow cells, we identified epigenetic targets whose disruption could promote or inhibit SIINFEKL expression and presentation (Fig. 2A). For example, sgRNAs targeting H2-K1, Tap1, Tap2, B2 m and Tapbp were enriched in the SIINFEKLlow population (Fig. 2B), consistent with their established role in positively regulating antigen presentation. In contrast, sgRNAs targeting Atf7ip were enriched in the SIINFEKLhigh population (Fig. 2B–D), indicating a role in negatively regulating antigen expression and presentation. Indeed, both SQ expression and SIINFEKL presentation were significantly increased in KP-IE2 cells upon Atf7ip disruption (Fig. 2E–G). Moreover, CpG methylation analysis indicated that the CMV promoter remained hypermethylated upon Atf7ip disruption (Fig. 2H and I), suggesting that Atf7ip deficiency functions downstream of the DNA methylation to reverse tumor antigen silencing.
Figure 2.

Atf7ip regulates SQ expression and presentation. A, Strategy of epigenetic CRISPR screen to identify epigenetic regulators of antigen expression and presentation. B, Volcano plot illustrating genes whose disruption enhanced (red) or inhibited (blue) presentation of SIINFEKL. C, Illustration of the top 10 candidates from B. D, Normalized read counts of Atf7ip sgRNAs in SIINFEKL 15% high and 15% low populations. E, RT-PCR of SQ transcripts. F, FACS of SIINFEKL presentation. G, Quantification of SIINFEKL presentation in F. H, Animation of CpG methylation on SQ promoter KP-IE2-C1 (the single clone of KP-IE2). I, Quantification of methylated CpG sites on SQ promoter. J, Representative MRI scans showing mouse lung tumors 4 weeks after transplant through tail vein injection in B6/J immunocompetent mice. K, Quantification of lung tumors volume in J (n = 3 mice/group), L, Survival curves for the immunocompromised nude mice (NU/NU) after being transplanted with control and Atf7ip-deficient KPIE2 cells (n = 3 mice/group). M, Survival curves for each group in the study (n = 9 mice/group). For survival data, results were plotted using Kaplan–Meier curve, and statistical significance was determined via a log-rank test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. NS, not significant.
To exclude the possibility that the regulatory effect of Atf7ip is CMV promoter specific, we analyzed an alternative model. A previous study showed that KP-LucOS lung cancer cells escaped immune surveillance by silencing tumor antigen expression in immunocompetent hosts (35). LucOS is a lentiviral vector expressing the T-cell antigen SIYRYYGL (SIY) and two antigens from ovalbumin-SIINFEKL (SIN, OVA257–264) and OVA323–339 (35) under the control of the UBC promoter (Supplementary Fig. S1A). We established immune escaped KP-LucOS cell line (IE) with silenced antigen expression (Supplementary Fig. S1B). Atf7ip inhibition significantly increased antigen expression in immune escaped IE cells (Supplementary Fig. S1C–S1E). These results indicate that in two distinct models, Atf7ip deficiency upregulates tumor antigen expression in the context of CMV, a strong viral promoter, and UBC, a mammalian promoter cloned from the ubiquitin c gene.
Atf7ip deficiency augments tumor immunogenicity and boosts antitumor immune responses
To further examine whether Atf7ip deficiency can restore the immunogenicity of KP-IE2 cells and stimulate an adaptive immune response, Atf7ip WT and Atf7ip-deficient KP-IE2 cells were injected into either B6/J immunocompetent mice or athymic nude (NU/NU) immunodeficient mice, which lack mature T cells and cannot mount many types of adaptive immune responses (36). Compared with Atf7ip WT tumors, Atf7ip-deficient tumors grew significantly slower in B6/J mice, contributing to prolonged survival benefit (Fig. 2J, K, and M). However, there was no survival benefit to nude mice grafted with Atf7ip-deficient KP-IE2 cells (Fig. 2L). Importantly, Atf7ip-deficient KP-IE2 cells showed only marginal change in proliferation in vitro (Supplementary Fig. S1F and S1G). These findings indicate that Atf7ip inhibition results in the restoration of immune surveillance in tumor cells in vivo, where the adaptive immune system plays a critical role.
Figure 3.

Atf7ip disruption stimulates antitumor immunity in vivo. A and B, Tumor growth curves of MC38 tumors with or without Atf7ip deficiency in NU/NU mice and B6/J mice (NU/NU: n = 8 mice/group, B6/J: n = 16 mice/group). C, Normalized tumor volume of B6/J mice at day 18. D, CD3 IHC staining of tumor samples at day 19. E, Quantification of CD3+ cells in D. F, Bar graph of CD8+ T-cell percentage in CD45+ cells in the tumor samples at day 19. G and H, Tumor growth curves of YUMM1.7 tumors with or without Atf7ip deficiency in NU/NU mice and B6/J mice (NU/NU: n = 8 mice/group, B6/J: n = 12 mice/group). I, Volcano plot illustrating retroelements that were upregulated (red) in Atf7i-deficient MC38 cells. J, Quantification of the number of ERV-derived antigens in G (adj. P < 0.05, fold change >2). K, RT-PCR of p15E and gp70 that encode tumor-associated ERV-derived antigens. L and M, Enrichment of genes associated with INTERFERON_ALPHA_RESPONSE (L) and INTERFERON_GAMMA_RESPONSE (M). N, RT-PCR of interferon-stimulated genes. O, Western blot of ERV sensors and interferon signaling pathway components. P, RT-PCR of Irf7 and Irf9. Q, Western blot of ERV sensors and interferon signaling pathway components. R, Enrichment of cancer testis antigen genes. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. NS, not significant.
To rule out the possibility that regulation of antigen expression and presentation by Atf7ip was due to an artificial effect from exogenous antigen induction, we expanded our study to a different cancer model by using the murine colorectal cancer cell line MC38, which was established from a colon tumor that developed in immunocompetent host (37). Atf7ip-deficient MC38 cells showed marginal reduction in proliferation in vitro (Supplementary Fig. S1H and S1I) and no change in cell apoptosis (Supplementary Fig. S1J and S1K). MC38 cells with or without Atf7ip disruption (Supplementary Fig. S1H) were subcutaneously injected into both nude mice and B6/J mice. Tumor progression with Atf7ip-deficient MC38 cells was significantly inhibited in B6/J mice but only marginally inhibited in nude mice (Fig. 3A–C). These data indicate that Atf7ip deficiency could stimulate an antitumor immune response through the adaptive immune system.
Indeed, IHC staining showed that there was a significant increase in CD3+ T-cell infiltration in Atf7ip-deficient tumors (Fig. 3D and E), which further supports that tumor cell–intrinsic Atf7ip deficiency enhances antitumor immune responses. Consistent with this observation, immune profiling showed a significant increase in infiltrated CD8+ T cells in Atf7ip-deficient tumors (Fig. 3F; Supplementary Fig. S2A). Moreover, the activation of cytotoxic CD8+ T cells was markedly enhanced in Atf7ip-deficient MC38 tumors (Supplementary Fig. S2B and S2C) and infiltrated CD4+ and CD8+ T cells were less exhausted (Supplementary Fig. S2D–S2G). T-bet is a key player in the generation of type 1 immunity in both helper and cytotoxic T cells and it is also a transcription factor that promotes the activation in CD8+ effector function (38). T-bet+ infiltrated CD4+ and CD8+ T cells were significantly increased in Atf7ip-deficient tumors (Supplementary Fig. S2H–S2K), also suggesting enhanced antitumor immunity. These results indicate that Atf7ip deficiency elevates tumor immunogenicity and the infiltration of activated T cells.
To explore whether from the effects of Atf7ip deficiency on antitumor immune responses are common to other cancers, we expanded our study to another tumor model, using YUMM1.7 cells, which recapitulate human melanoma because they have the BRAFV600E driver mutation, inactivating CDKN2A and PTEN mutations (16). YUMM1.7 cells with or without Atf7ip disruption (Supplementary Fig. S2L) were subcutaneously injected into both nude mice and B6/J mice. Tumor progression with Atf7ip-deficient YUMM1.7 cells was significantly inhibited in B6/J mice but only marginally inhibited in nude mice (Fig. 3G and H). Some sgAtf7ip-targeted YUMM1.7 tumors escaped immune control, as we previously observed with sgAtf7ip-targeted MC38 tumors. In both experiments, bulk populations of CRISPR–Cas9-disrupted cells were used to engraft recipient mice. Because of heterogeneity in CRISPR–Cas9 editing outcomes, bulk edited cell populations are composed of WT, partially disrupted, and fully disrupted genotypes. We hypothesized that cells with WT Atf7ip would have a selective advantage in vivo. To assess the editing outcomes before engraftment and at the endpoint of the experiment, we performed Sanger sequencing and TIDE analysis on in vivo sgAtf7ip-targeted YUMM1.7 tumor samples harvested from B6/J mice at day 17 and long-term in vitro cultured sgAtf7ip-targeted YUMM1.7 cells. We observed that most of in vivo sgAtf7ip-targeted tumor cells at day 17 had significantly more intact Atf7ip gene sequence and less out-of-frame Atf7ip gene sequence compared with long-term cultured sgAtf7ip-targeted cells (Supplementary Fig. S2M–S2P). This strongly supports that tumor cells lacking intact Atf7ip were rejected through immune surveillance. Collectively, these data indicate that Atf7ip deficiency consistently stimulated antitumor immune responses across different tumor models.
Atf7ip deficiency upregulates tumor antigen expression and interferon immune response
ERV-derived antigens are found in human tumors (39) and can be recognized by T cells (40). These antigens are potent tumor rejection antigens (41) and can potentially serve as immunotherapy targets (42–44). Atf7ip was identified as one of the determinants for provirus silencing in embryonic stem cells (45). Thus, we investigated whether Atf7ip deficiency could boost antitumor immunity by upregulating expression of ERV-derived antigens by performing RNA-seq of Atf7ip WT and Atf7ip-deficient MC38 cells and monitoring the change in ERV-derived antigens expression upon Atf7ip inhibition. The analysis indicated that Atf7ip deficiency significantly upregulated global expression of ERV-derived antigens (Fig. 3I and J). Specifically, the expression of the ERV-derived antigens gp70 and p15E was significantly increased upon Atf7ip inhibition (Fig. 3K). Double-strand RNAs (dsRNA), including ERVs, can be sensed by the immune system resulting in an interferon immune response (10, 11). Indeed, we found that upregulation of ERV expression in Atf7ip-deficient MC38 cells further activated interferon immune response (Fig. 3L and M). Key players in interferon signaling pathways, including Irf7 and Irf9, were upregulated in Atf7ip-deficient MC38 cells at both a transcriptional and translational level (Fig. 3N and O). This phenotype was recapitulated in Atf7ip-deficient YUMM1.7 cells (Fig. 3P and Q). These results indicate that Atf7ip deficiency might augment antitumor immune responses by promoting expression of ERV-derived antigens.
Several types of endogenous tumor antigens other than ERV-derived antigens have been described previously (41), and thus we investigated whether Atf7ip deficiency could cause changes to additional endogenous tumor antigens in tumor cells. For example, cancer testis antigens are another type of tumor endogenous antigen that have been applied in clinical trials for immunotherapy (46, 47). GSEA indicated global upregulation of CTs upon Atf7ip inhibition (ref. 48; Fig. 3R). In addition, changes in alternative splicing can produce neoepitopes (49, 50), and tumor-specific retained intron neoepitopes have been identified in both patient cancer and cell line–derived samples (51). Our GSEA indicated that Atf7ip deficiency inhibited spliceosome activity (Supplementary Fig. S2P and S2Q). There was a significant increase in RNA intron retention events in Atf7ip-deficient MC38 cells (Supplementary Fig. S2R). We also observed the downregulation of spliceosome assembly activities in ATF7IP low–expressing tumor samples in the TCGA lung adenocarcinoma dataset (Supplementary Fig. S2S and S2T). Collectively, these results indicate that Atf7ip deficiency promotes tumor antigen expression and activates antitumor immune responses.
Setdb1 is a partner of Atf7ip in the regulation of tumor antigen expression
To explore other key players that act alongside Atf7ip in regulating tumor antigen expression and presentation, we performed immunoprecipitation mass-spectrometry (IP-MS) of Atf7ip. We picked up 25 Atf7ip-binding partners and investigated whether their inhibition could modulate SQ transcription or presentation (Fig. 4A). Setdb1 was the only partner that not only interacted with Atf7ip but also regulated SQ expression and presentation (Fig. 4B–E; Supplementary Fig. S3A and S3B). Moreover, Setdb1 is one of the candidates whose inhibition promoted SIINFEKL expression and presentation in our initial genetic screen (Fig. 2B). Setdb1 is a histone H3K9 methyltransferases that works with HUSH complex for transcriptional silencing (52). However, our data showed that regulation of SQ presentation is independent of HUSH complex and other H3K9 methyltransferases (Supplementary Fig. S3C). In addition, Setdb1 deficiency significantly increased SIINFEKL expression in our KP-LucOS immune escaped tumor model (Supplementary Fig. S3D). These results indicate that Setdb1 might work with Atf7ip to regulate antigen expression.
Figure 4.

Atf7ip and Setdb1 work in a complex to regulate SQ expression and presentation. A, Strategy to identify Atf7ip-binding partners whose inhibition promotes both SQ transcription and SIINFEKL presentation. B, Coimmunoprecipitation of Atf7ip and Setdb1. C, RT-PCR of SQ transcripts. D, FACS of SIINFEKL presentation. E, Quantification of SIINFEKL presentation in D. F, Survival curve for NU/NU mice inoculated with WT or Setdb1-deficient KP-IE2 cells via tail vein injection (n = 3 mice/group). G, Survival curve for B6/J mice inoculated with WT or Setdb1-deficient KP-IE2 cells (n = 9 mice/group). H and I, RT-PCR of SQ transcripts after disrupting Setdb1 in Atf7ip knocked-out KP-IE2 cells. J and K, FACS of SIINFEKL presentation after disrupting Setdb1 in Atf7ip knocked-out KP-IE2 cells. L and M, RT-PCR of SQ transcripts after disrupting Atf7ip in Setdb1 knocked-out KP-IE2 cells. N and O, FACS of SIINFEKL presentation after disrupting Atf7ip in Setdb1 knocked-out KP-IE2 cells. P and Q, Western blot of Atf7ip and Setdb1 after depleting Setdb1 or Atf7ip in KP-IE2 cells (P) and MC38 cells (Q). R and S, Western blot of Atf7ip and Setdb1 after overexpressing Atf7ip or Setdb1 or both Atf7ip and Setdb1 in KP-IE2 cells (R) and MC38 cells (S). PCDH is empty vector. ***, P < 0.001; ****, P < 0.0001. NS, not significant.
To test whether Setdb1 disruption can restore the immunogenicity of KP-IE2 cells, KP-IE2 cells with or without Setdb1 deficiency were injected into both B6/J mice and nude mice (Supplementary Fig. S3E). We found no survival benefit in nude mice grafted with Setdb1-deficient KP-IE2 cells (Fig. 4F). However, Setdb1-deficient tumors grew significantly slower in B6/J mice, leading to significantly prolonged survival benefit (Fig. 4G). These results indicate that Setdb1 deficiency resulted in the restoration of immune surveillance to KP-IE2 cells in vivo. To understand whether there is a synergistic effect on regulating antigen expression and presentation when cells lack both Atf7ip and Setdb1, we generated Atf7ip and Setdb1 double knock-out KP-IE2 cell lines (Supplementary Fig. S3F). Simultaneous Atf7ip and Setdb1 deficiency showed no synergistic effect on SQ transcription and presentation (Fig. 4H–O), further supporting the idea that they work in the same pathway, and knocking out either one results in similar consequences. It has been reported that Atf7ip is essential for Setdb1 stability (53). Consistent with this, Atf7ip and Setdb1 proteins were positively correlated with each other in both KP-IE2 and MC38 cell lines (Fig. 4P and Q). Furthermore, overexpressing Atf7ip increased Setdb1 protein level and overexpressing Setdb1 increased Atf7ip protein level in the KP-IE2 cell line (Fig. 4R). The elevation of their protein levels was further enhanced when both Atf7ip and Setdb1 were overexpressed together in both KP-IE2 and MC38 cell lines (Fig. 4R and S). Thus, we hypothesize that Atf7ip and Setdb1 work in a complex to control tumor antigen expression and presentation.
Setdb1 deficiency augments antitumor immune responses
In our previously published in vivo epigenetic CRISPR screen, Setdb1 was identified as an epigenetic target that, upon loss, modulated the antitumor immune response in B6/J mice (ref. 15; Supplementary Fig. S4A and S4B). To investigate whether Setdb1 deficiency could also stimulate antitumor immunity in different tumor models, and based on what we observed in Atf7ip studies that clones with WT or partially disrupted genotypes could affect the tumor growth due to heterogeneity in CRISPR-Cas9 editing outcomes, we established Setdb1-deficient MC38 single-cell clones and pooled them together to avoid potential bias introduced by the clonal selection process (Supplementary Fig. S4C). Setdb1-deficient MC38 cells showed reduction in proliferation in vitro (Supplementary Fig. S4D), but no change in cell apoptosis (Supplementary Fig. S4E and S4F). MC38 cells and Setdb1-deficient MC38 cells were subcutaneously injected into both nude mice and B6/J mice. Greater inhibition in tumor growth was found in Setdb1-deficient MC38 tumor growth in B6/J mice compared with that in nude mice, indicating that Setdb1 deficiency induced an antitumor immune response (Fig. 5A–D). Moreover, the majority of Setdb1 disrupted tumors did not grow out at all, especially for cells generated using the first and third Setdb1 sgRNA (2 tumors formed out of 24 transplants for the first sgRNA; 2 tumors formed out of 24 transplants for the third sgRNA). Indeed, there was a significant increase in infiltrated CD3+ T cells in Setdb1-deficient tumors as assessed by IHC staining (Fig. 5E and F). Immune-profiling experiments also indicated an increase in infiltration of both CD4+ and CD8+ T cells in Setdb1-deficient tumors (Fig. 5G and H). Moreover, the activation of infiltrated T cells was markedly enhanced in Setdb1-deficient tumors (Supplementary Fig. S4G–S4N) and infiltrated T cells were less exhausted (Supplementary Fig. S4O–S4R). T-bet+ infiltrated T cells were significantly increased in Setdb1-deficient tumors, which also indicated enhanced antitumor immunity (Supplementary Fig. S4S–S4V).
Figure 5.

Setdb1 deficiency stimulates antitumor immunity in vivo. A–C, Tumor growth curves of MC38 tumors with or without Setdb1 deficiency in NU/NU mice and B6/J mice (NU/NU: n = 8 mice/group, B6/J: n = 8 mice/group). D, Normalized tumor volume of B6/J mice at day 18. E, CD3 IHC of tumor samples at day 19. F, Quantification of CD3+ cells in E. G and H, Bar graph of CD4+ (G) and CD8+ (H) T cells as a percentage of CD45+ cells in the tumor samples at day 19. I and J, Tumor growth curves of YUMM1.7 tumors with or without Setdb1 deficiency in nude (NU/NU) mice and B6 WT (B6/J) mice (NU/NU: n = 6 mice/group, B6/J: n = 12 mice/group). K, Volcano plot illustrating retroelements that are upregulated (red) in Setdb1-deficient MC38 cells. L, Quantification of the number of ERV-derived antigens in K (adj. P < 0.05, fold change >2). M, RT-PCR of p15E and gp70. N and O, Enrichment of genes associated with INTERFERON_ALPHA_RESPONSE (N) and INTERFERON_GAMMA_RESPONSE (O). P, RT-PCR of interferon-stimulated genes. Q, Western blot of ERV sensors and interferon signaling pathway components. R, RT-PCR of Irf7 and Irf9. S, Western blot of ERV sensors and interferon signaling pathway components. T, Overlap of upregulated genes in both Atf7ip KO and Setdb1 KO groups of MC38 cells. U, Enrichment of pathways associated with overlapping genes in T by Enrichr analysis tool. V, Enrichment of cancer testis antigen genes. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. NS, not significant.
We expanded our study to the YUMM1.7 tumor model. YUMM1.7 cells with or without Setdb1 disruption (Supplementary Fig. S4W) were subcutaneously injected into both nude mice and B6/J mice. Tumor progression with Setdb1-disrupted YUMM1.7 cells was significantly inhibited in B6/J mice but not in nude mice (Fig. 5I and J). Consistent with data from our previous Atf7ip-disrupted YUMM1.7 in vivo studies, YUMM1.7 tumors eventually grew out in immunocompetent mice injected with sgSetdb1-targeted cells. In addition, we observed that most in vivo sgSetdb1-targeted tumor cells at day 17 had significantly more intact Setdb1 gene sequence and less out-of-frame Setdb1 gene sequence compared with cells from long-term cultured sgSetdb1-targeted cells (Supplementary Fig. S4X and S4Y). This supports that tumor cells lacking intact Setdb1 were rejected through immune surveillance, which phenocopied our observations with tumor cells lacking intact Atf7ip. Collectively, these data indicate that Setdb1 deficiency consistently stimulated antitumor immune responses across different tumor models.
Setdb1 deficiency upregulates tumor antigen expression and interferon response
To analyze changes in expression of ERV-derived antigens upon Setdb1 disruption, we performed RNA-seq for Setdb1 WT and Setb1-deficient MC38 cells. The analysis indicated that Setdb1 deficiency significantly upregulated global expression of ERV-derived antigens (Fig. 5K and L). Expression of ERV-derived gp70 and p15E were significantly increased upon Setdb1 disruption (Fig. 5M).
Upregulation of the expression of ERV-derived antigens further activated interferon immune responses (Fig. 5N and O). Key players in interferon signaling pathways, including Irf7 and Irf9, were upregulated in Setdb1-deficient MC38 cells, as demonstrated at the transcriptional and translational level (Fig. 5P and Q). This phenotype was recapitulated in Setdb1-deficient YUMM1.7 cells (Fig. 5R and S). In addition, loss of Setdb1 and Atf7ip shared a similar impact on the transcriptome of MC38 cells (Fig. 5T). Enrichr analysis showed that the top three upregulated signaling pathways upon loss of Setdb1 and Atf7ip are interferon signaling pathways (Fig. 5U).
GSEA indicated global upregulation of CTs upon Setdb1 disruption (Fig. 5V), which phenocopied Atf7ip disruption. GSEA also indicated Setdb1 deficiency inhibited spliceosome activity (Supplementary Fig. S5A and S5B). There was also a significant increase in intron retention events in Setdb1-deficient MC38 cells (Supplementary Fig. S5C). Moreover, we observed the downregulation of spliceosome assembly activities in SETDB1 low–expressing tumor samples in the TCGA lung adenocarcinoma dataset (Supplementary Fig. S5D and S5E). This abnormal spliceosome activity and mRNA alternative splicing could be due to changes in the Atf7ip–Setdb1 network. The analysis of Atf7ip IP-MS by STRING database indicates that a significant number of Atf7ip-binding partners play a role in mRNA alternative splicing (Supplementary Fig. S5F and S5G). These results indicate that Setdb1 is necessary to suppress tumor antigen expression and its inhibition stimulates antitumor immunity.
Chromatin remodeling promotes ERV-derived antigen upregulation
H3K9 trimethylation (H3K9me3) is a histone modification associated with heterochromatin that contributes to gene downregulation by forming large repressive domains on the chromosomes (54). Because Setdb1 catalyzes the formation of H3K9me3 (55) and H3K9me3 levels decreased upon Atf7ip or Setdb1 inhibition (Fig. 4P and Q), we hypothesized that global upregulation of ERV-derived antigens upon Atf7ip or Setdb1 disruption was due to loss of H3K9me3. To test this hypothesis, we performed H3K9me3 ChIP-seq in WT, Atf7ip-deficient, and Setdb1-deficient MC38 cells and monitored differences in H3K9me3 in regions encoding ERV-derived antigens. The analysis showed that Atf7ip or Setdb1 deficiency significantly downregulated H3K9me3 deposition (blue color) in regions encoding ERV-derived antigens and the number of regions encoding ERV-derived antigens that showed H3K9me3 was significantly smaller (red color), indicating the overall genomic wide loss of H3K9me3 in ERVs region upon Atf7ip or Setdb1 inhibition (Fig. 6A–D). In addition, loss of Setdb1 and Atf7ip had similar impacts on H3K9 demethylation of regions encoding ERV-derived antigens (Fig. 6E). These results suggest that global upregulation of expression of ERV-derived antigens was due to loss of H3K9me3 deposition in the regions encoding these caused by Setdb1 deficiency, considering that Atf7ip disruption also compromises the function of Setdb1.
Figure 6.

Chromatin remodeling enables ERV-derived antigen upregulation upon Atf7ip or Setdb1 disruption. A, Volcano plot illustrating H3K9me3 of retroelements in MC38 cells with or without Atf7ip from differential H3K9me3 ChIP-binding peaks analysis. B, Quantification of the number of ERV-derived antigens with significant (adj. P < 0.05) higher binding peaks in A. C, Volcano plot illustrating H3K9me3 of retroelements in MC38 cells with or without Setdb1 from differential H3K9me3 ChIP-binding peaks analysis. D, Quantification of the number of ERV-derived antigens with significant (adj. P <0.05) higher binding peaks in C. E, Overlapped relative number of H3K9 demethylated ERV-derived antigens in both Atf7ip KO and Setdb1 KO groups of MC38 cells. F, Intra-TAD interaction changes in Atf7ip-deficient (sgAtf7ip) versus WT (sglacZ) MC38 cells. Atf7ip deficiency is associated with increased intra-TAD interaction (red, 918) and decreased intra-TAD interaction (blue, 40). G, Intra-TAD interaction changes in Setdb1-deficient (sgSetdb1) versus WT (sglacZ) MC38 cells. Setdb1 deficiency was associated with increased intra-TAD interaction (red, 786) and decreased intra-TAD interaction (blue, 94). H, Cumulative distribution function (CDF) plot illustrating the distribution of ERV expression associated by TADs that have increased interaction (red) and decreased interaction (blue) from WT MC38 cells to Atf7ip-deficient MC38 cells. I, CDF plot illustrating the distribution of ERV expression associated by TADs that have increased interaction (red) and decreased interaction (blue) from WT MC38 cells to Setdb1-deficient MC38 cells.
We further performed Hi-C for chromatin conformation capture analysis in WT, Atf7ip-deficient, and Setdb1-deficient MC38 cells. Genome-wide increase in the Hi-C signal was observed in both Atf7ip-deficient and Setdb1-deficient cells (Fig. 6F and G). Intra-TADs in the deficient groups exhibited more significantly upregulated elements than significantly downregulated ones, in comparison with the WT control group. The overall increase in the Hi-C signal also had functional impacts, measured by the RNA expression intensity of ERV-derived antigens (Fig. 6H and I). Those ERV-derived antigens placed in the differentially upregulated domains had higher signals overall than those in the differentially downregulated domains (Fig. 6H and I). This finding was corroborated by the signal of H3K9me3 repressive chromatin marker at the loci encoding ERV-derived antigens.
Low ATFIP or SETDB1 expression is associated with the hot tumor immune microenvironment
To explore the enhanced antitumor immunity resulting from Atf7ip or Setdb1 deficiency in a clinical setting, we interrogated public cancer transcriptomic data in TCGA. Pan-cancer analysis showed that either ATF7IP or SETDB1 expression strongly negatively correlated with antigen processing and presentation, interferon response signaling and T-cell–mediated immunity and cytotoxicity pathways (Fig. 7A and B), potentially confirming our results from murine tumor models.
Figure 7.
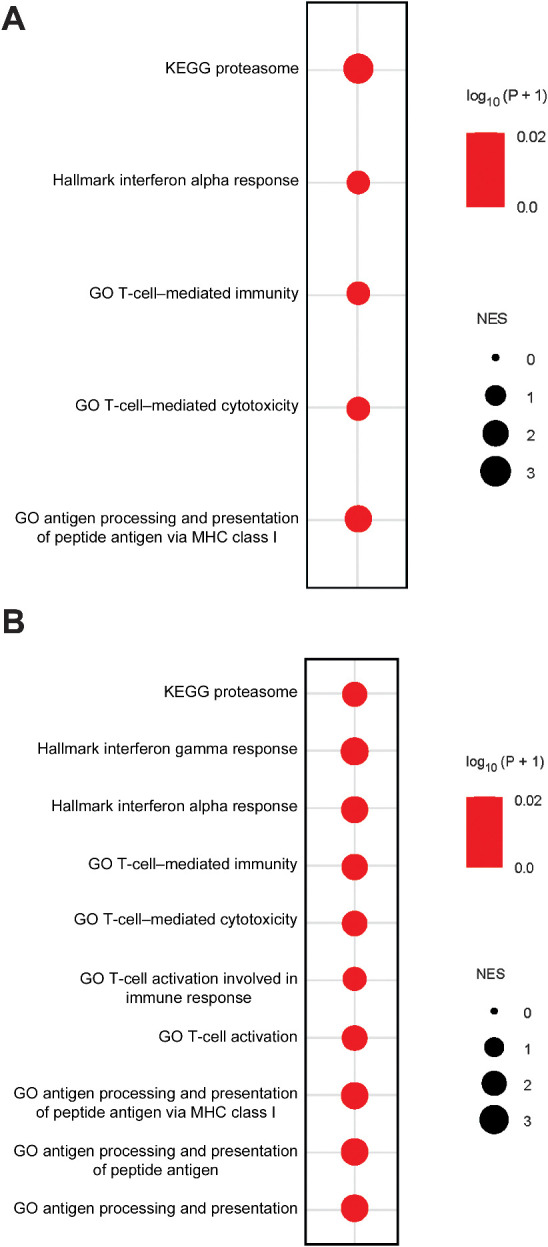
Low ATFIP or SETDB1 expression is associated with the hot tumor immune microenvironment in patients with cancer. A, Bubble plot showing multiple GSEA of the indicated pathways in a pan-cancer analysis of the TCGA dataset. NES >0: The gene set is enriched in ATF7IP low–expressing cancer samples (bottom 25%) compared with ATF7IP high–expressing cancer samples (top 25%). B, Bubble plot showing multiple GSEA of the indicated pathways in a pan-cancer analysis of the TCGA dataset. NES >0: The gene set is enriched in SETDB1 low–expressing cancer samples (bottom 25%) compared with SETDB1 high–expressing cancer samples (top 25%).
In summary, these results support the notion that Atf7ip or Setdb1 deficiency restrains tumor progression through increasing tumor immunogenicity, stimulating antitumor immunity, and enabling immune surveillance (Supplementary Fig. S6). Thus, targeting Atf7ip and/or Setdb1 might be a potential novel immunotherapeutic strategy for patients with cancer.
Discussion
Tumors can escape immune surveillance by losing the capacity to present antigens to effector cells of the immune system (5). We established a syngeneic murine tumor model that spontaneously adapted to immune pressure in vivo by silencing the model antigen SQ over a period of 10 months, modeling one process of human cancer evolution (6), in which the promoter of genes containing neo-antigenic mutations were silenced by an epigenetic mechanism. A cell line derived from tumors that escaped immune pressure, KP-IE2, was used in a genetic screen to identify epigenetic factors mediating presentation of SQ-derived peptides. We discovered that Atf7ip and its interacting partner Setdb1 are required for maintenance of the silenced state. Genetic disruption of either gene in tumor cells increased antigen presentation and induced an interferon response, leading to rejection of tumor engraftment with no other intervention, suggesting potential therapeutic strategies to overcome tumor immune evasion.
Atf7ip is a multifunctional nuclear protein, mediating heterochromatin formation and gene silencing in several contexts (53). Setdb1 is a histone methyltransferase that catalyzes the formation of H3K9me3 (55). Aberrant SETDB1 activity has been implicated to have potential pro-oncogenic function in the development of various types of cancer (56). In this study, we systemically investigated the role that Atf7ip and Setdb1 play in enforcing tumor antigen silencing, and identified their potential application in cancer immunotherapy.
A contemporary study by Griffin and colleagues (57) reported results of an in vivo screen for genes that are selectively required for tumor engraftment in the context of immune checkpoint blockade. The authors screened two independent syngeneic models, LLC and B16, identifying several candidate genes with sgRNAs depleted in the context of immune checkpoint blockade, including a common requirement for Setdb1. Although sgRNAs targeting Atf7ip were present in the library, they were not significantly depleted in any screen; however, the authors did identify Setdb1 as a hit in both cell lines. Several potential biological and technical differences could explain why Atf7ip was not identified by Griffin and colleagues in their primary screens. Griffin and colleagues screened two different cell lines and demonstrated divergent results for most hits. It is therefore possible that Atf7ip is required to maintain silencing in KP, MC38, and YUMM1.7 cells, but not in LLC or B16 cells. Technical factors may also explain differences in our findings; the screens were performed with different libraries, in different contexts, and with different types of selective pressure. In addition, our screen was specific to exploring epigenetic regulators playing a role in antigen expression and presentation. Because of the technical limitation of establishing endogenous tumor antigen models from immune evasion, we used an exogenous antigen model under the control of different promoters (CMV or UBC), which might not fully mimic endogenous promoters in the genome. Therefore, it would be worth establishing an endogenous tumor antigen model and further investigating novel epigenetic factors that regulating antigen expression and presentation.
Another significant difference is that in our study, immune checkpoint blockade was not required for tumor rejection in any secondary screen. Although we observed tumor outgrowth in mice engrafted with heterogeneous populations of Atf7ip knockout cells for both the MC38 and YUMM1.7 models, we subsequently determined that cells with intact Atf7ip efficiently populated the tumor. In subsequent experiments with Setdb1 disruption, we showed that clonal knockout cell lines were rejected for 2 out of 3 sgRNAs tested. It is possible that immune checkpoint blockade was required in Griffin and colleagues to control outgrowth of tumors with intact Setdb1, as knockout efficiency or clonal selection was not reported. Another possible explanation is context-specific differences in interferon induction. We found that antitumor immune responses promoted by Setdb1 deficiency were due to upregulation from both tumor antigen expression and interferon signaling, whereas Griffin and colleagues did not observe any significant interferon response in their model systems. Because five independent syngeneic tumor models are represented between these two complementary studies; it would be of significant future interest to cross validate our findings in these and further syngeneic models to understand better the heterogeneity and generalizability of these observations.
ERV-derived antigens are a type of cancer-specific antigen, which are silenced in normal tissues. Previous studies have shown that ERV-derived antigens can activate adaptive immunity and contribute to immune response against cancer cells (10, 11, 58). We observed a significant upregulation in the expression of ERV-derived antigens upon Atf7ip and Setdb1 disruption and subsequent antitumor immune responses in immunocompetent hosts. Although a previous study indicated that Atf7ip and Setdb1 regulated provirus silencing in embryonic stem cells (45), our study shows that Atf7ip and Setdb1 play an important role in modifying the tumor immune microenvironment by promoting expression of ERV-derived antigens. In addition, our immune profiling showed a significant expansion of T-bet+ infiltrated T cells in Atf7ip and Setdb1-deficient tumors and a remarkedly elevated Th1 antitumor immune response. Previous work shows that under antigen-specific stimulation conditions, T-bet is required for the differentiation of naïve CD8 T cells into effector CTLs (38). These results support the existence of enhanced antigen stimulation to T cells in Atf7ip or Setdb1-deficient tumor cells. It would be interesting to investigate whether other types of immune cells in the tumor microenvironment are changed upon intrinsic inhibition of Atf7ip or Setdb1 in tumor cells, like dendritic cells, macrophages and B cells, all of which can play a critical role in antigen presentation.
One class of epigenetic therapy, DNA methyltransferase inhibitor treatment mostly upregulates short interspersed nuclear elements (SINE, for instance, Alu retroelements), rather than long interspersed nuclear elements and DNA encoding ERV-derived antigens in patient-derived colorectal cancer cells (59). SINE (Alu retroelements) upregulation also results in activated interferon responses (59). These data indicate that different epigenetic inhibition strategies may induce interferon responses through different mechanisms in different cellular contexts.
We note that antigen SQ expression and presentation was reversed without significant change in the methylation of its promoter upon Atf7ip deficiency in the immune escaped KP-IE2 cell line. These results suggest that Atf7ip and Setdb1 might function downstream of DNA methylation. We did not perform further immune evasion mechanism studies in our immune escaped tumor model because our goals were focused on investigating immunotherapeutic targets that can reverse immune evasion, but future work should examine this. In addition, our data suggested the existence of euchromatin to heterochromatin transition in the SQ genomic region and the silencing of antigen SQ expression during tumor immune escape. Therefore, we hypothesize that the loss of Atf7ip might reverse the transition from heterochromatin to euchromatin in the SQ genomic region. It would be necessary to perform ATAC-seq to confirm the chromatin transition in SQ genomic region upon Atf7ip disruption in the future. Moreover, Atf7ip regulates H3K9me3, which plays a transcriptionally repressive function. Therefore, it would be worth performing H3K9me3 ChIP-seq to investigate whether there is H3K9me3 reduction in the SQ genomic region upon Atf7ip disruption in future studies.
Although TCGA pan-cancer analysis showed that ATF7IP or SETDB1 expression strongly negatively correlated with antigen-processing and presentation, interferon response signaling and T-cell–mediated immunity and cytotoxicity pathways, it would be worth dissecting the association between ATF7IP and SETDB1 and immune regulatory pathways based on immunogenic and nonimmunogenic types of cancer. In addition, given that disruption of Atf7ip or Setdb1 promoted expression of ERV-derived antigens in murine tumor models, it would be very interesting to investigate whether ATF7IP or SETDB1 expression also inversely correlates to expression of ERV-derived antigens in TCGA datasets. Moreover, to further explore more clinical relevance to our study, it would be worth examining the correlation between ATF7IP or SETDB1 expression of tumor samples and patients' response to immune checkpoint inhibitors in the future.
Collectively, we performed an epigenome-focused CRISPR screen and identified ATF7IP's and SETDB1's role in regulating antitumor immunity. Functional and mechanistic studies showed that tumor cell–intrinsic Atf7ip or Setdb1 deficiency promotes antigen expression and presentation, especially for ERV-derived antigens and intron-retained mRNAs. Upregulation of antigen expression and presentation increases T-cell infiltration and activation, leading to enhanced antitumor immune responses (Supplementary Fig. S6). Atf7ip and Setdb1 deficiency exerts significant inhibition on tumor growth in immunocompetent host. Thus, ATF7IP and Setdb1 inhibition might serve as potential novel immunotherapeutic strategies for patients with cancer.
Authors' Disclosures
H. Hu reports a patent for SETDB1 and ATF7IP as biomarker pending. H. Cho reports grants from NIH outside the submitted work. D. Schadendorf reports grants, personal fees, and nonfinancial support from Bristol Myers Squibb, Roche, MSD, Novartis, and Amgen outside the submitted work. K.A. Schalper reports grants from NIH/NCI, Navigate Biopharma, Tesaro/GlaxoSmithKline, Moderna Inc., Takeda, Surface Oncology, Pierre Fabre Research Institute, Merck Sharpe & Dohme, Bristol Myers Squibb, AstraZeneca, Ribon Therapeutics, and Eli Lilly and personal fees from Celgene, Ono Pharmaceuticals, AbbVie, Shattuck Labs, Pierre Fabre, EMD Serono, Clinica Alemana de Santiago, Genmab, PeerView, Fluidigm, Takeda/Millenium Pharmaceuticals, Merck Sharpe & Dohme, Bristol Myers Squibb, Agenus, GLG, and Torque Therapeutics outside the submitted work. V. Velcheti reports personal fees from Bristol Myers Squibb, Foundation Medicine, Merck, EMD Serono, Novartis, and AstraZeneca outside the submitted work. F. Li reports a patent for use of ATF7IP and SETDB1 as biomarker pending. K.-K. Wong reports grants from Tvardi Therapeutics, Mirati Therapeutics, Takeda, Bristol Myers Squibb, Merus, Alkermes, Ansun, Dracen, and Janssen and personal fees from Allorion, AZ, Genocea, Preclude, Recursion, Epiphanes, and Merck outside the submitted work; a patent for SETDB1 and ATF7IP as biomarker pending; and is an equity holder in G1 Therapeutics. No disclosures were reported by the other authors.
Acknowledgments
The authors thank the NYU School of Medicine Animal Resources Facility staff for their support of the animal studies; Genome Technology Center for NGS of CRISPR screen samples, RNA-seq, ATAC-seq, ChIP-seq, and Hi-C-seq; Applied Bioinformatics Laboratories for bioinformatics analyses; Proteomics Laboratory for IP-MS service and analysis; Biospecimen Research & Development and Experimental Pathology Research Laboratory for IHC staining service; Cytometry & Cell Sorting Laboratory for cell sorting service; and Preclinical Imaging Laboratory for providing MRI equipment. We thank Dr. Marcus Bosenberg for sharing the YUMM1.7 cell line for this study. K.-K. Wong is supported by NIH Research Project Grant Program (RO1CA248896, RO1CA219670, and RO1CA215471).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Contributions
H. Hu: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Khodadadi-Jamayran: Software, investigation, methodology. I. Dolgalev: Software, investigation. H. Cho: Software, investigation. S. Badri: Software, investigation. L.A. Chiriboga: Investigation. B. Zeck: Investigation. M. Lopez De Rodas Gregorio: Investigation. C.M. Dowling: Visualization. K. Labbe: Project administration. J. Deng: Visualization. T. Chen: Project administration. H. Zhang: Visualization. P. Zappile: Software, investigation. Z. Chen: Software, investigation. B. Ueberheide: Investigation, methodology. A. Karatza: Visualization. H. Han: Investigation. M. Ranieri: Visualization. S. Tang: Investigation. G. Jour: Visualization. I. Osman: Resources. A. Sucker: Resources. D. Schadendorf: Resources. A. Tsirigos: Visualization. K.A. Schalper: Resources. V. Velcheti: Resources. H.Y. Huang: Investigation. Y. Jin: Resources. H. Ji: Resources. J.T. Poirier: Data curation, formal analysis, supervision, visualization, methodology, writing–review and editing. F. Li: Conceptualization, data curation, formal analysis, supervision, investigation, visualization, methodology, writing–original draft. K.-K. Wong: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res 1970;13:1–27. [DOI] [PubMed] [Google Scholar]
- 2. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–70. [DOI] [PubMed] [Google Scholar]
- 3. Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology 2017;150:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S, et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res 2019;7:1148–61. [DOI] [PubMed] [Google Scholar]
- 5. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012;482:405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019;567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non–small cell lung cancer. Cancer Discov 2017;7:852–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019;36:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun T, Li Y, Yang W, Wu H, Li X, Huang Y, et al. Histone deacetylase inhibition upregulates MHC class I to facilitate cytotoxic T lymphocyte-mediated tumor cell killing in glioma cells. J Cancer 2019;10:5638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA, including endogenous retroviruses. Cell 2015;162:974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roulois D, Yau HL, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015;162:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Terracina KP, Graham LJ, Payne KK, Manjili MH, Baek A, Damle SR, et al. DNA methyltransferase inhibition increases efficacy of adoptive cellular immunotherapy of murine breast cancer. Cancer Immunol Immunother 2016;65:1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates antitumor immunity and enables checkpoint blockade. Cell 2018;174:549–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer 2021;2:503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li F, Huang Q, Luster TA, Hu H, Zhang H, Ng WL, et al. In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov 2020;10:270–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res 2016;29:590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31:2382–3. [DOI] [PubMed] [Google Scholar]
- 27. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lazaris C, Kelly S, Ntziachristos P, Aifantis I, Tsirigos A. HiC-bench: comprehensive and reproducible Hi-C data analysis designed for parameter exploration and benchmarking. BMC Genomics 2017;18:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 2012;9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 2013;49:503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 2017;171:1259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate antitumor immunity. Curr Opin Immunol 2012;24:207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, et al. CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discov 2020;10:1912–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, et al. Endogenous T-cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011;19:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pelleitier M, Montplaisir S. The nude mouse: a model of deficient T-cell function. Methods Achiev Exp Pathol 1975;7:149–66. [PubMed] [Google Scholar]
- 37. Corbett TH, Griswold DP Jr, Roberts BJ, Peckham JC, Schabel FM Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 1975;35:2434–9. [PubMed] [Google Scholar]
- 38. Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T-cell function regulated by T-bet. Proc Natl Acad Sci U S A 2003;100:15818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol 2014;192:1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. TIL therapy broadens the tumor-reactive CD8(+) T-cell compartment in melanoma patients. Oncoimmunology 2012;1:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kelderman S, Kvistborg P. Tumor antigens in human cancer control. Biochim Biophys Acta 2016;1865:83–9. [DOI] [PubMed] [Google Scholar]
- 42. Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest 2008;118:1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kershaw MH, Hsu C, Mondesire W, Parker LL, Wang G, Overwijk WW, et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res 2001;61:7920–4. [PMC free article] [PubMed] [Google Scholar]
- 44. Bronte V, Cingarlini S, Apolloni E, Serafini P, Marigo I, De Santo C, et al. Effective genetic vaccination with a widely shared endogenous retroviral tumor antigen requires CD40 stimulation during tumor rejection phase. J Immunol 2003;171:6396–405. [DOI] [PubMed] [Google Scholar]
- 45. Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015;163:230–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 2015;21:1019–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aruga A, Takeshita N, Kotera Y, Okuyama R, Matsushita N, Ohta T, et al. Phase I clinical trial of multiple-peptide vaccination for patients with advanced biliary tract cancer. J Transl Med 2014;12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun 2016;7:10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frankiw L, Baltimore D, Li G. Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol 2019;19:675–87. [DOI] [PubMed] [Google Scholar]
- 50. Park J, Chung YJ. Identification of neoantigens derived from alternative splicing and RNA modification. Genomics Inform 2019;17:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, et al. Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 2018;36:1056–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, et al. GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015;348:1481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep 2016;17:653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Becker JS, Nicetto D, Zaret KS. H3K9me3-dependent heterochromatin: Barrier to cell fate changes. Trends Genet 2016;32:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, et al. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell 2003;12:475–87. [DOI] [PubMed] [Google Scholar]
- 56. Strepkos D, Markouli M, Klonou A, Papavassiliou AG, Piperi C. Histone methyltransferase SETDB1: a common denominator of tumorigenesis with therapeutic potential. Cancer Res 2020;8:525–34. [DOI] [PubMed] [Google Scholar]
- 57. Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021;595:390–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 2018;3:e121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mehdipour P, Marhon SA, Ettayebi I, Chakravarthy A, Hosseini A, Wang Y, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020;588:169–73. [DOI] [PubMed] [Google Scholar]