

Abstract
Since 2017, three CD19 directed chimeric antigen receptor (CAR T-cell) therapies have been approved for relapsed/refractory aggressive large B cell lymphomas (LBCL) after two lines of therapy, axicabtagene ciloleucel, tisagenlecleucel and lisocabtagene maraleucel. Recently, three prospective phase 3 randomized clinical trials were conducted to define the optimal second line treatment by comparing each of the CAR T-cell products to the current standard of care: ZUMA-7 for axicabtagene ciloleucel, BELINDA for tisagenlecleucel and TRANSFORM for lisocabtagene maraleucel. These three studies, while largely addressing the same question, had different outcomes with ZUMA-7 and TRANSFORM demonstrating a significant improvement with CD19 CAR T-cell therapy in second line compared to SOC, while BELINDA did not show any benefit. The US FDA has now approved axicabtagene ciloleucel for LBCL that is refractory to first-line chemoimmunotherapy or relapses within 12 months of first-line chemoimmunotherapy. Following the reporting of these practice changing studies, a group of experts convened by the American Society for Transplantation and Cellular Therapy now provides a comprehensive review of the three studies, emphasizing potential differences, and share perspectives on what these results mean to clinical practice in this new era of treatment of B-cell lymphomas.
Introduction
Since 2017, three CD19 directed chimeric antigen receptor (CAR T-cell) therapies have been approved for relapsed/refractory aggressive large B cell lymphomas (LBCL) after two lines of therapy, axicabtagene ciloleucel (axi-cel, Kite/Gilead), tisagenlecleucel (tisa-cel, Novartis) and lisocabtagene maraleucel (liso-cel, Bristol Myers Squibb) 1–3. The three products have never been compared in a randomized fashion but differences in construct design, manufacturing nuances, and toxicity have influenced clinical implementation 4–7. The promising responses observed with these products raised the consideration for earlier use of CAR T-cell therapy, ahead of autologous hematopoietic cell transplant (auto-HCT), in the treatment paradigm for LBCL. To address this important question, three concurrent prospective phase 3 randomized clinical trials (RCT) were conducted to define the optimal second line treatment by comparing each of the CAR T-cell products to the current standard of care: ZUMA-7 for axicabtagene ciloleucel, BELINDA for tisagenlecleucel and TRANSFORM for lisocabtagene maraleucel 8–10. These three studies, while largely addressing the same question, had differences in study design that may have resulted in meaningful differences in outcomes, which are destined to influence clinical implementation of these results.
With preliminary release of positive results for two of the trials and anticipated FDA approval of CAR T cells for LBCL in second line, the American Society for Transplantation and Cellular Therapy (ASTCT) convened a group of experts in the fall of 2021 to provide a comprehensive review of the three studies and share perspectives on what these results mean to clinical practice as we implement the use of CD19 directed CAR T-cell products in this new era of treatment of B cell lymphomas (Box 1).
Box 1.
List of questions addressing the impact of randomized trials of CAR T cells in second line for LBCL
| FAQ 1 | What is the current standard of care for patients with LBCL in second line? |
| FAQ 2 | What are the key results of the three randomized trials of CAR T-cells in second line therapy? |
| FAQ 3 | What are additional results of the three randomized trials of CAR T-cells in second line therapy that should be considered? |
| FAQ 4 | What are the differences between the three studies’ designs and how may that have impacted outcomes? |
| FAQ 5 | If the randomized studies in second line show a difference in PFS/EFS but not in OS, should that change practice? |
| FAQ 6 | Do these trials justify a one size fits all approach in second line treatment of early relapsing aggressive B cell lymphomas? |
| FAQ 7 | If patients get CAR T-cells in second line, what should be the SOC in third line, auto-HCT vs allo-HCT vs other? |
| FAQ 8 | If CAR T cells are the SOC in second line, how will we approach patients referred to the transplant/cell therapy center having already started salvage and having established a CR or PR? |
| FAQ 9 | How should we interpret results from studies in the second line setting when frontline therapies change? |
| FAQ 10 | Since Auto-HCT is currently available in more centers than CAR T cells, will that affect access to care if the SOC changes? |
| FAQ 11 | Is the benefit-cost ratio better for CAR T cells or Auto-HCT in second line therapy of DLBCL? What are potential economic considerations of shifting CAR T to 2nd line? |
| FAQ 12 | Which components should be included in the next series of landmark CAR T-cell trials for frontline? |
Q1. What is the current standard of care for patients with LBCL in second line?
For almost three decades the standard of care for second line therapy for relapsed/refractory diffuse large B-cell lymphoma (DLBCL) has been platinum-based salvage chemotherapy followed by high dose therapy and auto-HCT consolidation for patients who were medically eligible and achieved a partial remission (PR) or complete remission (CR). This standard was based on the improved five-year event free survival (EFS, 46% vs 12%, with events defined as relapse, disease progression or death from any cause) and overall survival (OS, 53% vs 32%) seen in the PARMA study comparing auto-HCT to further cycles of salvage chemotherapy in patients achieving a CR or PR to the initial salvage attempt (randomization happening after establishing response to salvage therapy) 11. Of note, 64% of patients relapsing after an initial CR responded to salvage chemotherapy, but only 21% of the patients with primary refractory disease had response to salvage.
This has become even more of an issue in the current era of rituximab-based frontline chemoimmunotherapy (CIT). The pivotal randomized phase 3 CORAL study identified patients who relapsed within 12 months of initial diagnosis and those with age adjusted international prognostic index (IPI) of > 2 as having an inferior response rate, EFS and OS, regardless of salvage regimen 12. In the CORAL study, overall response rate (ORR, including CR or PR) was 62–64% with the two salvage options and only 51% patients went on to receive auto-HCT after response to salvage chemotherapy 12, 13. Although more patients are cured up front, and those that relapse >12 months after initial diagnosis still have a 45% 3-year EFS with salvage chemotherapy and auto-HCT, the CORAL study highlighted the poor outcomes with SOC in the subgroup of patients with primary refractory disease and/or relapse within 12 months of initial diagnosis. This high-risk subgroup was also confirmed in the NCIC-CTG LY.12 study with only 25% of patients with primary refractory disease responding to salvage chemotherapy and proceeding to auto-HCT 14. It is however noteworthy that a retrospective Center for International Blood and Marrow Transplant Research (CIBMTR) analysis showed durable disease control with 44% 3-year PFS despite early rituximab failure within 12 months, underscoring the fact that in the subset of early failure patients with responsive disease to salvage attempts, auto-HCT can provide durable control 15. While representing retrospective real-world data reported to the registry, CIBMTR results were in line with the 40% 3-year EFS with auto-HCT in this early relapse subgroup with responsive disease in the CORAL study and 45% 4-year EFS seen after auto-HCT in this subgroup in the NCIC-CTG LY.12 study 12, 14. Of note, the 12-months definitions differ between the CORAL and CIBMTR studies (12 months from diagnosis) compared to the NCIC-CTG LY.12 study and three RCT (12 months from end of treatment). Hence a small proportion of patients enrolled (~15%) in the three RCT would be considered low-risk by CORAL study criteria. It should also be noted that those who achieve complete response to salvage (CR2) and especially those achieving a negative post-salvage PET scan (PET-CR) have the best outcomes after auto-HCT (4-year PFS 64% with PET-CR vs 32% with positive PET) 16. Similar results were seen in another recent CIBMTR study where auto-HCT provided durable disease control in patients with primary refractory DLBCL who respond to salvage therapies 17. However, these results need to be seen in the context of the 75–80% failure rate for patients with primary refractory disease or early relapse that are unable to move on to auto-HCT after standard salvage chemotherapy, substantiating this subgroup as an area of unmet need. Furthermore, while CIBMTR data is collected prospectively, it only includes patients who received a cell infusion and who were reported to the registry, introducing potential selection biases.
Studies have shown that the addition of rituximab (CIBMTR), rituximab maintenance (CORAL) or iodine-131 tositumomab (BMT-CTN 0401) to standard conditioning chemotherapy and auto-HCT did not improve outcomes of auto-HCT using standard BEAM (carmustine, etoposide, cytarabine, melphalan) 13, 18, 19. However, it is unclear if some of the newer immunotherapies like bispecific antibodies or antibody drug conjugates may be able to induce deep enough remissions in early relapsed/refractory disease to move to auto-HCT or to combine with conditioning to make responses last longer. Are the durable remissions seen after auto-HCT in the early relapsed/refractory subgroup simply a result of better chemosensitivity and better lymphoma biology in those patients, or can we achieve similar results from auto-HCT as long as we can obtain a CR or PR using alternative methods like immunotherapy prior to auto-HCT 20–22? These questions remain to be determined.
Q2. What are the key results of the three randomized trials of CAR T-cells in second line therapy?
The three RCT asked a similar question in patients with lymphoma that was refractory or relapsed within 12 months of first line therapy: Is EFS superior with CAR T-cells or standard salvage CIT followed by auto-HCT 8–10? Not only were there differences in study design and endpoint definitions between the three trials, but overall outcomes were also different, with two trials, ZUMA-7 and TRANSFORM, reporting positive results in favor of the CAR T-cell arm for the primary outcome, EFS, while BELINDA did not show any difference between the CAR T-cell and controls arms (Table 1). Of note, with a median follow-up of 6 to 25 months, none of the trials have demonstrated a significant difference in OS at this time.
Table 1.
Clinical outcomes of the three trials.a
| TRIAL | ZUMA-7 | BELINDA | TRANSFORM | |||||
|---|---|---|---|---|---|---|---|---|
| Axi-Cel | SOC | Tisa-Cel | SOC | Liso-Cel | SOC | |||
| N=180 | N=179 | N=162 | N=160 | N=92 | N=92 | |||
| Patient disposition | ||||||||
| CAR T infusion (%) | 94 | N/A | 96 | N/A | 98 | N/A | ||
| Bridging (%) | 36b | N/A | 83 | N/A | 63 | N/A | ||
| 1 cycle (%) | N/A | N/A | 36 | N/A | 58 | N/A | ||
| > 1 cycle (%) | N/A | N/A | 48 | N/A | 5 | N/A | ||
| Median days to infusionc | 29 | N/A | 52 | N/A | 34 | N/A | ||
| ASCT (%) | N/A | 36 | N/A | 33 | N/A | 47 | ||
| Crossover to CAR T (%) | N/A | 56 | N/A | 51 | N/A | 55 | ||
| Efficacy | ||||||||
| Median follow-up (months) | 25 | 25 | 10 | 10 | 6 | 6 | ||
| ORR (%) | 83 | 50 | 46 | 43 | 86 | 48 | ||
| CR (%) | 65 | 32 | 28 | 28 | 66 | 39 | ||
| EFS median (months) | 8.3 | 2 | 3 | 3 | 10.1 | 2.3 | ||
| PFS median (months) | 14.7 | 3.7 | NR | NR | 14.8 | 5.7 | ||
| OS median (months) | Not reached | 35.1 | NR | NR | Not reached | 16.4 | ||
| Toxicityd | ||||||||
| Grade 3+ CRS (%) | 6 | N/A | 5 | N/A | 1 | N/A | ||
| Grade 3+ ICANS (%) | 21 | N/A | 2 | N/A | 4 | N/A | ||
| Deathse (n) | 64 | 78 | 52 | 45 | 13 | 24 | ||
| Progressive disease (n) | 47 | 64 | 42 | 32 | 7 | 13 | ||
| Other (n) | 17 | 14 | 10 | 13 | 6 | 11 | ||
Abbreviations: N/A: not applicable; NR: not reported
The only bridging allowed on ZUMA-7 was glucocorticoids.
Median days to infusion is reported as days from randomization to infusion for ZUMA-7 and TRANSFORM and days from leukapheresis to infusion for BELINDA (see text for details).
Toxicity was graded per Lee et al 25 for CRS and CTCAE for neurological toxicity.
Median duration of follow-up differed between trials as indicated.
A total of 865 patients were treated on the three trials, including 359 on ZUMA-7, 322 on BELINDA and 184 on TRANSFORM. Similar rates of patients randomized to the CAR T-cell arm were infused with axi-cel (94%), tisa-cel (96%) and liso-cel (98%), respectively. Among patients randomized to the CAR T-cell arm, 36% treated with axi-cel received bridging therapy consisting of glucocorticoids only, while the two other trials allowed bridging chemotherapy, which was administered to 83% and 63% of patients receiving tisa-cel and liso-cel, respectively. A higher percent of patients on the SOC arm proceeded to auto-HCT on TRANSFORM (47%) compared to ZUMA-7 (36%) and BELINDA (33%). Similar rates of crossover to CAR T-cell therapy were observed in the three studies, 56%, 51% and 55% on ZUMA-7, BELINDA, and TRANSFORM, respectively.
In the ZUMA-7 trial, at a median follow-up of 24.9 months, the median EFS was 8.3 vs 2.0 months for axi-cel vs. SOC, and the 24-month EFS was 41% and 16%, respectively (HR for death or event 0.40, 95% CI 0.31 – 0.51, p<0.001) 8. ORR was 83% (CR 65%) in the axi-cel arm and 50% (CR 32%) in the SOC arm. In an interim analysis, estimated 2-year OS was 61% in the axi-cel arm and 52% in the SOC arm (HR for death 0.73, 95% CI 0.53–1.01). Based on patient reported outcomes (PRO), axi-cel was associated with an improvement in quality of life (QOL) at Day 100 and faster recovery to pretreatment QOL compared with SOC 23. In the TRANSFORM trial, with a median follow-up of 6.2 months, median EFS was 10.1 vs. 2.3 months for liso-cel and SOC, respectively (HR 0.349, p<0.0001) 10. ORR was 86% (CR 66%) in the liso-cel arm and 48% (CR 39%) in the SOC arm. Estimated 12-month OS was 79% months in the liso-cel arm and 64% in the SOC arm. Liso-cel also showed favorable improvement in PRO compared with SOC 24. In contrast, in the BELINDA trial, median EFS was 3.0 months in both groups (HR for death or event 1.07, 95% CI 0.82–1.40, p=0.61) 9. ORR was 46.3% (CR 28%) in the tisa-cel arm and 42.5% (CR 28%) in the SOC arm. OS was not formally analyzed in the absence of an EFS benefit. On April 1, 2022, the FDA approved axi-cel for adult patients with LBCL that is refractory to first-line CIT or relapses within 12 months of first-line therapy. The NCCN guidelines (v. 2.2022) were updated in March 2022 to include axi-cel as the indicated treatment for patients who have primary refractory disease or relapse within 12 months and are eligible for CAR T-cell therapy. It is anticipated that approval of liso-cel in second line may follow later this year.
The use of CAR T cells has been associated with specific early toxicities, CRS and ICANS 25–31. Patients treated with axi-cel had a 6% incidence of grade ≥ 3 CRS and 21% had grade ≥ 3 neurotoxicity 8. Consistent with observations from the original registration studies in third line 1–3, these rates were higher than those observed in patients treated with liso-cel (1% grade ≥ 3 CRS; 4% grade ≥ 3 neurotoxicity) 10 and tisa-cel (5% grade ≥ 3 CRS; 2% grade ≥ 3 neurotoxicity) 9. All three studies used the original Lee criteria for CRS grading 25, and CTCAE grading for neurological toxicity, rather than the ASTCT consensus criteria routinely used in current clinical practice 26. While not in exact agreement, the grading systems used on these trials were closer to the ASTCT consensus grading than the original Penn grading for CRS or CARTOX for neurotoxicity 27. Beyond CRS and ICANS, longer follow-up of the trials will be needed to document some of the late effects of CAR T cells 32–37.
Q3. What are additional results of the three randomized trials of CAR T-cells in second line therapy that should be considered?
The CORAL study identified a subgroup of patients with lower EFS and OS 12, 13. In the pooled retrospective SCHOLAR-1 data, patients with progressive disease or stable disease as best response after >4 cycles of first-line chemotherapy or 2 cycles of later line chemotherapy and those who relapsed within 12 months from auto-HCT were labelled as refractory DLBCL and objective response to next line of therapy in this cohort was 26% with 7% CR 38. Keeping the above in context, ZUMA-7, BELINDA and TRANSFORM enrolled patients with a similar pattern of aggressive disease status including LBCL refractory (lack of CR) to first-line therapy or relapsed within 12 months of first-line CIT including a CD20 monoclonal antibody and an anthracycline 8–10. Patients also needed to be eligible for an auto-HCT. With approval by the FDA of at least one CD19 CAR T-cell therapy in second line in the setting of limited OS data, an important consideration will be to see if the trial results would apply to all patients with the above criteria or if a select group of patients would benefit the most.
Activated B-cell-like (ABC) cell of origin large B cell lymphomas have poorer overall outcomes compared to germinal center B-cell-like (GCB) in the chemotherapy era 39. Patients with double-hit, triple-hit (MYC, BCL-2 and BCL-6 translocations) and double-expressor (high MYC and BCL2 protein expression without an underlying translocation) lymphomas are known to have a poorer outcome with chemotherapy and auto-HCT in a salvage setting. From the CORAL data, 4-year PFS and OS were 18% and 29% in patients with MYC rearrangement versus 42% and 62% in MYC negative patients 40. In a more recent retrospective study of 117 patients who underwent auto-HCT between 2000 and 2013 from Dana Farber and City of Hope, 4-year PFS for those with neither double-hit/double-expressor was 59%, double-expressor 48% and double-hit 25% while 4-year OS was 67%, 56% and 25%, respectively 41. Within studies on CAR T-cells published thus far, albeit with a shorter follow-up and absence of direct comparison, these differences appear less dramatic 1–3. A subgroup analysis of ZUMA-7 showed favorable EFS in both GCB and ABC lymphoma, although the later only composed <10% of the patients 8. Similarly, patients with both GCB and ABC lymphoma had superior outcomes with liso-cel compared to SOC 10. EFS was also superior with axi-cel in double-hit, triple-hit and double-expressor lymphoma, which together composed almost 50% of the cohort. This suggests that CAR T cells may overcome some of the adverse prognostic factors for lymphoma identified in the pre-CAR T era. However, sufficiently powered studies comparing CAR T-cell versus salvage chemotherapy/auto-HCT are needed to specifically address this question. A number of factors have been shown to predict responses to CAR T cells in the third line setting, including the IPI and co-morbidities, as well as disease characteristics 42–47. One area that deserves further investigation is the identification of biomarkers for safety and toxicity of CAR T-cells in the setting of large multicenter cohorts 48.
One population of particular interest is patients over 65. All three RCT included around 30% of patients in this age group 8–10. Older patients and those who may not be considered candidates for auto-HCT due to comorbidities can potentially receive CAR T-cell therapy, and some patients are already being treated in second line with CAR T-cell therapy in these circumstances. While response to salvage, rates of auto-HCT and OS were similar in patients >60 and ≤60 years in subgroup analysis of CCTG LY.12 trial, NRM was 4% for salvage chemotherapy and 8% for auto-HCT at 100 days for age >60 years (versus 1% for each for age ≤60 years) 49. In the post-hoc subgroup analysis of ZUMA-1, which followed strict inclusion criteria for organ function, CAR T-cell expansion, ORR, CR rate, cytopenias, infections, and CRS were not higher in age >65 versus compared to age ≤65 years 50. However, neurotoxicity grade ≥3 was higher in older patients (44%) compared to the younger cohort (28%), mostly manifesting as delirium and encephalopathy. Forty-two percent of patients treated on the TRANSFORM trial were over 65 years 3. Both efficacy and safety after treatment with liso-cel were similar in those patients compared to those <65 years 10. In a retrospective single institution study using commercial axi-cel and tisa-cel, incidence of CRS, ICANS, hematological toxicity and intensive care admissions were similar in older (≥65 years) and younger (<65 years) cohort, despite a high comorbidity index in the former 51. It should be noted that, while NRM at 100 days and 1 year in older patients undergoing auto-HCT has been found to be comparable to younger patients, higher rates of cardiovascular toxicities have been observed as well as increased risk of disease progression or death 52. In clinical practice, inclination towards auto-HCT for older patients is lower as also witnessed in the ZUMA-1 subgroup analysis 50. Combined with ongoing efforts (prophylactic and treatment) to abrogate CAR T-cell toxicity and, given the relatively low incidence of severe CRS and ICANS with liso-cel, CAR T-cell therapy may become more feasible for the elderly or frail population, not deemed fit for auto-HCT.
Q4. What are the differences between the three studies’ designs and how may that have impacted outcomes?
Given differences in outcomes between the three trials, a key question is what drove those differences. Was it differences in study design, patient populations, CAR T-cell product, other factors, or a combination of factors? It is critical to try and understand those differences, particularly as we implement the results of these trials into clinical practice.
Table 2 outlines the study designs of the three trials. Inclusion criteria were similar across the studies as they enrolled patients with LBCL with disease that was refractory or relapsed within 12 months of 1st line therapy. All patients had to be transplant eligible. BELINDA and TRANSFORM also included primary mediastinal B-cell lymphoma and follicular lymphoma grade 3B. Finally, as ZUMA-7 only allowed glucocorticoids as bridging, the study excluded patients with requirement for urgent therapy due to tumor mass effects. As noted above, the inclusion criteria were similar to the NCIC-CTG LY.12 study but differed from the CORAL study where time of diagnosis rather than end of 1st line therapy was used to define the 12 months timepoint 12, 14. Disease assessment was done according to Lugano criteria 53. The primary endpoint for all three trials was EFS. However, the definitions of EFS varied among the 3 trials (Table 3). In addition, patients on BELINDA had a PET scan at 6 weeks that did not count as an event but may have impacted clinical decision making. Another difference among the trials was how crossover was handled on the SOC arm. Both BELINDA and TRANSFORM included crossover on trial; while crossover was not built into ZUMA-7, patients on the SOC arm ended up being treated with CAR T-cells, including many with axi-cel. As noted above, crossover rates were similar.
Table 2.
Comparison of the study design between the three trials.a
| TRIAL | ZUMA-7 | BELINDA | TRANSFORM | ||
|---|---|---|---|---|---|
| Inclusion Criteria | |||||
| Histology | LBCL | LBCL, PMBL, FL grade 3B | LBCL, PMBL, FL grade 3Bb | ||
| Inclusion criteria | Refractory or relapsed within 12 months of 1st line | Refractory or relapsed within 12 months of 1st line | Refractory or relapsed within 12 months of 1st line | ||
| Study Design | |||||
| Primary endpointc | EFS | EFS after week 12 | EFS | ||
| Crossoverd | Off-study | Allowed | Allowed | ||
| Treatments | |||||
| CAR T Arm | |||||
| CAR T Product | Axi-Cel | Tisa-Cel | Liso-Cel | ||
| Vector | CD19/CD28 Gamma retrovirus | CD19/4-1BB Lentivirus | CD19/4-1BB Lentivirus | ||
| Cell selection | No | T-cell selection | CD4:CD8 in 1:1 ratio | ||
| CAR T cell dose | 2 × 106/kg | 0.6 – 6 × 108 Median 2.9 × 108 |
1 × 106/kg | ||
| Lymphodepletion | Flu 30 mg/m2 Cy 500 mg/m2 × 3days |
Flu 25 mg/m2 Cy 250 mg/m2 × 3days or Benda 90 mg/m2 × 2 days |
Flu 30 mg/m2 Cy 300 mg/m2 × 3 days |
||
| Bridging | Steroids only | Allowed | Allowed | ||
| Control Arm | |||||
| Salvage | 2nd line CIT | 2nd line CIT 3rd line allowed |
2nd line CIT | ||
Abbreviations: Benda: bendamustine; CY: cyclophosphamide; CIT: chemoimmunotherapy; FL: Follicular lymphoma; FLU: fludarabine; LBCL: Large B-cell lymphoma; PMBL: primary mediastinal B-cell lymphoma.
TRANSFORM was the only study that allowed inclusion of patients with secondary CNS involvement.
EFS definition was different among trials (see text for details).
Crossover was part of the design in 2 trials (BELINDA and TRANSFORM), but not in ZUMA-7 although patients could get commercial or other investigational CAR T cells.
Table 3.
Definitions of EFS on the three trials.
| Trial | EFS definition |
|---|---|
| ZUMA-7 | Time from randomization to the earliest date of disease progression according to the Lugano classification 53, the commencement of new therapy for lymphoma, death from any cause, or a best response of stable disease up to and including the response on the day 150 assessment after randomization) according to blinded central review. |
| BELINDA | Time from randomization to stable or progressive disease at or after the week 12 assessment by the independent review committee according to the Lugano criteria. |
| TRANSFORM | Time from randomization to death due to any cause, progressive disease, failure to achieve CR or PR by 9 weeks post randomization, or start of new antineoplastic therapy, whichever occurs first. |
In addition to differences in CAR T-cell products, the intensity of the lymphodepletion also varied, with tisa-cel being given after the lowest dose of lymphodepletion. Although some studies suggest that higher intensity lymphodepletion may lead to better T-cell expansion 54, 55, the doses of CAR T-cells and lymphodepletion used in the trials were the same as those used in the pivotal trials and currently used routinely in clinical practice in 3rd line. A significant difference in the control arm was the fact that patients treated on BELINDA and TRANSFORM were allowed to receive up to two lines of salvage CIT, while ZUMA-7 trials only allowed one line, and would have considered patients needing more than one line as having reached an endpoint. This approach in BELINDA and TRANSFORM was based on the fact that, in the CORAL study, 30% of patients who did not respond to the 1st line of salvage successfully proceeded to auto-HCT after 2nd line of salvage. Of 62 patients who went on to receive auto-HCT on BELINDA, 10 required a second line of salvage. On TRANSFORM, 12 patients switched salvage regimen, including 5 due to inadequate response.
Patient characteristics across the 3 trials are described in Table 4. Overall, the age of patients (median 58–60) and percent of patients over 65 (28–39%) were similar across the trials. All the trials had a slightly higher percent of patients with ECOG PS1 in the CAR T arm (43–48%) compared to the SOC arm (38–44%). There were more patients with stage III-IV disease on ZUMA-7 (79%) and TRANSFORM (71%) than BELINDA (64%). There was an imbalance in the distribution of IPI in the BELINDA study with 65.4% of patients on the tisa-cel arm having IPI ≥ 2 vs. 57.5% on the SOC arm, due to an error in stratification in the initial phase of the study 9. This may also explain a higher percentage of high-grade LBCL including rearrangement of MYC with BCL2 or BCL6 or both on that arm (20 vs. 12% for SOC). More patients on ZUMA-7 were classified as having GCB (58%). Fewer patients on BELINDA had primary refractory disease (62%) compared to ZUMA-7 (74%) and TRANSFORM (73%). Finally, while there was a difference in median OS on the SOC arm between ZUMA-7 (35.1 months) and TRANSFORM (16.4 months), the median follow-up is shorter for the latter and there are very few patients with a follow-up that far out. Therefore, longer follow up is needed to determine whether patients enrolled on the ZUMA-7 trial had a better prognosis than TRANSFORM.
Table 4.
Patient Characteristics.a
| TRIAL | ZUMA-7 | BELINDA | TRANSFORM | ||||
|---|---|---|---|---|---|---|---|
| Axi-Cel | SOC | Tisa-Cel | SOC | Liso-Cel | SOC | ||
| N=180 | N=179 | N=162 | N=160 | N=92 | N=92 | ||
| Age | |||||||
| Median (range – IQR for TRANSFORM) yr | 58 (21–80) | 60 (26–81) | 59.5 (19–79) | 58 (19–77) | 60 (54–68) | 58 (42–65) | |
| ≥ 65 yr – no. (%) | 51 (28) | 58 (32) | 54 (33) | 46 (29) | 36 (39) | 25 (27) | |
| Male – no. (%) | 110 (61) | 127 (71) | 103 (64) | 98 (61) | 44 (48) | 61 (66) | |
| ECOG PS 1 – no. (%) | 85 (47) | 79 (44) | 70 (43) | 65 (41) | 44 (48) | 35 (38) | |
| Disease stage – no. (%) | |||||||
| I or II | 41 (23) | 33 (18) | 55 (34) | 62 (39) | 24 (26) | 29 (31) | |
| III or IV | 139 (77) | 146 (82) | 107 (66) | 98 (61) | 68 (74) | 63 (68) | |
| Second-line aaIPI 2–3 – no. (%) | 82 (46) | 79 (44) | NR | NR | 36 (39) | 37 (40) | |
| Second-line IPI ≥ 2 – no. (%) | NR | NR | 106 (65) | 92 (58) | NR | NR | |
| Disease type – no. (%) | |||||||
| DLBCL | 126 (70) | 120 (67) | 101 (62) | 112 (70) | 53 (58) | 49 (53) | |
| High-grade BCL NOS | 0 | 1 (1) | 7 (4) | 8 (5) | - | - | |
| High-grade BCL including rearrangement of MYC with BCL2 or BCL6 or both | 31 (17) | 25 (14) | 32 (20) | 19 (12) | 22 (24) | 21 (23) | |
| Not confirmed or missing data | 18 (10) | 28 (16) | - | - | - | - | |
| Other | 5 (3) | 5 (3) | 22 (14) | 21 (13) | 17 (18) | 22 (24) | |
| Molecular subgroup – no. (%) | |||||||
| GCB | 109 (61) | 99 (55) | 46 (28) | 63 (39) | 45 (49) | 40 (43) | |
| ABC | 16 (9) | 9 (5) | 52 (32) | 42 (26) | 21 (23) | 29 (320 | |
| Unclassified | 17 (9) | 14 (8) | 3 (2) | 7 (4) | 25 (27) | 23 (25) | |
| N/A | 10 (6) | 16 (9) | NR | NR | N/A | N/A | |
| Missing data | 28 (16) | 41 (23) | NR | NR | 1 (1) | 0 | |
| Prognostic marker – no. (%) | |||||||
| Double or triple hit | 31 (17) | 25 (14) | NR | NR | 22 (24) | 21 (23) | |
| Double expressor | 57 (32) | 62 (35) | NR | NR | NR | NR | |
| MYC rearrangement | 15 (8) | 7 (4) | NR | NR | NR | NR | |
| Not applicable | 74 (41) | 70 (39) | NR | NR | NR | NR | |
| Missing data | 3 (2) | 15 (8) | NR | NR | NR | NR | |
| Response to 1st line therapy – no. (%) | |||||||
| Primary refractory | 133 (74) | 131 (73) | 107 (66) | 107 (67) | 67 (73) | 68 (74) | |
| Relapse ≤ 12 months | 47 (26) | 48 (27) | - | - | 25 (27) | 24 (26) | |
| Relapse < 6 months | NR | NR | 30 (19) | 32 (20) | NR | NR | |
| Relapse 6–12 months | NR | NR | 25 (15) | 21 (13) | NR | NR | |
| Bone marrow involvement – no. (%) | 17 (9) | 15 (8) | NR | NR | 9 (10) | 13 (14) | |
| Elevated LDH – no. (%) | 101 (56) | 94 (53) | NR | NR | 10 (11) | 11 (12) | |
Abbreviations: NR: not reported.
While there are certainly differences between ZUMA-7 and TRANSFORM, the most striking difference is with BELINDA. A number of factors may explain the negative outcome in the study. Clearly the definition of EFS and the fact that patients on the SOC arm could receive 2 different lines of CIT prior to auto-HCT, similar to TRANSFORM but different from ZUMA-7, makes it hard to compare results head-to-head and may account for at least some of the differences. Whether other factors such as the imbalance in high-risk patients (based on IPI or molecular markers) in the tisa-cel cohort, more GCB in ZUMA-7 and less primary-refractory disease in BELINDA also impact outcomes is harder to assess. One critical difference appears to have been the vein-to-vein time, which was longest for patients treated on the BELINDA trial, with a median of 52 days (IQR 43–61) from leukapheresis to infusion, and a marked difference between US (median 41 days; IQR 36–45) and non-US patients (median 57 days; IQR 50–63) 9. This was in contrast to a median time from randomization to infusion of 29 days (IQR 27–34) for axi-cel on ZUMA-7 and 34 days (IQR 31‒36) for liso-cel on TRANSFORM (median of 36 days (IQR 34–41) for leukapheresis to infusion) 8, 10. This is particularly relevant since 26% and 14% of patients had evidence of progressive disease on the PET scan at 6 weeks in the tisa-cel and SOC arms, respectively. The lower response rates (both ORR and CR) seen with tisa-cel compared to axi-cel and liso-cel were mostly consistent with results in the 3rd line setting. It is interesting to note that, in the case of tisa-cel, patients treated on BELINDA had lower ORR (46%) and CR (28%) compared to those treated on the original JULIET trial (ORR: 52%; CR: 40%). The lower CR rate, in particular, may be a significant factor contributing to the overall outcome of the trial. Rates of CRS and ICANS consistent with prior data for the three products, with 21% of patients treated with axi-cel experiencing grade 3 or more neurotoxicty compared to much lower rates with the other two CAR T-cell products. It should also be noted that the EFS in the control arms were similar in the three RCT, 2 to 3 months, roughly corresponding to first radiographic assessments after salvage treatment (i.e. median EFS achieved in these studies even before auto-HCT step, indicating known poor response rates to salvage treatments in high-risk patients). Finally, PFS, which may be a more useful measure of comparison given the differences in EFS definitions, was not provided for the BELINDA study.
In sum, while there may be patient selection bias and differences in the patients’ risk for early progression in the three RCT, the longer manufacturing time and lower response rates seen with tisa-cel (including lower than in JULIET), combined with some study design issues such as definition of EFS for the primary endpoint and allowing 2 salvage regimens prior to auto-HCT on BELINDA are likely key reasons that study did not show a benefit for CAR T-cells.
Q5. If the randomized studies in second line show a difference in PFS/EFS but not in OS, should that change practice?
The three prospective RCT used EFS as the primary endpoint, although with slightly different definitions of EFS as noted above 8–10. When it comes to the study design in general, should we always require OS benefit to be established first? Do the current results support change in practice, and if so, how broad should that change of practice be in second line?
When asking whether EFS is a valid endpoint in lymphoma studies, it is difficult to generalize across all randomized studies and, due to lack of direct evidence supporting this, the panel members did not have consensus on this issue. The second line treatment goal for relapsed LBCL is considered curative 12, but patients who are not cured now have a growing number of other treatment options in third line and beyond. These are likely to prolong survival and make demonstration of OS benefit challenging in a randomized trial, especially with a shorter follow-up. When these three RCT were designed, it was presumed that patients with treatment failure on the SOC arm would have access to CAR T-cells in third line as part of a crossover or standard of care. Ultimately, over 50% of patients on the SOC arm on these trials received cellular therapy 8–10. While EFS as defined by the ZUMA-7 and TRANSFORM protocols could have been affected by investigator’s bias (i.e. commencement of new therapy for lymphoma was considered an event), the EFS in SOC arms across all of the 3 trials was similar 8–10. This is, however, a reflection of the known poor ORR to salvage CIT in this patient population 12.
Considering factors like crossover and multiple subsequent treatment options for patients enrolled on these RCT, an OS benefit may never be shown, or may take a long time to demonstrate. It is unlikely that treatment related toxicity would negatively affect the OS in the CAR T-cell therapy arms with longer follow-up. The toxicity profile of CAR T-cells from third line and beyond has been well established and there is no reason to suspect that it would be significantly different in the second line setting 31, 56–58. While some remain hesitant to label CAR T-cell therapy as curative in LBCL, the long term CAR T-cell therapy data in patients in third line and beyond are consistent with curative potential of this modality with ongoing responses at 5 years from a single infusion of the cell product 56–58. The demonstrated EFS improvement with axi-cel and liso-cel in second line represents a portion of lymphoma patients who may not require additional lines of therapy. While OS would be the ideal endpoint and ultimately the OS benefit may emerge with longer follow-up, EFS appears to be a reasonable surrogate for cure when examining second line therapy for these patients with high-risk lymphoma. Axi-cel is now approved by the FDA and recommended in second line in the most recent NCCN guidelines.
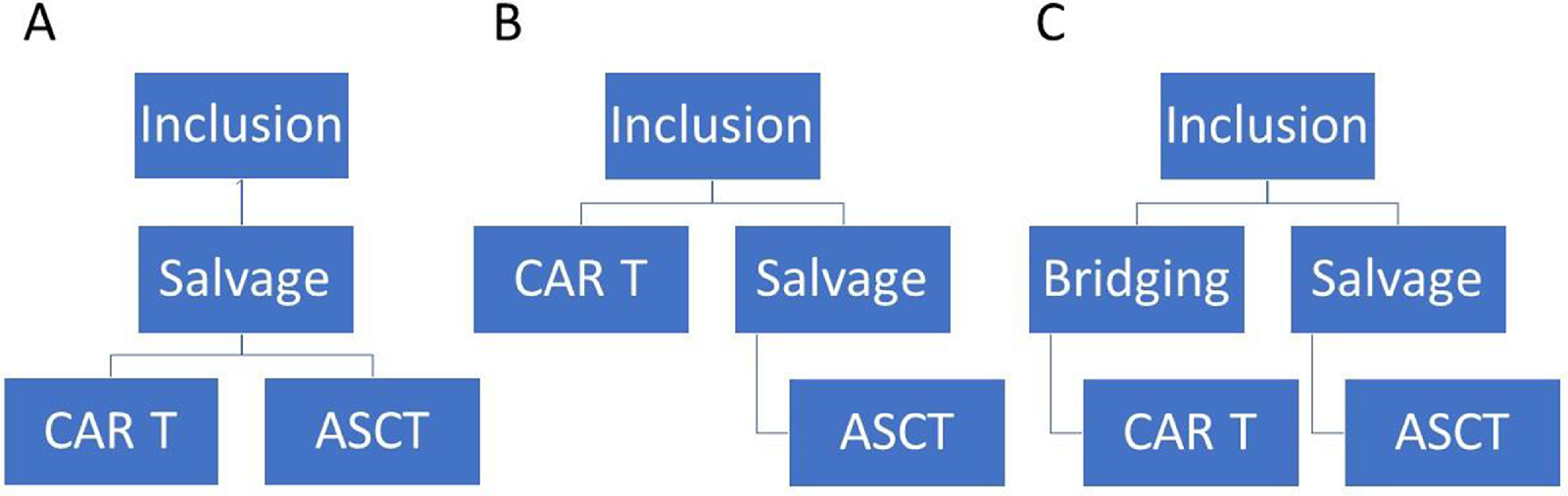
An important consideration in this context is the question that was being asked by the trials. Were the studies meant to compare CAR T to auto-HCT in second line, or more globally, CAR T to the “package” of salvage CIT and auto-HCT? We could consider three potential study designs that could have been used in second line RCT (Figure 1). One approach, which represents a true comparison between CAR T-cell therapy and auto-HCT, would randomize patients treated with 2–3 cycles of salvage CIT to receive either CAR T-cells or auto-HCT (Figure 1A). The second approach would randomize patients directly to either receive CAR T-cells or salvage CIT followed by auto-HCT in responding patients (Figure 1B). This is essentially the design of ZUMA-7 where the only bridging therapy allowed was corticosteroids. Finally, a third approach would allow patients on the CAR T-cell arm to get CIT considered as bridging therapy rather than salvage, meaning that lack of response would not preclude getting CAR T-cells, while it would be considered an event on the SOC arm (Figure 1C). This design was used in TRANSFORM and BELINDA. In practical terms, the use of EFS as an endpoint meant that patients on the SOC arm with disease that progressed while receiving salvage therapy were considered to have had an event. Essentially, another way to consider the two arms is a comparison between CAR T cell therapy and salvage CIT. While we can debate the merits of the various study designs and advocate for one as the optimal design, ultimately the regulatory authorities will examine the studies that were performed and the results of those studies. As noted, the FDA has approved axi-cel in second line therapy based on the inclusion criteria of ZUMA-7. A key question that will remain is how broadly we can apply the results of the trials to clinical practice, and what the role will be of auto-HCT in this setting. Considerations of short and long-term toxicity, cost-effectiveness, and access to care will also need to be addressed. These issues are discussed in more details in the following sections.
Figure 1.
Potential study designs (see text for details).
Q6. Do these trials justify a one size fits all approach in second line treatment of early relapsing aggressive B cell lymphomas?
After the anticipated FDA approval of some of the CD19 directed CAR constructs in second line treatment of aggressive B-cell lymphomas, the clinicians will soon have to apply these results into actual clinical practice. Transplant and cell therapy physicians will need to assess if findings of these trials justify a one size fits all approach for all aggressive B cell lymphomas with early treatment failure, especially considering the discordant primary endpoint results of these trials (EFS benefit in two out of three RCT), and the lack of a clear OS benefit to date. We caution against extrapolating these trial findings to aggressive B cell lymphoma with late treatment failure (> 1 year after diagnosis). In addition, as mentioned above in FAQ 1, patients relapsing 6–12 months after finishing frontline treatments do not meet CORAL study’s definition of early treatment failure, but are in line with the definition used in the NCIC-CTG LY.12 study and were included in the three RCT comparing CAR to SOC. Careful consideration of CAR vs. SOC treatment in such patients would be warranted. Furthermore, a majority of patients treated on all three trials had primary refractory disease, and no separate analysis was provided in the relatively smaller subset of patients who relapsed 6–12 months. As a result, the trials likely do not fully address this issue. As discussed in FAQ 8 below, patients who arrive at transplant or cell therapy programs after already demonstrating a response (CR or PR) to salvage CIT may do well with auto-HCT consolidation and, for appropriate candidates, this option must be discussed with the patients.
There are many high-risk clinical scenarios where results of these three very important randomized trials immediately impact practice. Patients with primary refractory disease (progression or stable disease as best response to frontline treatments) 38, those with so-called DHL or THL 41, and high-risk disease by REFINE study criteria (at least 2 out of following risk factors: 1. primary progression on frontline treatments, 2. presence of myc rearrangement, 3. high IPI score at relapse) 59, even after achieving response to salvage treatment have low rates of durable disease control with auto-HCT, and could be ideal candidates for CAR T-cells in second line treatment. In addition, many early relapsing patients due to advanced age and comorbid conditions may not be auto-HCT candidates regardless of other biological risk factors. CAR T-cell therapy can be feasible in patients not considered optimal candidates for high dose therapy and represents another clinical scenario where results of these randomized trials will be key in refining clinical practice.
Q7. If patients get CAR T-cells in second line, what should be the SOC in third line, auto-HCT vs allo-HCT vs other?
Although patients had longer EFS with both axci-cel and liso-cel CAR T-cell therapy compared to salvage CIT +/− auto-HCT as second line therapy for early relapse/progression of DLBCL 8, 10, only about 40% remained in remission at 2 years following second line axi-cel in ZUMA-7 8, which means that the majority of patients relapse after second line CAR T and may benefit from further salvage or consolidation therapy 60. However, moving CAR T-cell therapy to second line may pose a challenge for those that don’t achieve a CR or long-lasting remission, as there is limited data on what to do after CAR T-cell failure 61–64. Currently available standard of care salvage therapy options include polatuzumab vedotin (with bendamustine/ rituximab), tafasitamab (with lenalidomide), selinexor, loncastuximab tesirine, chemotherapy, auto-HCT, and allogeneic (allo)-HCT 65. However, some of these options are only available for patients that have not experienced CD19 antigen loss at relapse, including tafasitamab (anti-CD19 monoclonal antibody) and loncastuximab (anti-CD19 antibody drug conjugate) 66, 67. In addition, some of the most encouraging data comes from clinical trials of bispecific antibodies and CAR T-cells recognizing alternative targets (e.g. CD22 or CD20) or dual targets, so clinical trial enrollment is encouraged until these become available 68–73. However, it is unclear if targeted therapies will lead to long term remissions in patients that have already failed CD19 CAR T-cell therapy, and post-CAR cytopenias may be prohibitive in some patients 35, 36. As a result, consolidation or salvage with either auto-HCT or allo-HCT may be considered in eligible patients that relapse or do not achieve a CR after CD19 CAR T-cells, especially if clinical trials of novel therapies are not available 74–76. In some patients with prolonged cytopenias post CAR T cells, it is unlikely that stem cell mobilization for auto-HCT will be feasible.
In an effort to better understand outcomes after CAR T-cell failure, the US Lymphoma CAR T-Cell Consortium reported retrospective data on subsequent therapies in 136 patients following relapse after axi-cel 77. Thirty-six % had progressed after CR, 31% after PR, 28% were primary refractory to axi-cel, and 30% were CD19 negative at relapse. Median OS was only 6 months from the time of progression, and only 8 of 136 (6%) ultimately went on to salvage allo-HCT, 3 of whom remained in CR. Although checkpoint inhibitor-based therapy initially seemed most effective (46% responders among 28 patients), only 18% achieved a CR with median PFS only 88 days and OS 11 months. Responses were even less durable for lenalidomide, radiation, and chemotherapy (median 48–58 days). The Fred Hutchinson Cancer Research Center (FHCRC) also reported poor outcomes for 61 patients after post-CAR T-cell relapse with median OS 3.8 months for early relapse (within 30 days) and 9.3 months for later relapses after 30 days 78. Only 5 (8%) went on to receive allo-HCT with two of those in ongoing remission over a year out.
Another strategy has been re-infusion of CAR T-cells to improve suboptimal response. In the ZUMA-1 trial, axi-cel re-infusion was allowed for those who achieved at least a PR for at least 3 months, and out of 13 patients re-infused, 7 (53%) achieved a response (CR in 4 and PR in 3), but the median duration of response was only 81 days 79. The FHCRC also published data on re-infusion of CD19 CAR T-cells in 21 patients with NHL, and despite a dose increase in 82% of patients, CR was only achieved in 19% with median duration of response 6 months 80.
Allo-HCT may ultimately be necessary in many patients that relapse after CAR T-cell therapy, as allo-HCT is the only therapy proven to be curative following failure of auto-HCT (other than CAR T-cells), and failure after second line auto-HCT may be considered the closest surrogate to failure after second line CAR T-cells until we have further data in this population. In the CIBMTR analysis of 503 DLBCL patients relapsing after auto-HCT and receiving a subsequent allo-HCT, 31% were able to achieve long term PFS at 3 years 81. Reduced intensity conditioning was not inferior to myeloablative but less toxic, and inferior outcomes were noted in chemo-refractory disease. Prediction algorithms may also be used to identify patients at high risk for failure of allo-HCT 82. It is noted that combinations of novel agents and salvage therapy followed by allo-HCT may also be utilized for those in relapse to try to increase the odds of achieving a durable remission. However, there is currently no consensus on the role of and timing of allo-HCT in relapsed/refractory lymphoma. Because of poor outcomes at progression after CAR T-cells, one logical next step may be consolidation with auto or allo-HCT in those not achieving CR at 3 months; nevertheless, it is noted that about 22% of patients with stable disease at 3 months will later improve and go on to experience durable responses without further intervention after axi-cel according to Zuma-1, so carefully designed trials are necessary to evaluate the impact of an intervention for these patients 83, 84.
Q8. If CAR T cells are the SOC in second line, how will we approach patients referred to the transplant/cell therapy center having already started salvage and having established a CR or PR?
Historically, presence of chemo-responsive disease to salvage therapy, as defined by either CR or PR assessed by radiographic imaging, has been the key factor in determining a patient with relapsed DLBCL’s eligibility for auto-HCT. In addition, the depth of metabolic response to salvage treatments on PET (or PET/CT) imaging is a known prognostic predictor of auto-HCT outcomes in aggressive B-cell lymphoma. Sauter et al. retrospectively analyzed auto-HCT outcomes of relapsed/refractory DLBCL relative to the depth of metabolic response to salvage therapy using the Deauville 5-point scale 20. At 3 years, patients achieving a Deauville response of 1 to 3 (or complete metabolic response; N=81) to salvage experienced superior PFS and OS rates of 77% and 86%, respectively, compared with patients achieving Deauville 4 (partial metabolic response; N=48) (49% and 54%, respectively) (P < .001). These observations were subsequently validated by a large retrospective CIBMTR analysis that only included relapsed/refractory DLBCL achieving a PET positive PR prior to auto-HCT (N=240) and demonstrated an adjusted 5 year PFS 41% for both patients with or without early failure of rituximab-based frontline CIT 21. Collectively both these studies show that high-dose therapy and auto-HCT consolidation is curative for approximately 45% of patients with DLBCL despite achieving only a PR after salvage therapy.
To our knowledge, no alternative consolidation strategy (including CAR T-cell therapy) has been proven superior to auto-HCT consolidation in DLBCL with demonstrable chemo-responsive disease after salvage attempts. A recent CIBMTR analysis compared auto-HCT (N=266) vs. CAR T-cell (N=145) consolidation in relapsed/refractory DLBCL achieving a PR as best response after salvage 22. In the univariable analysis, the 2-year PFS (52% vs. 42%; p=0.1) and the rate of 100-day non-relapse mortality (4% vs. 2%; p=0.3) were not different between the 2 cohorts but consolidation with auto-HCT was associated with a lower rate of relapse/progression (40% vs. 53%; p=0.05) and a superior OS (69% vs. 47% ; p=0.004) at 2-years. In the multivariable regression analysis, treatment with auto-HCT was associated with a significantly lower risk of relapse/progression rate (HR=1.49; p=0.01) and a superior OS (HR=1.63; p=0.008). In addition, subgroup analyses of patients with early failure of frontline treatments, those with > 2 prior treatment lines and a propensity score matching were in line with overall analysis. As noted above, one needs to consider the potential biases and limitations of registry analyses. Nevertheless, there are examples in which registry data has subsequently been confirmed in prospective randomized trials, such as the use of bone marrow vs. peripheral blood stem cells in myeloablative allogeneic stem cell transplantation 85, 86.
Recently results of three RCT in the subset of patients with aggressive LBCL with early treatment failure, comparing salvage therapy followed by auto-HCT consolidation in responding patients versus proceeding directly with CAR-T treatment were reported 8–10. However, since these trials randomized patients before salvage treatments were initiated, they were not designed to address the management of patients with DLBCL already achieving either a CR or PR in response to salvage therapies. This question nonetheless remains highly relevant in clinical practice, as patients with relapsed DLBCL are often referred to transplant or cell therapy programs after starting salvage therapy or may receive such treatments as a bridge. With CAR T-cell treatment approval as second line therapy, caution must be exercised in routinely offering this treatment to otherwise fit patients, with established chemo-responsive disease after salvage because: (a) auto-HCT is curative for up to half of such patients with generally low risk of NRM, (b) no data have established superiority of CAR T-cell therapy over auto-HCT in this setting, (c) and cost considerations that may favor auto-HCT in this setting 87. A potential counterargument is the fact that there is emerging data that patients who receive CAR T-cells in CR have excellent outcomes 88. While these data require validation in larger series and longer follow-up, it does suggest that we may one day consider CAR T-cells as an option in patients with chemosensitive disease after salvage CIT. Finally, another potential consideration is the fact that, while CAR T-cell therapy can salvage patients relapsing after auto-HCT, the reverse sequence is often not feasible. It should be noted that over 50% of patients in the SOC on all three RCTs ended up crossing over, including some after auto-HCT. If the preliminary evidence of a potential OS benefit for CAR T-cells is confirmed with longer follow-up, the lower overall treatment burden of proceeding to CAR T-cells in second line, with associated emerging data on a QOL benefit as well as cost-effectiveness, decisions about switching to “CAR T-cells” in a patient who has already received a 1st cycle of salvage CIT will have to be looked at carefully.
Q9. How should we interpret results from studies in the second line setting when frontline therapies change?
Several randomized trials are comparing new combinations to the established frontline standards like R-CHOP or R-EPOCH. While most of these trials failed to show clear benefit, a recently presented randomized study showed a modest improvement in PFS in patients with high risk DLBCL for polatuzumab+R-CHP over R-CHOP 89. It remains unclear if polatuzumab+R-CHP will become a new standard for these patients, but we can debate how the change in the frontline standard could impact interpretation of the CAR T-cell studies from the second line. In this particular case, it is unlikely that patients with disease refractory or progressing after polatuzumab containing frontline treatment will have different outcomes with CAR T-cells compared to those who failed traditional therapies like R-CHOP. Considering the modest improvement in PFS in frontline, there may be less patients requiring second line treatment. However, polatuzumab is a form of targeted chemotherapy and it is unlikely that using polatuzumab+R-CHP in frontline would diminish the EFS benefit of anti-CD19 CAR T-cells, which use a different target and have a unique mechanism of cell killing. On the other hand, should CD19-targeting agents become a component of new standards for frontline treatment of LBCL in the future, there could be a potential impact on the efficacy of the anti-CD19 CAR T-cell therapies in the second line and additional data may need to be obtained.
Q10. Since Auto-HCT is currently available in more centers than CAR T cells, will that affect access to care if the SOC changes?
With CD19 CAR T-cells approved as a second line therapy, questions remain regarding the broader access to such complex and costly therapy. Nearly 94% of Americans live within three hours driving distance of an Auto-HCT center 90, but there are less than half as many centers approved to administer CAR T-cells as auto-HCT, making auto-HCT more accessible compared to CAR T-cell therapy. In addition, data on the use of CAR T-cells reported to the CIBMTR Cellular Immunotherapy Data Resource (CIDR) identifies health care disparities, with African American patients representing 5.6% of patients treated between 2016 and 2021 91, 92. In a recent analysis of demographics of patients treated on 7 pivotal CART clinical trials, African Americans constituted only 3% of the enrolled patients 93.
CAR T-cell manufacturing capacity is a limiting factor for wider access as well. One of the main bottlenecks to scaling up CAR T-cell manufacturing is that demand for GMP-grade lentivirus has far outstripped supply 94. In fact, some cell therapy companies have acquired their own lentiviral manufacturing facilities. Bioprocessing protocols are being improved to address global GMP-grade lentiviral shortages, but clinicians should consider manufacturing timelines for available products at their center when choosing between auto-HCT and CAR T-cell therapy. Out-of-specification (OOS) CAR T-cell products are another potential concern in manufacturing logistics. Interestingly, auto-HCT serves as a historical example of OOS product efficacy. Hematopoietic stem cell products are frequently OOS (due to low CD34+ cells numbers or viability, bacterial contamination, etc.), but nonconforming products are often infused into patients due to dire clinical scenarios 95–97. As in auto-HCT, it is likely that the benefits outweigh the risks of OOS CAR T-cell products. One retrospective analysis showed that tisa-cel products that were OOS due to low T cell viability (<80% per release specifications) did not show statistically significant decreases in CR, PFS, or OS or increased safety risks compared to in-specification CAR T-cells 98, 99 The decision to administer OOS CAR T-cells is ultimately up to the patient’s physician, and multiple factors regarding product efficacy, safety, and logistical feasibility must be considered.
Finally, the regulatory processes and accreditation by FACT will need to be streamlined to facilitate the widespread adoption of CAR T-cell as a second-line therapy. FACT has established standards for immune effector cells (IEC), including CAR T-cells, that outline the infrastructure needed to enable the safe administration of CAR T-cells and regulate post-treatment monitoring and reporting of patient outcomes 100. Sites administering CAR T-cells must be prepared to handle manufacturing and administration logistics that are more complicated than those of standard chemotherapy. CAR T-cells have a specific toxicity profile (including most notably CRS and ICANS) that requires resources from a wide array of healthcare teams and, as such, requires a robust clinical infrastructure 30, 100–102. IEC accreditation is separate from hematopoietic progenitor cell (HPC) transplantation accreditation, allowing the creation of stand-alone CAR T-cell programs or expanding upon existing HPC programs to include IEC therapies. However, there is significant overlap between IEC and HPC guidelines and infrastructure; therefore, centers that are accredited for auto-HCT may have much of the required infrastructure in place for CAR T-cells as well.
Q11. Is the benefit-cost ratio better for CAR T cells or ASCT in second line therapy of DLBCL? What are potential economic considerations of shifting CAR T to 2nd line?
The costs of CAR T-cell therapy can be separated into two distinct categories, cost of the product itself and cost of the care delivery. In the standard pricing model for DLBCL, the product cost is fixed, though in the ALL indication, an outcomes-based pricing model is used for tisa-cel in which there is no charge for the CAR T-cell if there is not an adequate response to therapy. While companies have not followed this paradigm to date for the lymphoma indications, there would be a significant impact to the total therapy cost if they did 103. The referenced model in the 3rd line setting also showed the variability in the second component based on the rates of CRS and ICANS, and thereby need for ICU admission and overall length of stay. In more direct observation, two US centers have presented data on resource utilization in the first months after CAR T-cell therapy 104, 105. In addition, a micro-costing study of the TRANSCEND trial showed increased cost with higher grade CRS 106. Models also show the differential in costs when patients are treated in the inpatient versus outpatient setting 107. Rates of CRS and ICANS in the 2nd line setting were not radically different from the clinical trials in the 3rd line setting or real-world analyses. Therefore, it is likely that the impact on resource utilization and the cost of care delivery in the 2nd line setting from the time of lymphodepletion will be similar and by extension products with lower toxicity profiles, which can be given in the outpatient setting, will have a less overall economic burden on the health care system 108, 109.
Additional economic considerations in the second line setting include access to care, reimbursement, duration and type of bridging therapy, and cost of long-term complications. Currently, approximately 100 centers in the US provide CAR T-cell therapy. In the current model of salvage therapy followed by high dose therapy and auto-HCT for patients with chemosensitive disease, a non-CAR T-cell provider can start the salvage therapy as soon as deemed clinically necessary. With CAR T-cell therapy, the timing of referral to a CAR T-cell provider, insurance approval, and leukapheresis have to be accounted for 110. Insurance approval particularly for commercial payers can take several weeks and without improvements in those workflows, patients will either have to have therapy prior to leukapheresis (and such costs would need to be included in the costs of care) or will have delays in care overall, which can impact the clinical benefit. Furthermore, insurance and reimbursement models are country-specific, and each would have to evaluate the feasibility. Vein-to-vein time (i.e. leukapheresis to infusion) will also determine the need for bridging therapy, which is potentially of higher cost with newer more targeted agents vs. lower cost platinum-based salvage. In the US, payer mix and reimbursement may also impact centers’ ability to expand capacity and volume of products infused. Frequently changing coding options add to the complexity (https://www.astct.org/practice/practice-resources/car-t-therapy-reimbursement-resources). Finally, a portion of patients have prolonged cytopenias and B cell aplasia, potentially requiring IVIG use 35, 111, 112. In an earlier setting, the total impact of more patients requiring frequent monitoring and higher cost therapy would have to be included in accounting.
In the 3rd line setting, the Institute for Clinical and Economic Review model based on early clinical trial data in 2018 found an incremental cost effectiveness ratio (ICER) of $136 000/QALY gained for axi-cel compared to chemotherapy for DLBCL (Tice 2018 https://icer-review.org/wp-content/uploads/2017/07/ICER_CAR_T_Final_Evidence_Report_032318.pdf). Thresholds of $100,000 – $150,000 per quality adjusted life year have been suggested as the cutoff for cost-effectiveness in the US 113. A further markov model including both axi-cel and tisa-cel showed variations in the ICER based on the assumed 5-year PFS for each product 87. In the optimistic scenarios (40% PFS for axi-cel and 35% PFS for tisa-cel), the ICERs were $129,000/QALY and $168,000/QALY, respectively. Importantly, they concluded that the use of CAR T-cells for all eligible patients would add $10 billion to US health care costs, but that both products could meet $150,000/QALY thresholds depending on true efficacy. Models in the second line setting presented at the 2022 Tandem meeting reported ICERs ranging from $87,026/QALY 114 to $2,687,607/QALY 115, and therefore additional analyses of each model’s assumptions is needed. It should be noted that the latter study uses data from CAR T-cells in 3rd line for modeling purposes, while the study by Perales et al 114 uses data directly from ZUMA-7 with assumptions based on long-term follow-up and a potential significant OS benefit. In addition, a significant effect is due to the fact that 56% of patients on the SOC arm crossed over, similar to what was observed in the other 2 RCT, resulting in the fact many of the patients alive long-term in that arm also ended up receiving CAR T-cells.
Value based care involves evaluation of the patient experience, cost, and processes in addition to the clinical benefit. The recent studies have shown the increased PFS for specific patients with DLBCL who had primary refractory disease or progressed within one year of treatment when compared to standard salvage therapy. Other measures of benefit including PRO and quality of life remain under investigation and have been reported in abstract form 23, 24. The overall cost-benefit ratio is likely to be in an acceptable range in certain situations, but further research is needed to confirm the patients most likely to benefit taking into consideration country-specific resource limitations.
Q12. Which components should be included in the next series of landmark CAR T-cell trials for frontline?
The results of the three RCT comparing second generation autologous CAR T-cells with SOC have led to approval of CAR T-cells in second line, with the caveats discussed above 8–10. Large RCT are typically very expensive and take significant time to complete. As a result, while these trials were in the design and testing phase, a number of new CAR T-cells directed against CD19 and other targets have reached clinical trials. It is easy to argue that the single arm phase 2 pivotal trials were more impactful than the phase 3 RCTs because they provided timely data that was helpful to design safety protocols, reliable enough to secure regulatory approvals in record time, and provided early efficacy outcomes that convinced investigators and companies to develop new CAR T-cell products. There is now a growing list of 3rd generation autologous CARs, armored CARs, CARs with suicide switch, and several allogeneic or off-the-shelf products with and without HLA restriction that are also being developed 116–119. These newer and other future products need to be tested not only to understand where they fit in the growing list of cellular therapy products in the treatment of relapsed lymphoma, but they also need to be tested in the upfront setting. It is not realistic to expect all of these to be put to RCT as was done for ZUMA-7, BELINDA and TRANSFORM for reasons stated above.
The following are some important considerations for future clinical trials of CAR constructs in the treatment of lymphoma.
Carefully conducted single arm phase 2 trials in the upfront setting that select for patients who are intolerant of full dose upfront anthracycline based chemotherapy (e.g. due to comorbidities or age), or those at higher risk of treatment failure and relapse (e.g. high IPI, adverse risk cytogenetics) will be helpful. Outcomes of interest, which are reliable and report earlier such as ORR, are of most interest. Other outcomes like PFS, which also report relatively early, may be preferable compared to OS.
Propensity score matching and matching-adjusted indirect comparison (MAIC) can be utilized in the post-hoc setting to compare different trials. Though the information obtained is dependent on how well the matching is done, it is an inexpensive way to get comparisons done.
PRO and quality of life surveys are important to capture during the clinical trial process as they can provide valuable input from the patient’s perspective.
In summary, with the emergence of several broad categories of CAR T-cells for lymphoma, single arm phase 2 trials that incorporate outcomes that report relatively early are far more important as the information will be helpful to rapidly evaluate the myriad of products and find where they fit. The RCT design, because of the expense and longer time to report, may be of more relevance to determine between rather than within categories of the various types of CAR T cells.
Highlights.
CD19 CAR T-cells have been shown to be highly effective in relapsed/refractory LBCL
Three randomized phase 3 trials were conducted in 2nd line by comparing CAR T-cells to the current SOC
Two trials showed improved EFS in patients treated in 2nd line with CAR T-cells
We review the 3 trials and share perspectives on implications for clinical practice
Footnotes
Disclosures
Miguel-Angel Perales reports honoraria from Abbvie, Astellas, Bristol-Myers Squibb, Celgene, Equilium, Incyte, Karyopharm, Kite/Gilead, Merck, Miltenyi Biotec, MorphoSys, Novartis, Nektar Therapeutics, Omeros, OrcaBio, Takeda, VectivBio AG, and Vor Biopharma. He serves on DSMBs for Cidara Therapeutics, Medigene, Sellas Life Sciences, and Servier, and the scientific advisory board of NexImmune. He has ownership interests in NexImmune and Omeros. He has received research support for clinical trials from Incyte, Kite/Gilead, Miltenyi Biotec, and Novartis.
Larry D. Anderson, Jr reports honoraria from consulting and scientific advisory board activity from BMS, Celgene, Janssen, Amgen, GSK, Oncopeptides, Karyopharm, and AbbVie. He serves on a DSMB for Prothena and has received institutional research funding for clinical trials from BMS, Celgene, Janssen, GSK, and AbbVie.
Tania Jain reports Institutional research support from CTI Biopharma, Syneos Health, Incyte; Consultancy with Targeted Healthcare Communications; Advisory board participation with Care Dx, Bristol Myers Squibb, Incyte, and CTI.
Saad Kenderian: Patents and Royalties: Novartis, Humanigen, MustangBio, Mettafogre; Research Funding: Novartis, Gilead/Kite, BMS/Juno, Humanigen, Morphosys, Lentigen, Tolero, Viracta/Sunesis, Leahlabs; DSMB: Humanigen; Consultancy: Novartis, Torque, Calibr; SAB: Kite, Juno, Novartis, Humanigen.
Olalekan Oluwole reports consultancy and scientific advisory board for: Pfizer, Kite, Gilead, AbbVie, Janssen, TGR therapeutics, Novartis and curio science. Institution funding from Kite, Pfizer, Daichi Sankyo. Honoraria from Pfizerand Gilead.
Gunjan Shah reports research support for clinical trials to the institution from Janssen, Amgen, and Beyond Spring.
Jakub Svoboda reports institutional research support from AstraZeneca, Merck, Incyte, Bristol-Myers Squibb, Pharmacyclics, TG Therapeutics, Seattle Genetics, Adaptive and consultancy fees/honoraria from ATARA, AstraZeneca, Adaptive, Seattle Genetics, Bristol-Myers Squibb, Incyte.
Mehdi Hamadani reports: Research Support/Funding: Takeda Pharmaceutical; ADC Therapeutics; Spectrum Pharmaceuticals; Astellas Pharma. Consultancy: Incyte Corporation; ADC Therapeutics; Omeros, Verastem, MorphoSys, Kite, Genmab, SeaGen, Gamida Cell, Novartis, Legend Biotech. Speaker’s Bureau: Sanofi Genzyme, AstraZeneca, BeiGene, ADC Therapeutics. DMC: Myeloid Therapeutics, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. 2019;380:45–56. [DOI] [PubMed] [Google Scholar]
- 3.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–852. [DOI] [PubMed] [Google Scholar]
- 4.Jain T, Bar M, Kansagra AJ, et al. Use of Chimeric Antigen Receptor T Cell Therapy in Clinical Practice for Relapsed/Refractory Aggressive B Cell Non-Hodgkin Lymphoma: An Expert Panel Opinion from the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant. 2019;25:2305–2321. [DOI] [PubMed] [Google Scholar]
- 5.Bachanova V, Perales MA, Abramson JS. Modern management of relapsed and refractory aggressive B-cell lymphoma: A perspective on the current treatment landscape and patient selection for CAR T-cell therapy. Blood Rev. 2019:100640. [DOI] [PubMed] [Google Scholar]
- 6.Mohty M, Gautier J, Malard F, et al. CD19 chimeric antigen receptor-T cells in B-cell leukemia and lymphoma: current status and perspectives. Leukemia. 2019;33:2767–2778. [DOI] [PubMed] [Google Scholar]
- 7.Mohty M, Dulery R, Gauthier J, et al. CAR T-cell therapy for the management of refractory/relapsed high-grade B-cell lymphoma: a practical overview. Bone Marrow Transplant. 2020;55:1525–1532. [DOI] [PubMed] [Google Scholar]
- 8.Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N Engl J Med. 2022;386:640–654. [DOI] [PubMed] [Google Scholar]
- 9.Bishop MR, Dickinson M, Purtill D, et al. Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. N Engl J Med. 2022;386:629–639. [DOI] [PubMed] [Google Scholar]
- 10.Kamdar M, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. The Lancet. 2022;399:2294–2308. [DOI] [PubMed] [Google Scholar]
- 11.Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333:1540–1545. [DOI] [PubMed] [Google Scholar]
- 12.Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gisselbrecht C, Schmitz N, Mounier N, et al. Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30:4462–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32:3490–3496. [DOI] [PubMed] [Google Scholar]
- 15.Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemoimmunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2014;20:1729–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armand P, Welch S, Kim HT, et al. Prognostic factors for patients with diffuse large B cell lymphoma and transformed indolent lymphoma undergoing autologous stem cell transplantation in the positron emission tomography era. Br J Haematol. 2013;160:608–617. [DOI] [PubMed] [Google Scholar]
- 17.Bal S, Costa LJ, Sauter C, Litovich C, Hamadani M. Outcomes of Autologous Hematopoietic Cell Transplantation in Diffuse Large B Cell Lymphoma Refractory to Firstline Chemoimmunotherapy. Transplant Cell Ther. 2021;27:55 e51–55 e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagadeesh D, Majhail NS, He Y, et al. Outcomes of rituximab-BEAM versus BEAM conditioning regimen in patients with diffuse large B cell lymphoma undergoing autologous transplantation. Cancer. 2020;126:2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vose JM, Carter S, Burns LJ, et al. Phase III randomized study of rituximab/carmustine, etoposide, cytarabine, and melphalan (BEAM) compared with iodine-131 tositumomab/BEAM with autologous hematopoietic cell transplantation for relapsed diffuse large B-cell lymphoma: results from the BMT CTN 0401 trial. J Clin Oncol. 2013;31:1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sauter CS, Matasar MJ, Meikle J, et al. Prognostic value of FDG-PET prior to autologous stem cell transplantation for relapsed and refractory diffuse large B-cell lymphoma. Blood. 2015;125:2579–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah NN, Ahn KW, Litovich C, et al. Is autologous transplant in relapsed DLBCL patients achieving only a PET+ PR appropriate in the CAR T-cell era? Blood. 2021;137:1416–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shadman M, Pasquini M, Ahn KW, et al. Autologous transplant vs chimeric antigen receptor T-cell therapy for relapsed DLBCL in partial remission. Blood. 2022;139:1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elsawy M, Chavez JC, Avivi I, et al. Patient-Reported Outcomes in a Phase 3, Randomized, Open-Label Study Evaluating the Efficacy of Axicabtagene Ciloleucel (Axi-Cel) Versus Standard of Care Therapy in Patients with Relapsed/Refractory Large B-Cell Lymphoma (ZUMA-7). Blood. 2021;138:430–430. [Google Scholar]
- 24.Abramson JS, Solomon SR, Arnason JE, et al. Improved Quality of Life (QOL) with Lisocabtagene Maraleucel (liso-cel), a CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy, Compared with Standard of Care (SOC) As Second-Line (2L) Treatment in Patients (Pts) with Relapsed or Refractory (R/R) Large B-Cell Lymphoma (LBCL): Results from the Phase 3 Transform Study. Blood. 2021;138:3845–3845. [Google Scholar]
- 25.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625–638. [DOI] [PubMed] [Google Scholar]
- 27.Pennisi M, Jain T, Santomasso BD, et al. Comparing CAR T-cell toxicity grading systems: application of the ASTCT grading system and implications for management. Blood Adv. 2020;4:676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santomasso BD, Park JH, Salloum D, et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer discovery. 2018;8:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022;22:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanez L, Alarcon A, Sanchez-Escamilla M, Perales MA. How I treat adverse effects of CAR-T cell therapy. ESMO Open. 2020;4:e000746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson CA, Locke FL, Ma L, et al. Real-world Evidence of Axicabtagene Ciloleucel for the Treatment of Large B-Cell Lymphoma in the United States. Transplant Cell Ther. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chakraborty R, Hill BT, Majeed A, Majhail NS. Late Effects after Chimeric Antigen Receptor T cell Therapy for Lymphoid Malignancies. Transplant Cell Ther. 2021;27:222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cordeiro A, Bezerra ED, Hirayama AV, et al. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol Blood Marrow Transplant. 2020;26:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wudhikarn K, Pennisi M, Garcia-Recio M, et al. DLBCL patients treated with CD19 CAR T cells experience a high burden of organ toxicities but low nonrelapse mortality. Blood Adv. 2020;4:3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain T, Knezevic A, Pennisi M, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood Adv. 2020;4:3776–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rejeski K, Perez A, Sesques P, et al. CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood. 2021;138:2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meir J, Abid MA, Abid MB. State of the CAR-T: Risk of Infections with Chimeric Antigen Receptor T-Cell Therapy and Determinants of SARS-CoV-2 Vaccine Responses. Transplant Cell Ther. 2021;27:973–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. [DOI] [PubMed] [Google Scholar]
- 40.Cuccuini W, Briere J, Mounier N, et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood. 2012;119:4619–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrera AF, Mei M, Low L, et al. Relapsed or Refractory Double-Expressor and Double-Hit Lymphomas Have Inferior Progression-Free Survival After Autologous Stem-Cell Transplantation. J Clin Oncol. 2017;35:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Recio M, Wudhikarn K, Pennisi M, et al. The International Prognostic Index Is Associated with Outcomes in Diffuse Large B Cell Lymphoma after Chimeric Antigen Receptor T Cell Therapy. Transplant Cell Ther. 2021;27:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho C, Svoboda J. Predicting Outcomes following Anti-CD19 CAR T Cell Therapy in Aggressive B Cell Lymphomas. Transplant Cell Ther. 2021;27:195–196. [DOI] [PubMed] [Google Scholar]
- 44.Kittai AS, Huang Y, Gordon M, et al. Comorbidities Predict Inferior Survival in Patients Receiving Chimeric Antigen Receptor T Cell Therapy for Diffuse Large B Cell Lymphoma: A Multicenter Analysis. Transplant Cell Ther. 2021;27:46–52. [DOI] [PubMed] [Google Scholar]
- 45.Pennisi M, Sanchez-Escamilla M, Flynn JR, et al. Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood Adv. 2021;5:3397–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shouval R, Alarcon Tomas A, Fein JA, et al. Impact of TP53 Genomic Alterations in Large B-Cell Lymphoma Treated With CD19-Chimeric Antigen Receptor T-Cell Therapy. J Clin Oncol. 2022;40:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith M, Dai A, Ghilardi G, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paczesny S, Pasquini MC, Pavletic SZ, et al. Blueprint for the discovery of biomarkers of toxicity and efficacy for CAR T cells and T-cell engagers. Blood Adv. 2021;5:2519–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davison K, Chen BE, Kukreti V, et al. Treatment outcomes for older patients with relapsed/refractory aggressive lymphoma receiving salvage chemotherapy and autologous stem cell transplantation are similar to younger patients: a subgroup analysis from the phase III CCTG LY.12 trial. Ann Oncol. 2017;28:622–627. [DOI] [PubMed] [Google Scholar]
- 50.Neelapu SS, Jacobson CA, Oluwole OO, et al. Outcomes of older patients in ZUMA-1, a pivotal study of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. 2020;135:2106–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin RJ, Lobaugh SM, Pennisi M, et al. Impact and safety of chimeric antigen receptor T-cell therapy in older, vulnerable patients with relapsed/refractory large B-cell lymphoma. Haematologica. 2021;106:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dahi PB, Lee J, Devlin SM, et al. Toxicities of high-dose chemotherapy and autologous hematopoietic cell transplantation in older patients with lymphoma. Blood Adv. 2021;5:2608–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curran KJ, Margossian SP, Kernan NA, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fabrizio VA, Boelens JJ, Mauguen A, et al. Optimal fludarabine lymphodepletion is associated with improved outcomes after CAR T-cell therapy. Blood Adv. 2022;6:1961–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuster SJ, Tam CS, Borchmann P, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021;22:1403–1415. [DOI] [PubMed] [Google Scholar]
- 57.Chong EA, Ruella M, Schuster SJ, Lymphoma Program Investigators at the University of P. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med. 2021;384:673–674. [DOI] [PubMed] [Google Scholar]
- 58.Jacobson C, Locke FL, Ghobadi A, et al. Long-Term (≥4 Year and ≥5 Year) Overall Survival (OS) By 12- and 24-Month Event-Free Survival (EFS): An Updated Analysis of ZUMA-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients (Pts) with Refractory Large B-Cell Lymphoma (LBCL). Blood. 2021;138:1764–1764. [Google Scholar]
- 59.Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol. 2017;92:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed N, Kumar A, Kharfan-Dabaja MA, et al. ASTCT Committee on Practice Guidelines Survey on Evaluation & Management of Diffuse Large B-cell Lymphoma after failure of Chimeric antigen receptor T cell therapy (CAR-T) therapy. Transplant Cell Ther. 2022. [DOI] [PubMed] [Google Scholar]
- 61.Logue JM, Chavez JC. How to Sequence Therapies in Diffuse Large B-Cell Lymphoma Post-CAR-T Cell Failure. Current treatment options in oncology. 2021;22:112. [DOI] [PubMed] [Google Scholar]
- 62.Byrne M, Oluwole OO, Savani B, Majhail NS, Hill BT, Locke FL. Understanding and Managing Large B Cell Lymphoma Relapses after Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant. 2019;25:e344–e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shadman M, Gauthier J, Hay KA, et al. Safety of allogeneic hematopoietic cell transplant in adults after CD19-targeted CAR T-cell therapy. Blood Adv. 2019;3:3062–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Imber BS, Sadelain M, DeSelm C, et al. Early experience using salvage radiotherapy for relapsed/refractory non-Hodgkin lymphomas after CD19 chimeric antigen receptor (CAR) T cell therapy. Br J Haematol. 2020;190:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheson BD, Nowakowski G, Salles G. Diffuse large B-cell lymphoma: new targets and novel therapies. Blood cancer journal. 2021;11:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salles G, Duell J, Gonzalez Barca E, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21:978–988. [DOI] [PubMed] [Google Scholar]
- 67.Caimi PF, Ardeshna KM, Reid E, et al. The AntiCD19 Antibody Drug Immunoconjugate Loncastuximab Achieves Responses in DLBCL Relapsing After AntiCD19 CAR-T Cell Therapy. Clinical lymphoma, myeloma & leukemia. 2021. [DOI] [PubMed] [Google Scholar]
- 68.Schuster SJ, Bartlett NL, Assouline S, et al. Mosunetuzumab Induces Complete Remissions in Poor Prognosis Non-Hodgkin Lymphoma Patients, Including Those Who Are Resistant to or Relapsing After Chimeric Antigen Receptor T-Cell (CAR-T) Therapies, and Is Active in Treatment through Multiple Lines. Blood. 2019;134:6–6.31273004 [Google Scholar]
- 69.Bannerji R, Allan JN, Arnason JE, et al. Odronextamab (REGN1979), a Human CD20 × CD3 Bispecific Antibody, Induces Durable, Complete Responses in Patients with Highly Refractory B-Cell Non-Hodgkin Lymphoma, Including Patients Refractory to CAR T Therapy. Blood. 2020;136:42–43. [Google Scholar]
- 70.Hutchings M, Mous R, Clausen MR, et al. Subcutaneous Epcoritamab Induces Complete Responses with an Encouraging Safety Profile across Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Subtypes, Including Patients with Prior CAR-T Therapy: Updated Dose Escalation Data. Blood. 2020;136:45–46. [Google Scholar]
- 71.Baird JH, Frank MJ, Craig J, et al. CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR-refractory large B-cell lymphoma. Blood. 2021;137:2321–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shah NN, Johnson BD, Schneider D, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26:1569–1575. [DOI] [PubMed] [Google Scholar]
- 73.Spiegel JY, Patel S, Muffly L, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dholaria B, Savani BN, Huang XJ, Nagler A, Perales MA, Mohty M. The evolving role of allogeneic haematopoietic cell transplantation in the era of chimaeric antigen receptor T-cell therapy. Br J Haematol. 2021;193:1060–1075. [DOI] [PubMed] [Google Scholar]
- 75.Dreger P Allogeneic stem cell transplant in non-Hodgkin lymphomas: Still an indication? Hematol Oncol. 2021;39 Suppl 1:100–103. [DOI] [PubMed] [Google Scholar]
- 76.Dreger P, Dietrich S, Schubert ML, et al. CAR T cells or allogeneic transplantation as standard of care for advanced large B-cell lymphoma: an intent-to-treat comparison. Blood Adv. 2020;4:6157–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spiegel JY, Dahiya S, Jain MD, et al. Outcomes of patients with large B-cell lymphoma progressing after axicabtagene ciloleucel therapy. Blood. 2021;137:1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chow VA, Gopal AK, Maloney DG, et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am J Hematol. 2019;94:E209–E213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Locke FL, Bartlett NL, Jacobson CA, et al. Retreatment (reTx) of patients (pts) with refractory large B-cell lymphoma with axicabtagene ciloleucel (axi-cel) in ZUMA-1. Journal of Clinical Oncology. 2020;38:8012–8012. [Google Scholar]
- 80.Gauthier J, Bezerra ED, Hirayama AV, et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood. 2021;137:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol. 2016;174:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hamadani M, Gopal AK, Pasquini M, et al. Allogeneic transplant and CAR-T therapy after autologous transplant failure in DLBCL: a noncomparative cohort analysis. Blood Adv. 2022;6:486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lekakis LJ, Moskowitz CH. The Role of Autologous Stem Cell Transplantation in the Treatment of Diffuse Large B-cell Lymphoma in the Era of CAR-T Cell Therapy. Hemasphere. 2019;3:e295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. 2012;367:1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eapen M, Logan BR, Confer DL, et al. Peripheral blood grafts from unrelated donors are associated with increased acute and chronic graft-versus-host disease without improved survival. Biol Blood Marrow Transplant. 2007;13:1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol. 2019;37:2105–2119. [DOI] [PubMed] [Google Scholar]
- 88.Wudhikarn K, Alarcon Tomas A, Brower J, et al. Outcomes of Aggressive B Cell Lymphoma Patients with No Evidence of Measurable Disease at the Time of CD19 Chimeric Antigen Receptor T Cell Therapy: The Experience from the CAR T Cell Consortium. Blood. 2021;138:2843–2843. [Google Scholar]
- 89.Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N Engl J Med. 2022;386:351–363. [DOI] [PubMed] [Google Scholar]
- 90.Delamater PL, Uberti JP. Geographic access to hematopoietic cell transplantation services in the United States. Bone Marrow Transplant. 2016;51:241–248. [DOI] [PubMed] [Google Scholar]
- 91.Moskop A, Hu ZH, Pasquini MC. Current uses of CAR T cell Therapies in the US: CIDR summary slides, 2020. Vol 20222020. [Google Scholar]
- 92.Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and Racial Disparity in Chimeric Antigen Receptor T Cell Therapy Access. Transplant Cell Ther. 2022. [DOI] [PubMed] [Google Scholar]
- 93.Al Hadidi S, Schinke C, Thanendrarajan S, Zangari M, van Rhee F. Enrollment of Black Americans in Pivotal Clinical Trials Supporting Food and Drug Administration (FDA) Chimeric Antigen Receptor (CAR)-T Cell Therapy Approval in Hematological Malignancies. Blood. 2021;138:566–566. [Google Scholar]
- 94.Labbe RP, Vessillier S, Rafiq QA. Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives. Viruses. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Almeida ID, Schmalfuss T, Rohsig LM, Goldani LZ. Autologous transplant: microbial contamination of hematopoietic stem cell products. Braz J Infect Dis. 2012;16:345–350. [DOI] [PubMed] [Google Scholar]
- 96.Dal MS, Tekgunduz E, Cakar MK, et al. Does microbial contamination influence the success of the hematopoietic cell transplantation outcomes? Transfusion and apheresis science : official journal of the World Apheresis Association : official journal of the European Society for Haemapheresis. 2016;55:125–128. [DOI] [PubMed] [Google Scholar]
- 97.Jacobs MR, Good CE, Fox RM, Roman KP, Lazarus HM. Microbial contamination of hematopoietic progenitor and other regenerative cells used in transplantation and regenerative medicine. Transfusion. 2013;53:2690–2696. [DOI] [PubMed] [Google Scholar]
- 98.Chong EA, Levine BL, Grupp SA, et al. CAR T cell viability release testing and clinical outcomes: is there a lower limit? Blood. 2019;134:1873–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chong EA, Gerson JN, Landsburg DJ, et al. Outcomes in Aggressive B-Cell Non-Hodgkin Lymphomas with Anti-CD19 CAR T-Cell (CTL019) Products Not Meeting Commercial Release Specifications. Blood. 2019;134:594–594. [Google Scholar]
- 100.Maus MV, Nikiforow S. The Why, what, and How of the New FACT standards for immune effector cells. Journal for immunotherapy of cancer. 2017;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perica K, Curran KJ, Brentjens RJ, Giralt SA. Building a CAR Garage: Preparing for the Delivery of Commercial CAR T Cell Products at Memorial Sloan Kettering Cancer Center. Biol Blood Marrow Transplant. 2018;24:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yanez L, Sanchez-Escamilla M, Perales MA. CAR T Cell Toxicity: Current Management and Future Directions. Hemasphere. 2019;3:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hernandez I, Prasad V, Gellad WF. Total Costs of Chimeric Antigen Receptor T-Cell Immunotherapy. JAMA oncology. 2018;4:994–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shah GL, Park J, Sauter CS, et al. Thirty Day Resource Utilization after Chimeric Antigen Receptor (CAR) T Cell Infusion for Hematologic Malignancies. Biology of Blood and Marrow Transplantation. 2019;25:S38–S39. [Google Scholar]
- 105.Mian A, Wei W, Hill BT, et al. Resource Utilization and Factors Prolonging Hospitalization for Patients with Refractory and Relapsed B-Cell Lymphoma Receiving Axicabtagene Ciloleucel (Axi-cel). Biology of Blood and Marrow Transplantation. 2020;26:S44–S45. [Google Scholar]
- 106.Siddiqi T, Garcia J, Dehner C, et al. Estimation of the Resource Utilization and Costs of Cytokine Release Syndrome Observed in the Transcend-NHL Clinical Trial: A Micro-Costing Study. Blood. 2018;132:319–319. [Google Scholar]
- 107.Lyman GH, Nguyen A, Snyder S, Gitlin M, Chung KC. Economic Evaluation of Chimeric Antigen Receptor T-Cell Therapy by Site of Care Among Patients With Relapsed or Refractory Large B-Cell Lymphoma. JAMA Netw Open. 2020;3:e202072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alexander M, Culos K, Roddy J, et al. Chimeric Antigen Receptor T Cell Therapy: A Comprehensive Review of Clinical Efficacy, Toxicity, and Best Practices for Outpatient Administration. Transplant Cell Ther. 2021;27:558–570. [DOI] [PubMed] [Google Scholar]
- 109.Wakase S, Teshima T, Zhang J, et al. Cost Effectiveness Analysis of Tisagenlecleucel for the Treatment of Adult Patients with Relapsed or Refractory Diffuse Large B Cell Lymphoma in Japan. Transplant Cell Ther. 2021;27:506 e501–506 e510. [DOI] [PubMed] [Google Scholar]
- 110.Geethakumari PR, Ramasamy DP, Dholaria B, Berdeja J, Kansagra A. Balancing Quality, Cost, and Access During Delivery of Newer Cellular and Immunotherapy Treatments. Curr Hematol Malig Rep. 2021;16:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wudhikarn K, Palomba ML, Pennisi M, et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood cancer journal. 2020;10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baird JH, Epstein DJ, Tamaresis JS, et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood Adv. 2021;5:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neumann PJ, Cohen JT, Weinstein MC. Updating cost-effectiveness--the curious resilience of the $50,000-per-QALY threshold. N Engl J Med. 2014;371:796–797. [DOI] [PubMed] [Google Scholar]
- 114.Perales M-A, Kuruvilla J, Snider JT, et al. LBA2 - Cost-Effectiveness of Axicabtagene Ciloleucel As Second-Line Therapy for Patients Large B-Cell Lymphoma (LBCL) in the United States. Transplantation and Cellular Therapy. 2022;28:S475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kelkar AH, Jacobson CA, Abel GA, et al. 427 - Cost-Effectiveness of CD19 Chimeric Antigen Receptor T Cell Therapy Versus Autologous Hematopoietic Cell Transplantation for Diffuse Large B Cell Lymphoma in First Relapse. Transplantation and Cellular Therapy. 2022;28:S332–S333. [Google Scholar]
- 116.Globerson Levin A, Riviere I, Eshhar Z, Sadelain M. CAR T cells: Building on the CD19 paradigm. Eur J Immunol. 2021;51:2151–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mansilla-Soto J, Eyquem J, Haubner S, et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Khurana A, Lin Y. Allogeneic Chimeric Antigen Receptor Therapy in Lymphoma. Current treatment options in oncology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goldenson BH, Hor P, Kaufman DS. iPSC-Derived Natural Killer Cell Therapies - Expansion and Targeting. Frontiers in immunology. 2022;13:841107. [DOI] [PMC free article] [PubMed] [Google Scholar]