

Abstract
Despite antiretroviral therapy, HIV-1 persists as proviruses integrated into the genomic DNA of CD4+ T cells. The mechanisms underlying the persistence and clonal expansion of these cells remain incompletely understood. Cases have been described in which proviral integration can alter host gene expression to drive cellular proliferation. Here, we review observations from other genome-integrating human viruses to propose additional putative modalities by which HIV-1 integration may alter cellular function to favor persistence, such as by altering susceptibility to cytotoxicity in virus-expressing cells. We propose that signals implicating such mechanisms may have thus far been masked by the preponderance of defective and/or non-reactivatable HIV-1 proviruses but may be revealed by focusing on the integration sites of intact proviruses with expression potential.
Keywords: Virus integration, host-virus DNA interactions, virus-induced cell proliferation, virus-host chimeric RNA, insertional mutagenesis
Clonal Expansion of the HIV-1 Reservoir and the Roles of Proviral Integration Sites
Combination antiretroviral therapy (ART), the mainstay for the treatment of human immunodeficiency virus (HIV-1) infection, halts viral replication but does not eliminate cells with integrated HIV-1 provirus (see Glossary). A pool of these cells persists as a long-lived HIV-1 reservoir, which can reseed viral replication upon ART cessation [1]. With time on ART, these reservoirs become dominated by expanded clones of infected cells (identifiable based on the sharing of unique integration sites and TCR sequences), which differ in their abilities to survive and proliferate [2–4]. The mechanisms underlying the relative fitness of specific clones remain poorly understood, and closing this knowledge gap is a research priority towards the goal of curing HIV-1 infection [5]. Alterations in cellular function driven by the proviral integration itself comprises a potentially stable and diverse source of fitness heterogeneity amongst clones. However, such functional consequences have thus far been limited to integrations that drive proliferation, and these appear to make only minor contributions to total proviral landscapes [6–11]. Outside of these cases, are clonal dynamics governed exclusively by normal CD4+ T-cell biology [12–15]? Or, might other types of HIV-1 integration-mediated alterations in clonal fitness (beyond proliferation) contribute to clonal persistence [7,9–11,16]? Literature on Hepatitis B virus (HBV), human papillomavirus (HPV), Human T-lymphotropic virus type 1 (HTLV-1), and Epstein-Barr virus (EBV) — viruses whose integration can induce changes in host gene transcription by mechanisms overlapping with HIV-1 [17–30]— might yield clues. HBV and HPV integrations have been shown to accumulate in genes that influence various cellular functions in addition to proliferation [18,20,22,27,31–35], with recent studies implicating immune evasion through integration site-driven overexpression of T-cell inhibitory ligands [34,35]. HIV-1 is the subject of intense cytotoxic T-cell (CTL) pressure, which persists to a degree on ART [36,37], and although many proviruses are latent in ART-treated individuals, some expression continues [38]. Ex vivo studies of in vivo infected human CD4+ T cells provide evidence that some HIV-1 reservoir cells may resist CD8+ cytotoxic T cell (CTL)-mediated elimination via the overexpression of prosurvival genes including BCL2 and BIRC5 [39–41]; however, to our knowledge, no reports have linked HIV-1 provirus modulation of host genes, to resistance to CTL-mediated killing. As described below, complexities in the HIV-1 proviral landscape create a scenario whereby only a small fraction of proviruses - encompassing those most relevant to viral rebound - may be subject to immune selection or to viral cytopathicity. Thus, we propose that leveraging new approaches to focus on this subset of proviruses may reveal additional modalities by which integration sites alter cell function to enable persistence of the HIV-1 reservoir.
Proviruses Disrupt Host Gene Expression in Diverse Ways
We approach the topic of comparative functional consequences of genomic integration across different viruses, by first reviewing the diverse modalities by which these can affect host gene expression (Figure 1, Key Figure). The most commonly reported integration site-dependent effects observed in HIV-1 are (1) HIV-1 promoter or enhancer insertion resulting in host gene activation [6,8,9,16], (2) virus-host chimeric transcription resulting in chimeric RNA with potentially altered expression and function [6,16], (3) activation of cryptic host splice sites [8,16], and (4) transcriptional interference [7,16]. While yet-to-be observed for HIV-1 infection, other well characterized molecular effects of viral integration are (5) 3′-untranslated region (UTR) substitution [42–44], (6) the remodeling of the epigenetic landscape (Box 1) [19,24–26], (7) the induction of genomic instability [27,28,32], as well as (8) host gene disruption (Figure 1) [23,29,30,45].
Figure 1 Key figure. Overview of putative integration site-dependent mechanisms that can affect host protein expression.
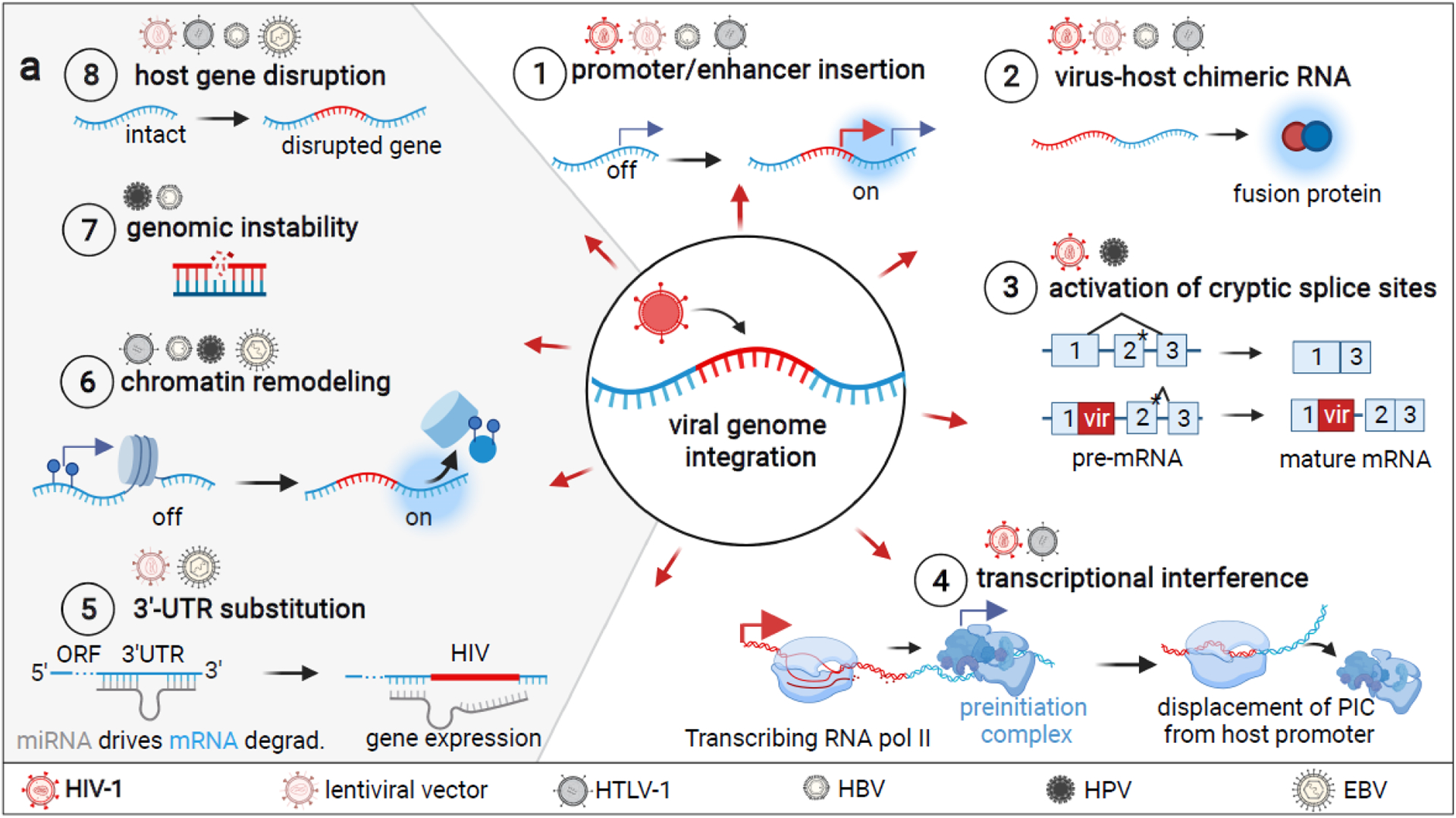
Un-demonstrated HIV-1 integration mechanisms are depicted in gray and viruses for which experimental evidence exists are depicted above each. 1) Integration of a viral promoter or enhancer can activate gene expression [6,8,9,16]. 2) Viral or host promoters may drive the transcription of chimeric virus-host RNA and translation of fusion proteins [6,16]. 3) Disruption of a splice site can result in the activation of a downstream cryptic splice site (denoted with *) that was previously disfavored for a stronger splice site [8,16]. This can result in the retention of introns in mRNA [16]. 4) Transcriptional interference of viral and host promoters can result in various scenarios in which only either cellular or viral transcription occurs [49,50], regardless of the orientation of the two promoters. Typically, it allows for transcription of the gene driven by the stronger promoter only [52]. 5) Genes can be upregulated by mRNA 3′ end substitution if a degradation-promoting 3′ mRNA end is disrupted by viral integration [42–44]. 6) Viral sequences interact with various host chromatin remodelers and may interact with other chromatin regions to impact 3D organization/accessibility [19,24–26]. 7) Integrations may entail single or double strand breaks that result in various mutations, including chromosomal translocations in the case of HBV [27,28,32]. 8) Viral integration may disrupt host genes via the insertion of premature STOP codons, poly-A sites, or frameshift mutations [23,29,32,45,64,102,103]. This figure was created with BioRender.com.
Box 1: Chromatin Remodeling resulting from Viral Integration.
It is unclear whether HIV-11 integration can alter the surrounding host epigenetic landscape — while the converse scenario of HIV-1, can construct the insertion into heterochromatic loci marked by H3K9me, resulting in silencing of the provirus [70]. This silencing has been shown to be mediated by the HUSH protein complex [70]. Since this human protein complex functions as an epigenetic regulator of endogenous and viral genes, it remains to be determined whether HUSH-mediated silencing of HIV-1 provirus can affect the transcription of surrounding host genes [70]. HIV-1 also lacks any conventional binding sites for chromatin modifying proteins, the presence of which would point towards a mechanistic role for HIV-1 integration in remodeling of host chromatin. For HPV, HBV, and HTLV-1, however, insertion-induced changes in chromatin organization are becoming increasingly evident [19,24–26]. The HBV, EBV, HPV, and HTLV-1 genomes contain binding sites for the transcriptional repressor and chromatin regulator CCCTC-Binding factor (CTCF) [26,73–75] that mediates the formation of loops in the human genome and establishes boundaries between hetero- and euchromatin [76]. Chromosome conformation capture (Hi-C) studies on human cervical cancer biopsies harboring HPV integrations into the recurrent integration gene (RIG) CCDC106, as well as HPV provirus harboring cell lines , showed changes within host genomic topologically associating domains (TADs) as well as altered TAD borders in the vicinity of the integration site [25,77,78]. Changes in nuclear architecture were associated with the formation of new virus–host DNA interactions including with host enhancer sequences [25,78]. Associated changes in gene expression included the transcriptional down- and upregulation of genes encoding tumor-suppressor (PEG3, KLF12) and proto-oncogene (CCDC106) functions in HPV harboring cell lines and cervical cancer biopsies [77–79]. Similarly assays revealed that HBV, upon integration, establishes 3D contact regions with cellular chromatin [80,81]. These contacts occur preferentially in transcriptionally active regions (as supported by RNA-seq studies) that contain enhancers and transcriptional start sites, and in CpG islands that are associated with genes that are differentially expressed in HBV-infected primary hepatocytes and the HBV-integrated cell line HepAD38 [80,81]. Moreover, HTLV-1 provirus-bound CTCF mediates the formation of virus-host gene loops that can block enhancer-promoter contacts, control HTLV-1 mRNA splicing, and can alter transcription and splicing at a distance of > 300kB from the integration site [19]. Based on the far-ranging changes in cellular transcription in other human viruses, such effects, if present, would likely be capable of contributing to altered cellular function.
Promoter/enhancer insertion
Host gene activation driven by the viral promoter and or enhancer can occur when a virus integrates in tandem-orientation, i.e. in the same orientation as the cellular gene, into a 5′ intron or upstream of a host gene [10,11,16]. In the case of HIV-1, the LTRs function as strong promoters that drive increased expression of host genes [8,9,16]. While diverse genes and HIV-1 insertional events have been identified as targets of HIV-1-driven host gene activation, one cellular consequence highlighted to date is the enhanced expansion of a minority of clones (cases identified thus far have been clones with defective proviruses), (Box 2). Promoter or enhancer insertion induced proliferation has been observed for lentiviral vectors [45], HBV [17], as well as HTLV-1 [19] (Box 2). Transduction of murine hematopoietic stem cells with lentiviral constructs prior to transfusion does not appear to significantly heighten the risk of malignancies in tumor-prone mice [46], suggesting that HIV-1 promoter or enhancer insertion alone is not substantially tumorigenic. A recent in vitro proof-of-concept study leveraged targeted insertion of the HIV-1 LTR into BACH2 to demonstrate that HIV-1 LTR integration can promote the proliferation of CD4+ T cells [7]. Taken together, HIV-1 LTR-driven transcription of cellular genes has been observed in the HIV-1 reservoirs of individuals across multiple studies, and this molecular mechanism might result in diverse cellular consequences [6,8,16].
Box 2: Proliferation as a Direct Consequence of HIV-1 Integration.
There is in vitro [7,9] and clinical [6,10,82] evidence that intronic HIV-1 integration into oncogenes can result in LTR-driven transcriptional activation that promotes cellular proliferation (Figure I). Integrations in specific recurrent integration genes (RIGs) drive the expansion of some CD4+ T cell clones within the HIV-1 reservoir [8,10,11,16,49,50]. Insertional mutagenesis has resulted in a case of STAT3-associated B cell lymphoma, where integration of a defective HIV-1 provirus upstream of the first exon of STAT3 resulted in cellular proliferation caused by 3′ LTR-driven STAT3 overexpression [9,82] and has likely contributed to T cell lymphoma development in at least six individuals [6]. LTR-driven transcription has also been observed for the RIGs BACH2 and STAT5B, resulting in overexpression [8]. CRISPR-Cas9-mediated in-tandem insertion of the viral LTR and major splice donor site into the frequently observed intronic site in BACH2 in primary CD4+ T cells, resulted in the proliferation of cells with a regulatory T cell-like RNA and protein expression pattern, demonstrating that LTR-driven activation of BACH2 resulted in CD4+ T cellular proliferation and differentiation [7]. Coherent with these studies, the most studied biological consequence of HTLV-1, HPV, and HBV integration is the proliferation of infected cells, associated with the overexpression of proto-oncogenes and downregulation of tumor suppressor genes (Figure I) [22,25,29,30,34,35,78,83,84]. For instance, HBV integration into TERT, the gene encoding the catalytic subunit of telomerase, can drive increased telomerase production and promote the survival of malignant cells [85,86]. HBV integration also targets and downregulates tumor-suppressor genes, such as p32 pathway components [29]. HPV integrations into introns and exons of RAD51B (RAD51 paralog B), a gene encoding a protein with DNA-repair and apoptotic function might indicate that this tumor-suppressor gene is disrupted by HPV integration [34]. For HIV-1, however, integration-dependent induction of proliferation appears to have a small overall contribution to clonal expansion [6,87], and has recently been reviewed [88]. Like BACH2, the other RIGs of HIV-1 —STAT5B, MKL2, MKL1, IL2RB, MYB and POU2F1 — show enrichment for HIV-1 integrations in the same orientation as host gene transcription and into specific introns in PLWH [4,8,10,87,89,90]. This indicates that HIV-1 integration into those RIGs might provide a fitness advantage to cells [4,8,10,87,89,90]. Additional evidence for the functional importance of HIV-1 RIGs comes from their overrepresentation in mouse models compared to in vitro [88,91,92], and the accumulation of cells with integrations in these seven genes over ART [4,8,10,87,89,90].
Figure I (in Box 2). Integration-induced Cellular Proliferation.
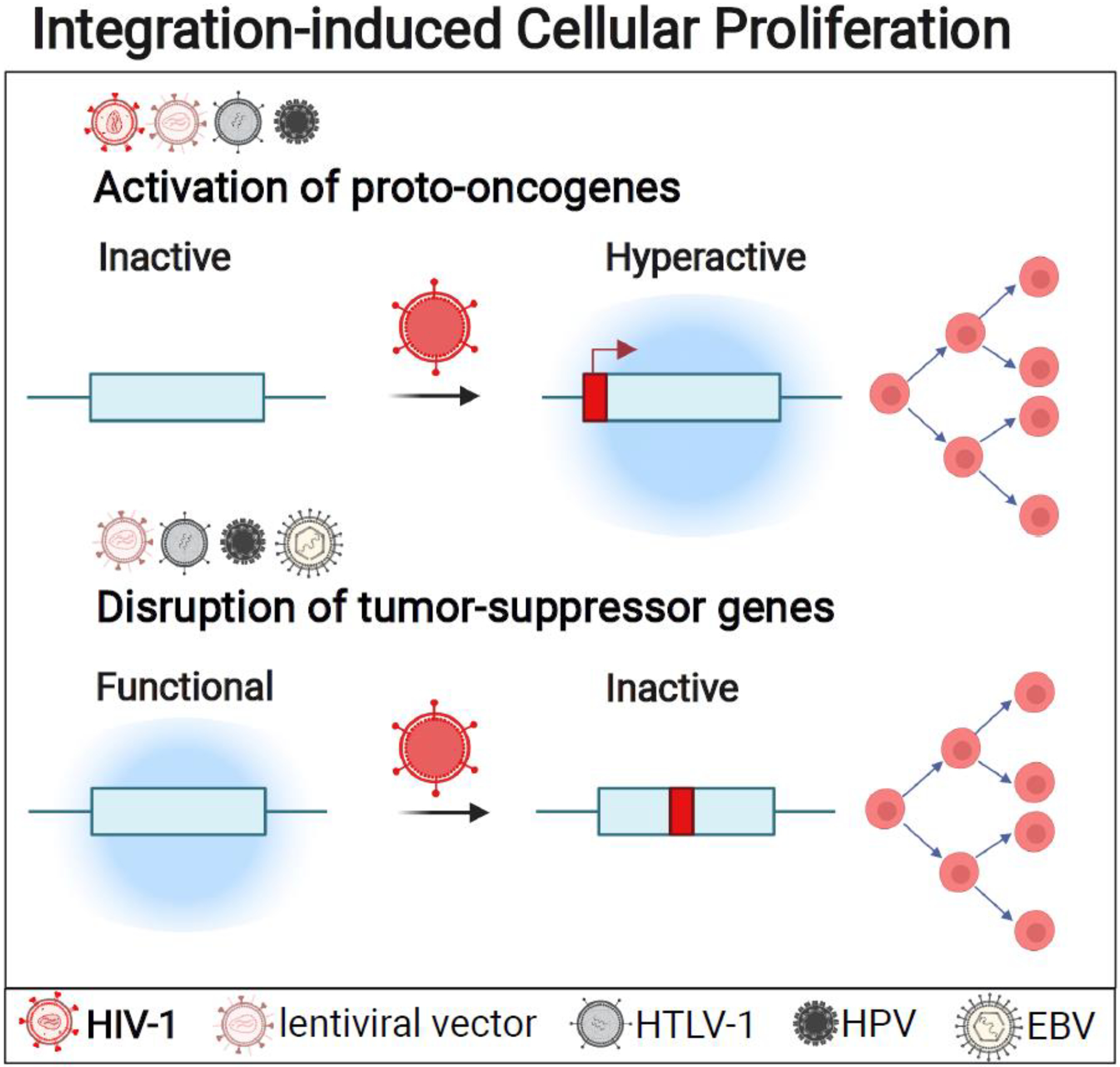
Genomic integration of HIV-1 [10,38,87,89,93], HBP [29], and HPV [34,35], HTLV-1 [23], and lentiviral vectors [45] can directly affect cellular proliferation by targeting oncogenes or tumor-suppressor genes. Viruses for which experimental evidence of integrating into proto-oncogenes as well as tumor-suppressor genes exists are listed above the respective class of genes. Recurrent integration genes (RIG)s for HIV-1 include the proto-oncogenes MKL2 [11], BACH2 [10,11], and STAT3 [6,9,82]. HBV RIGs include TERT, [85,86]. Viruses can also target and downregulate the expression of tumor-suppressor genes to disrupt death pathways, such as p53 downregulation by HBV [29], and RAD51B downregulation by HPV integration [34], suggesting that gene disruption may promote survival and growth of the infected cells. This figure was created with BioRender.com.
Virus-host chimeric RNAs
Viral as well as host promoter driven transcription can result in the generation of virus-host chimeric RNAs [16]. These chimeras may alter splicing, function, and expression of viral and host proteins [6,16,18]. Readthrough transcription driven by intronically integrated HIV-1 3′ LTR into downstream exons is abundant in CD4+ T-cells of people living with HIV-1 (PLWH) on ART, and has been observed in-tandem as well as in the opposite orientation relative to the host gene [16]. Less commonly, host promoters can drive readthrough host-virus chimeras into HIV-1 5′ LTR [16]. One such chimeric RNA was identified in one case of PLWH presenting with T cell lymphoma, for whom reverse transcription polymerase chain reaction (RT-PCR) identified a transcript in which exon 1 of STAT3 —a noncoding sequence, and thus unlikely to interfere with the expression of the downstream HIV-1 sequence— was spliced into the HIV-1 Tat sequence [6]. Thus, virus-host chimeric RNAs might allow for the functional expression of both viral as well as host genes [6]. In addition to HIV-1, virus-host chimeric RNAs have been observed for lentiviral vectors, as recently reviewed [45], as well as for HPV, and HBV [18,20,21]. For instance, chimeric transcripts of HPV spliced into human genes were detected in 12 out of 14 samples of invasive cervical cancer [20]. Hemi-nested RT-PCR assays of hepatocellular carcinoma (HCC) tumor samples from 90 HBV-infected individuals revealed the presence of chimeric transcripts of the coding sequence for HBV virus X protein (HBx) fused to LINE1 sequences in 23.3% of HBV-associated HCC tumors – confirmed by sequencing and by RT-PCR using probes that were virus-host junctions specific to the [18]. These HBx-LINE1 RNAs acted as long noncoding RNA (lncRNA)-like transcripts and promoted oncogenesis through activation of the Wnt/β-catenin signaling pathway [18]. Another chimeric RNA was translated into an HBV-host fusion protein that deregulated the cellular stress response to promote cell growth [21]. These findings demonstrate that fusion proteins may differ in function from viral or host gene products and can have significant impact on virus infected cells.
Activation of cryptic splice sites
A cryptic splice site is one that is not used in wild-type mRNA, but only selected as a result of a mutation elsewhere in a gene. Viral genomes, such as HIV-1, contain splice sites that can be joined to host mRNA. This can result in the activation of cryptic splice sites when a provirus disrupts a nearby canonical splice site [8,16]. In the HIV-1 genome, there are four splice donor and seven splice acceptor sites [47]. Host-driven splicing from host exons into the HIV-1 genome has been identified by integration site sequencing and corroborated by RT-PCR and RNAseq in human lymphomas and lymphoproliferative disorders from 13 PLWH [6,16]; most commonly, HIV-1-driven splicing from the HIV-1 major splice donor occurs into host exons, e.g. into BACH2, STAT5B and NFATC3 [8,16]. Recently, imitation of HIV-1 integration sites in introns of three cancer-associated genes in human Jurkat CD4+ T cells resulted in host intron retention and a remarkable increase in host-protein expression driven by the HIV-1 LTR [16]. Thus, various HIV-1-host splicing events can increase host protein expression. Moreover, HIV-1-induced aberrant splicing events in Jurkat T cells have resulted in N-terminal truncation of VAV1; moreover, in VAV1 cDNA transduction studies in NIH 3T3 fibroblasts the 5’ terminal truncation of VAV1 was previously linked to increased oncogenic potential [48]. This finding is relevant as it suggests that splicing of HIV-1 and host sequences might alter protein activity. Based on this literature, we suggest that splicing of HIV-1 and cellular sequences might result in instances of altered cellular protein function.
Transcriptional interference
Besides these activating effects, the insertion of the strong HIV-1 promoter can also negatively affect the transcription of host genes due to a set of phenomena collectively termed transcriptional interference [49,50], reviewed elsewhere [51]. Regardless of the orientation of the viral promoter relative to the host gene, HIV-1 promoters and host transcriptional promoters can interfere with each other’s transcription by dislodging the transcriptional preinitiation complex [51,52] (Figure 1a). Recent reports indicate that HIV-1 can integrate in-between exons six and seven of the proto-oncogene NFATC3 as well as between exon 5 and 6 of BACH2; these events disrupt transcription upstream of the HIV-1 integration site, and possibly represent the first accounts of “roadblock” transcriptional interference via HIV-1 insertion [7,16,52]. Hence, transcriptional interference events associated with HIV-1 integration may down-regulate host gene expression as well as viral expression, which has also been reported with HTLV-1 integration-mediated cis-perturbations of host gene transcription [23]. In the context of HIV-1 infection, transcriptional interference of actively transcribed provirus might affect diverse cellular processes.
3′-UTR substitution
Lentiviral transduction of the β-globin gene into bone marrow CD34+ cells that were used to cure a patient from β-thalassemia, unveiled a particularly rare effect of lentiviral sequence integration: 3′-UTR substitution [42]. Intronic insertion of the vector into HMGA2 formed the new 3′ end of the host mRNA, thus excluding the cellular 3′-UTR, a region that is usually targeted by the complementary cellular let7 microRNA for mRNA degradation [42]. Lentiviral insertion of the new viral “premature” polyadenylation site resulted in the exclusion of this downstream regulatory 3′-UTR sequence from the cellular mRNA in a myeloid progenitor, which allowed increased expression of HMGA2 protein in a fraction of vector-bearing nucleated blood cells that arose from the transfused bone marrow CD34+ cells; this in turn, promoted proliferation of the vector-harboring cells [43] (Box 2). Recently, a similar mechanism was reported for the effects of EBV on the expression of DOK1, a tumor-suppressor gene that is commonly mutated in human cancers [53]. DNA sequencing of Burkitt’s lymphoma cell lines as well as lymphoblastoid cell lines revealed a positive correlation between the presence of EBV and increased genetic variation in the 3’ UTR of DOK1; the presence of EBV was associated with significantly reduced copy numbers of DOK1 mRNA [44]. This suggested that EBV integration might disrupt the 3’ UTR of a tumor-suppressor gene to promote degradation rather than expression of DOK1. Therefore, these case reports can only hint at the diverse effects that HIV-1 integration might have on the mRNA expression and stability of various genes [54].
Genomic instability
To our knowledge, there are no indications of increased genomic instability due to lesions induced during HIV-1 integration, but there is growing evidence that HPV [27,33,55–57] and HBV [28,31,32] may directly cause high mutation rates in the vicinity of the viral integration site, affecting host gene expression [25,31,57,58]. DNA sequencing studies catalogued local sequence deletions but also chromosomal translocations in HBV-positive biopsies from chronic hepatitis patients [32] as well as HPV-integrant cell lines from human cervical and head and neck cancer samples [27]. However, HPV integrates fragmented DNA by different mechanisms that can result in genome amplification, which is in stark contrast to the integration mechanism of HIV-1 [25,59]. The mechanism of HPV integration can involve the generation of focal amplifications and rearrangements in cellular DNA, which is significantly more disruptive than HIV-1 integration [27]. HIV-1 integration merely involves the introduction of a 4–6 basepair sequence duplication flanking both ends of the integrated viral DNA as well as unpaired dinucleotides at the 5’ end of the proviral sequence [60]. Evidence that HPV, HBV, EBV integrations in human subjects are enriched at chromosome fragile sites (CFS), i.e. regions with an enhanced risk of DNA breakage and genomic instability, has been suggested to contribute to the genomic instability observed in these oncogenic viral infections [61,62]. However, a recently established database combining results from various studies determined the percentage of HIV-1 integrations into CFS integrations to be 42.2%, exceeding the percentage of integrations in CFS regions of the oncoviruses mentioned above (33.9% HBV, 37.5% HPV, 34.6% HTLV-1, 35.2% EBV) [61], which might suggest a potential role for HIV-1 integration in contributing to genomic instability. It is conceivable that the minor DNA lesions caused by the genomic integration of HIV-1 proviruses are sufficient to render those genic regions susceptible to further mutations [63].
Host gene disruption
HIV-1, as well as EBV, HTLV-1, and HBV, are found integrated at elevated frequencies into proto-oncogenes as well as tumor-suppressor genes [61]. Compared to other retroviruses, HIV-1 does not appear to frequently cause significant host gene disruption. However, there are reports of HTLV-1 and HIV-1-derived lentiviral vectors that abolish gene function at the integration site (reviewed elsewhere [23,45]); this suggests that lentiviral integration might result in such effects. HBV integration causes interchromosomal rearrangements, megabase-size telomeric deletions, chromosomal fusions, and dicentric chromosomes in over 8% of HCC samples [32,64]. Despite this wide-range mutational burden of HBV integration, this result was only identified as a cause of HCC in 2021 [32,64]. Indeed, HBV integration results in disruptions of p53 and other tumor-suppressor genes, which only became recently clear [29]. Similarly, sequencing of 177 EBV-associated nasopharyngeal carcinomas (NCP) by hybridization-based enrichment revealed that EBV integration into introns of genes involved in TNF-α-dependent apoptosis, TNFAIP3, PARK2, and CDK15, disrupted their expression, as evidenced by immunohistochemistry [65]. Knockdown studies have shown that the EBV recurrent integration gene (RIG) TNFAIP3 downregulates NF-κB [65]. Thus, further studies are needed to determine if HIV-1-mediated host gene disruption may have been equally overlooked and might be a factor contributing to cellular consequences that favor HIV-1 persistence. In sum, an arsenal of molecular mechanisms such as HIV-1 promoter or enhancer driven cellular gene expression, chimeric transcription, transcriptional interference with cellular transcription processes, as well as splicing of HIV-1 into cellular cryptic splice sites can allow HIV-1 integration to positively or negatively affect cellular transcription. We suggest that additional mechanisms such as 3′-UTR substitution, chromatin remodeling, genomic instability, and host gene disruptions (recently identified as mechanisms used by HTLV-1, EBV, HPV, and HBV), might also occur at HIV-1 integration sites.
Functional Consequences of Proviral Integration
On the one hand, the vast landscape of potential viral integration sites in the human genome, multiplied by the diverse mechanisms by which a provirus can influence gene expression, supports the idea that - given a large enough pool of infected cells - any aspect of infected-cell fitness may be subject to such influences. On the other hand, it is expected that a large majority of random integrations will not impact cellular fitness. The likelihood that rare advantageous integration sites will appreciably impact the overall landscape of infected cells is therefore a function of the nature of the selective pressure, the degree of the advantage, and of time. It stands to reason that integrations that drive proliferation of a given infected cell will be most prone to detection, and indeed this phenomenon has been well-established for a number of integrating viruses, including HIV-1. Evidence and further indications for an involvement of HIV-1 integration in cellular proliferation are summarized in Box 2.
HBV and HPV integrations have been shown to directly target genes that control various cellular functions other than proliferation, including survival, and can ward off CTL through overexpression of inhibitory ligands [34,35,65–67] (Box 3). Some of these HPV findings are quite recent, and were enabled by technological advances that shed new light on proviral integration landscapes. A similar scenario may hold for HIV-1, where the challenge to detecting integration sites that alter cell-intrinsic susceptibility to viral cytopathic effects or to CTL is two-fold: 1) Approximately 98% of HIV-1 proviruses in ART-treated individuals contain defects, such as large internal deletions [68]. Some defective proviruses can express viral products that may be directly cytopathic or enable recognition by CTL, but most cannot and are thus exempt from corresponding selective pressures [68,69]. 2) Many intact proviruses remain latent in ART-treated individuals, in some cases attributable to another key aspect of proviral context - integration into transcriptionally silent genomic regions [38,70] Such proviruses would similarly be exempt from the selection pressures under discussion. A recently developed technique termed ‘PRIP-seq’ - which enables parallel analysis of transcription, integration sites, and sequences of single proviruses - has revealed the existence of expanded clones with transcriptionally-active intact HIV-1 proviruses on ART [38]. This both supports the existence of pools of clonally-expanded cells that may be subject to ongoing immune selection on ART, and provides a platform to profile associated integration sites. The functional consequences of proviral integration sites revealed by such emerging approaches will also be most relevant to ongoing efforts to cure HIV-1 infection, given the potential of this subset of proviruses to give rise to viral rebound.
Box 3: Immune Evasion Resulting from Changes in Host Gene and Viral Expression Caused by Viral Integration.
Virus integration can alter cellular gene expression to contribute to immune evasion by two main pathways (Figure II):
(1). increase in pro-survival factors:
Integrations of HPV and HBV sequences can induce the expression of pro-survival factors. For HBV, the RIG FOXP2 [66,67], encodes a transcription factor that contributes to preventing inflammation and apoptosis [94]. Similarly, primary oropharyngeal cancer cells show an HPV integration hotspot in intron 10 of BRISC and BRCA1 A complex member 2 (BABAM2) [35], a gene that blocks tumor necrosis factor alpha (TNF-α)-induced apoptosis [95]. Another example is the ETS proto-oncogene 2 (ETS2), a tumor-suppressor gene that inhibits apoptosis and which is frequently targeted by HPV integrations in tumor tissues [34]. HPV integration in multiple individuals with different types of HPV-induced cancers has been observed into and near TNFα-induced protein 2 (TNFAIP2) [35], a gene encoding an angiogenic factor overexpressed in HPV+ cervical cancer, which promotes the viability of non-small cell lung cancer (NSCLC) cells by limiting the induction of caspase 3 [96].
(2). expression of co-inhibitory receptors to suppress cytotoxic T lymphocyte (CTL) function:
Recently, CD274, the gene encoding Programmed death-ligand 1 (PD-L1) was identified as a RIG for HPV [34,35]. Integration into this gene was more common in HPV-induced oropharyngeal cancers, and integration amplified host gene expression 5–10-fold [35]. Indeed, expression of PD-L1, on human tumor cells is a generally recognized tumor immune escape mechanism [97]. Ligation of PD-1 on tumor-infiltrating CTL limits their proliferative and cytotoxic activity [97].
In addition to changes in cellular expression, viral integration regulates viral gene expression and antigen presentation. HBV integration is also associated with viral protein surface expression and secretion, which has been linked to CD8+ T cell exhaustion and thus, viral persistence, despite integrated HBV being unable to replicate [98] (Figure II). Silent provirus escapes immune recognition, but also high viral protein expression can function as an immune evasion mechanism, contributing to the dysfunction of virus-specific CTL in the presence of persistent antigen; this has been observed for HPV integrations, and there is mounting evidence that the integration site is a crucial determinant of HIV-1 provirus expression [38]. Therefore, the effects of viral integration sites on host gene expression and viral transcription will need to be considered in parallel to conclude on the contributions of cellular and viral transcription on the persistence of a given cell.
Figure II in Box 3. Integration-induced Immune Evasion.
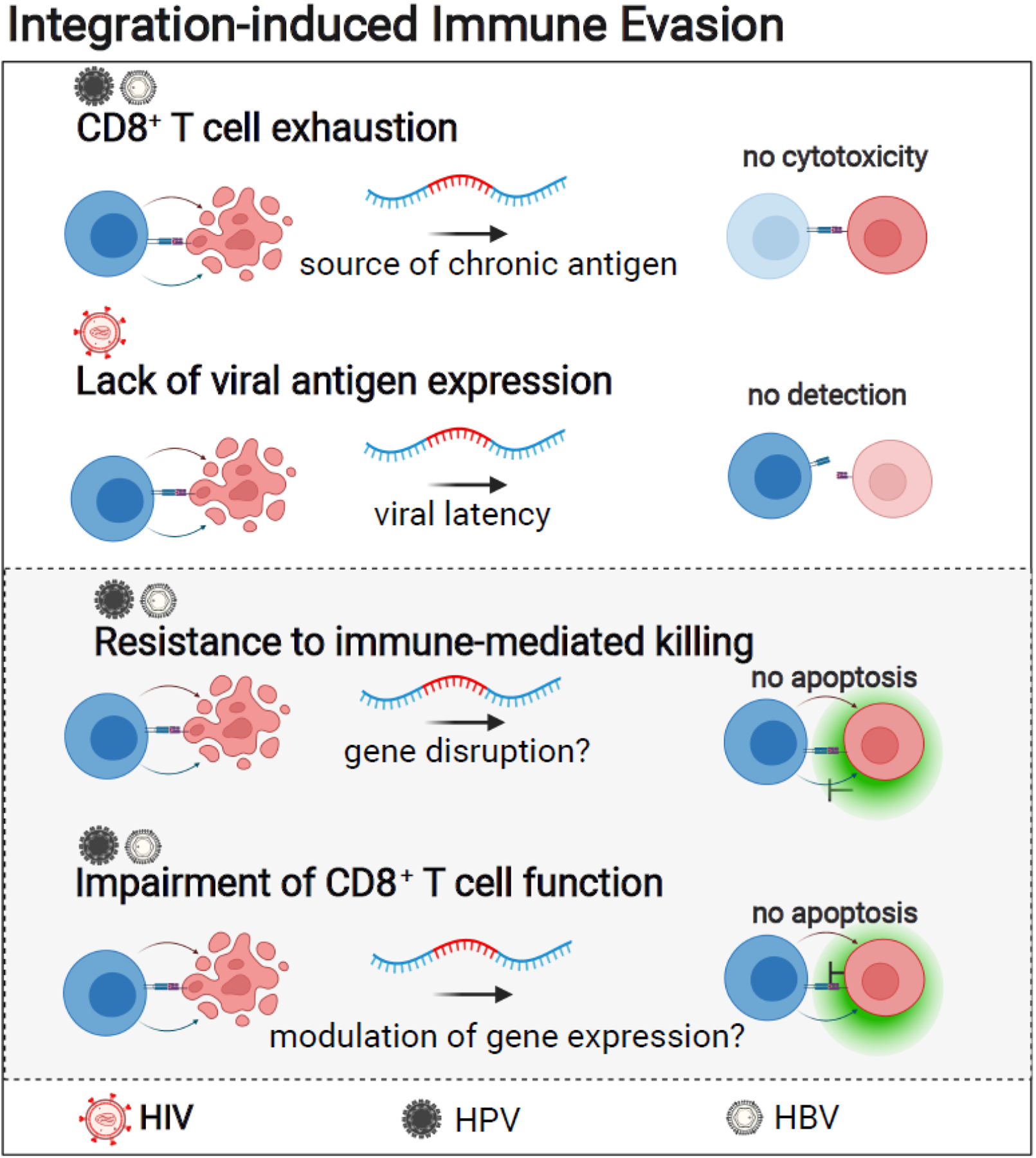
Genomic integration of HPV and HBV can target genes that control cellular functions related to immune evasion. Viruses for which experimental evidence exists for integration and transcriptional upregulation of genes involved in the indicated cellular functions are listed above each cellular mechanism. HBV, HPV, and EBV integration can upregulate the expression of anti-apoptotic proteins [34,35,65–67]. HBV integration can result in upregulation of FOXP2, a gene that encodes a transcription factor with anti-apoptotic functions [66,67]. Anti-apoptotic genes targeted by HPV include ETS2 [34] as well as BABAM2 [35], a gene that encodes an anti-apoptotic protein that blocks tumor necrosis factor alpha (TNF-α)-induced apoptosis [95]. Likewise, EBV integration can result in upregulation of an inhibitor of TNF-α-dependent apoptosis encoded by TNFAIP3 [65]. For HPV integration, an additional mechanism of immune evasion has been described; integration into CD274 results in upregulated encoded T-cell inhibitory receptor PD-L1 [34,35]. Expression of this inhibitory ligand functions as a generally recognized tumor immune escape mechanism that limits the proliferative and cytotoxic activity of cytotoxic T lymphocytes [97]. In addition to changes in host gene expression, changes in viral gene expression can contribute to immune evasion. Integration into less accessible chromatin regions might silence HIV-1 provirus and preclude antigen-presentation of viral epitopes. By contrast, integration and high amounts of viral protein expression of HBV sequences have been suggested to contribute to immune evasion by promoting T cell exhaustion of HBV-specific CD8+ T cells [98]. This figure was created with BioRender.com.
Concluding remarks
HIV-1 integration can cause changes in host cell gene expression, but it remains to be determined to what extent these effects support the clonal expansion and persistence of HIV-1 reservoir cells (Outstanding Questions Box). Functional contributions of genomic integrations to viral persistence might hold potential to affect HIV-1 therapy (Box 4). It is possible that HIV-1 integration can confer fitness advantages in ways as diverse as those that influence clonal dynamics in cancer – including mechanisms that impair immune recognition or activate survival/ antiapoptotic and proliferative pathways. Alternatively, the functional implications of integration may generally be too subtle to manifest, and reservoir clonal dynamics may instead be dominated by the relative propensity of a provirus to stay hidden in latency and by normal T-cell biology. However, recent evidence for intact and transcriptionally active provirus in six PLWH argue against the completeness of the former explanation [38,71], and the integration sites of these particular clones should be prioritized for further study. In contrast to the oncogenic viruses discussed here, any such HIV-1-infected clone would remain an exceptionally small fraction of a biological sample - complicating functional characterization (typically, only 10–1,000 cells per 106 CD4+ T cells harbor an intact provirus in an ART-treated individual)[72]. However, we propose that this can be approached in two ways: i) Adapting and applying emerging single-cell technologies to identify and directly characterize these rare cells, along with their integration sites or ii) Applying CRISPR-Cas9 or related approaches to recreate specific integration sites in a population of cells that can then be studied, as has been achieved for BACH2 and STAT5B integration sites [7]. We believe that the challenges inherent in these approaches are worth tackling given the central importance of understanding and counter-acting factors that favor clonal expansion towards the goal of curing HIV-1 infection.
Outstanding Questions Box.
How predominant are virus integration-site mediated effects on gene expression within the HIV-1 reservoir? It will be relevant to determine how many cells in a given individual rely on integration-dependent effects for their persistence.
To which extent does HIV-1 integration imprint infected cells with an epigenetic and transcriptional footprint? More research to determine the strength and maintenance of integration-dependent effects is warranted.
How much do the mechanisms of HIV-1 integration in the HIV-1 reservoir differ between PLWH? Does this heterogeneity need to be considered for HIV-1 cure strategies?
What is the contribution of HIV-1 integration-dependent changes in cellular gene expression to the proliferation and survival of HIV-1 infected cells?
Is HIV-1 affecting the host gene transcriptome by integration-dependent molecular mechanisms similarly to HPV, HBV, EBV, and HTLV-1?
Which repercussions, other than cellular proliferation, does HIV-1 integration cause?
Do HIV-1 integration-mediated effects increase their clinical relevance as the population of PLWH on ART ages? Other viruses that integrate into the human DNA only cause disease decades after integration, and diseases only affect a small number of chronically infected individuals.
Which cooperating factors support cellular consequences of integration site-dependent mechanisms? Given the rarity of malignancies caused by HIV-1 integration, it is highly likely that HIV-1 integration on its own is not sufficient to result in cellular consequences.
Can integration site-dependent mechanisms be detected before cellular consequences ensue? Certain integration sites of HPV, HBV, and HIV-1 have been linked to cellular proliferation. However, most of the viral integration sites are seeded early during infection. This might open the possibility of prophylactic integration site analysis to assess personalized medicine approaches.
Box 4: Possible Implications for the occurrence of HIV-1 Integration Sites.
If the integration of HIV-1 proviruses can functionally contribute to the abilities of reservoir-harboring clones to persist, these functional characteristics might form the basis of new putative therapeutic targets for treating HIV-1 infections. In the case that this is observed and converges upon a relatively narrow set of mechanisms, such therapies might be broadly applicable upon robust testing. Alternatively, various therapeutic approaches might be needed to target those persisting clones with a fitness advantage. The analysis of integration-dependent mechanisms at play in a specific HIV-1-positive individual might thus allow for individualized treatment approaches. To eradicate a clone that comprises a dominant fraction of the HIV-1 reservoir, therapies might conceivably be directed to the cells harboring a specific integration site, e.g. based on the overexpression of certain biomarkers (if known) or based on the integration site itself. The clinical consequences of HIV-1 integration are not yet part of an HIV-1 infection diagnosis but integration site analysis of PLWH is technically feasible and might perhaps become part of diagnosis in the future, if the prevalence of integration-induced effects (see Outstanding Questions), warrants such measures. Since the infections that result in the integration sites that make up the HIV-1 reservoir occur early upon HIV-1 acquisition [99], it might be possible to detect the integration site resulting in easy viral reactivation, clonal expansion, or even T cell lymphomas many years before viral reactivation or T cell lymphoma onset, although this remains conjectural. Recent work showed that HIV-1–1-driven aberrant transcription can be suppressed in vitro by CRISPR-dCas9-mediated inhibition of HIV-1 5′ LTR targeted CRISPR-CAS9 to specific regions in LTR regions [16]’ this exemplifies that excision of such sequences could drive aberrant host gene expression -- a technology that has since been refined and applied numerously, and to other HIV-1 regions [100,101].
The most commonly reported integration site-dependent effects observed in HIV-1 are (1) HIV-1 promoter or enhancer insertion resulting in host gene activation [6,8,9,16], (2) virus-host chimeric transcription resulting in chimeric RNA with potentially altered host gene expression and function [6,16], (3) HIV-1-induced activation of cryptic host splice sites [8,16], and (4) transcriptional interference [7,16]. While not yet observed for HIV-1, other well characterized molecular effects of viral integration are (5) 3′-untranslated region (UTR) substitution [42–44], (6) the remodeling of the epigenetic landscape [19,24–26], (7) the induction of genomic instability [27,28,32], as well as (8) host gene disruption (Figure 1) [23,29,30,45].
Significance box.
Integration of HIV-1 into the DNA of CD4+ T cells can alter cellular gene expression but the contribution of this mechanism to the clonal expansion and persistence of HIV-1 infected cells remains unclear.
Highlights Box.
HIV-1 provirus integration can affect host gene expression at the site of integration and directly induce benign but also in rare cases, malignant cellular proliferation
HIV-1 can affect cellular gene expression through promoter/enhancer insertion, the formation of virus-host chimeric RNAs, the activation of cryptic cellular splice sites, as well as transcriptional interference
Multiple human pathogenic viruses, including HTLV-1, HBV, EBV, and HPV, can affect human gene expression upon viral genomic integration by similar mechanisms as HIV-1
Diverse cellular consequences of viral integration-mediated changes in host gene expression have recently been observed for HTLV-1, HBV, EBV, and HPV, but not yet for HIV-1
Observations of diverse virus-induced changes in host gene expression in the context of other human viruses can serve as a signpost for diverse persistence mechanisms that remain to be elucidated in the context of HIV-1 infection
Acknowledgments
NL and RBJ are supported by the National Institute of Allergy and Infectious Diseases of the NIH under awards UM1AI164565 (to RBJ), R01AI165301, UM1AI164562, R01AI147845, and R01AI131798 (to RBJ). Awards UM1AI164565 and UM1AI164562 were also supported by the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Neurological Disorders and Stroke, the National Institute on Drug Abuse, and the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- CFS
Chromosome fragile sites; AT-rich sequences associated with a higher frequency of deletions, rearrangements, chromosomal translocations, and recombination.
- CpG islands
DNA regions in which a cytosine nucleotide is followed by a guanine nucleotide in a linear repetitive sequence of bases along the 5’ → 3’ direction of a DNA strand.
- Clone
group of cells within the HIV-1 reservoir that arose from clonal expansion of a single CD4+ T cell carrying integrated provirus; all cells within a clone can be identified based on their identical genome, including the HIV-1 integration site and the TCR sequence.
- EBV
Epstein-Barr virus; human herpesvirus with a dsDNA genome that infects B cells. EBV usually persists as a latent chromatized episome, but its full-length genome can also integrate into the human genome, which is observed in various EBV-associated malignancies
- Genomic instability
high frequency of mutations, which can refer to base-pair mutations but also large-scale changes in chromosomal structure.
- Hepadnavirus
Class of viruses with a DNA genome that transcribe their genome first into RNA before integrating a reverse-transcribed DNA copy of their genome into the DNA of the host cell. The best-characterized human pathogenic hepadnavirus is HBV.
- Hi-C
high-throughput genomic analysis method which captures 3D chromatin interactions.
- HIV-1 reservoir
Infected CD4+ T cells harboring intact or defective HIV-1 provirus that persist under ART; can give rise to viremia when therapy is interrupted.
- HBV
Hepatitis B virus; Hepadnavirus causing acute and chronic hepatitis B; can underlie hepatocellular carcinoma (HCC). Most people developing chronic hepatitis or HCC are infected at birth.
- HPV
Human papillomavirus; DNA viruses that, depending on the type, infect the human skin or mucosal epithelia; can cause various malignancies including cervical and oropharyngeal cancer. Most HPV types maintain viral DNA as circular episome copies, particularly invasive, high-risk HPV16 and HPV18; they integrate fragmented double stranded DNA (dsDNA) genome into the host cell in over 80% of HPV-positive cervical cancers.
- HLTV-1
Human T-cell lymphotropic virus type 1, deltaretrovirus; causes latent infection but can reactivate to cause various pathologies later in , including adult T-cell lymphoma in around 10% of infected individuals
- Intron retention
type of alternative splicing; keeps an intronic region as part of a spliced gene product.
- Insertional mutagenesis
Changes in gene sequence or expression caused by the insertion of a viral (or other foreign) sequence.
- Promoter or enhancer insertion
transcriptional activation of a cellular gene driven by a proviral promoter or enhancer inserted in proximity to the host gene.
- Provirus
Viral genome inserted into the genome of the host cell; for retroviruses, it also comprises the non-integrated reverse-transcribed dsDNA.
- Readthrough transcription
continues beyond the terminal site where RNA polymerase usually dissociates from the nascent RNA and DNA template.
- Recurrent integration gene (RIG)
enriched in proviral integration sites across studies and biological samples.
- “Roadblock” transcriptional interference
occurs in a protein-bound promoter that obstructs the progress of a transcription-elongation complex driving a transcriptional event initiated by another promoter.
- Topologically associating domains (TADs)
Chromatin domains encompassing DNA sequences that more frequently physically interact with one another than with DNA sequences from neighboring chromatin domains.
- Transcriptional interference
cis effect in which transcription impacts a second transcriptional process.
- Virus-host chimeric RNA
fused/hybrid virus-host cell transcripts regulated by either a viral or host promoter.
- 3′-UTR substitution
Mutation disrupting the 3′-UTR region of a gene, thus abolishing microRNA binding to 3′-UTR that would otherwise promote mRNA degradation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no conflict of interest.
References
- 1.Chun T-W et al. (1999) Re-emergence of HIV after stopping therapy. Nature 401, 874–875 [DOI] [PubMed] [Google Scholar]
- 2.Lee GQ et al. (2017) Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 127, 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bui JK et al. (2017) Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLOS Pathogens 13, e1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohn LB et al. (2015) HIV-1 Integration Landscape during Latent and Active Infection. Cell 160, 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seiki M et al. (1984) Nonspecific integration of the HTLV provirus genome into adult T-cell leukaemia cells. Nature 309, 640–642 [DOI] [PubMed] [Google Scholar]
- 6.Mellors JW et al. (2021) Insertional activation of STAT3 and LCK by HIV-1 proviruses in T cell lymphomas. Science Advances 7, eabi8795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christian ML et al. (2022) CRISPR/Cas9-Mediated Insertion of HIV Long Terminal Repeat within BACH2 Promotes Expansion of T Regulatory–like Cells. The Journal of Immunology 208, 1700–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cesana D et al. (2017) HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat Commun 8, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon JK et al. (2020) HIV proviral DNA integration can drive T cell growth ex vivo. Proceedings of the National Academy of Sciences 117, 32880–32882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner TA et al. (2014) HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345, 570–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maldarelli F et al. (2014) Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science at 345:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simonetti FR et al. (2021) Antigen-driven clonal selection shapes the persistence of HIV-1-infected CD4+ T cells in vivo. J Clin Invest 131, 145254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinzone MR et al. (2019) Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat Commun 10, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chomont N et al. (2009) HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15, 893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collora JA et al. (2022) Single-cell multiomics reveals persistence of HIV-1 in expanded cytotoxic T cell clones. Immunity. S1074–7613(22)00127–3. DOI: 10.1016/j.immuni.2022.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R et al. (2020) Single-cell transcriptional landscapes reveal HIV-1–driven aberrant host gene transcription as a potential therapeutic target. Science Translational Medicine 16, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sung W-K et al. (2012) Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44, 765–769 [DOI] [PubMed] [Google Scholar]
- 18.Lau C-C et al. (2014) Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 25, 335–349 [DOI] [PubMed] [Google Scholar]
- 19.Melamed A et al. (2018) The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis. Elife 7, e36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brant AC et al. (2020) Preferential expression of a HPV genotype in invasive cervical carcinomas infected by multiple genotypes. Genomics 112, 2942–2948 [DOI] [PubMed] [Google Scholar]
- 21.Muroyama R et al. (2022) Fusion HBx from HBV Integrant Affects Hepatocarcinogenesis through Deregulation of ER Stress Response. Virus Res . 315:198787. [DOI] [PubMed] [Google Scholar]
- 22.Warburton A et al. (2018) HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet 14, e1007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosewick N et al. (2017) Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis. Nat Commun 8, 15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melamed A et al. (2022) Selective clonal persistence of human retroviruses in vivo: Radial chromatin organization, integration site, and host transcription. Science Advances 8, eabm6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groves IJ et al. (2021) Short- and long-range cis interactions between integrated HPV genomes and cellular chromatin dysregulate host gene expression in early cervical carcinogenesis. PLoS Pathog 17, e1009875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satou Y et al. (2016) The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc Natl Acad Sci U S A 113, 3054–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akagi K et al. (2014) Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 24, 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii T et al. (2020) Analysis of HBV Genomes Integrated into the Genomes of Human Hepatoma PLC/PRF/5 Cells by HBV Sequence Capture-Based Next-Generation Sequencing. Genes 11, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collot-Teixeira S et al. (2004) Human tumor suppressor p53 and DNA viruses. Rev Med Virol 14, 301–319 [DOI] [PubMed] [Google Scholar]
- 30.Chakravorty S et al. (2019) Integrated Pan-Cancer Map of EBV-Associated Neoplasms Reveals Functional Host–Virus Interactions. Cancer Research 79, 6010–6023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao L-H et al. (2016) Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun 7, 12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Buuren N et al. (2022) Targeted long-read sequencing reveals clonally expanded HBV-associated chromosomal translocations in patients with chronic hepatitis B. JHEP Rep 4, 100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parfenov M et al. (2014) Characterization of HPV and host genome interactions in primary head and neck cancers. Proceedings of the National Academy of Sciences 111, 15544–15549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koneva LA et al. (2018) HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Molecular Cancer Research 16, 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Symer DE et al. (2022) Diverse tumorigenic consequences of human papillomavirus integration in primary oropharyngeal cancers. Genome Res 32, 55–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warren JA et al. (2020) The HIV-1 latent reservoir is largely sensitive to circulating T cells. Elife 9, e57246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas AS et al. (2017) T-cell responses targeting HIV Nef uniquely correlate with infected cell frequencies after long-term antiretroviral therapy. PLoS Pathog 13, e1006629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Einkauf KB et al. (2022) Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 185:266–282.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang S-H et al. (2018) Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest 128, 876–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren Y et al. (2020) BCL-2 antagonism sensitizes cytotoxic T cell-resistant HIV reservoirs to elimination ex vivo. J Clin Invest 130, 2542–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo H-H et al. (2018) Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4+ T Cells. Immunity 48, 1183–1194.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavazzana-Calvo M et al. (2010) Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467, 318–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng Y et al. (2008) Antiproliferative Effects by Let-7 Repression of High-Mobility Group A2 in Uterine Leiomyoma. Molecular Cancer Research 6, 663–673 [DOI] [PubMed] [Google Scholar]
- 44.Lee S et al. (2007) Dok1 expression and mutation in Burkitt’s lymphoma cell lines. Cancer Lett 245, 44–50 [DOI] [PubMed] [Google Scholar]
- 45.Bushman FD (2020) Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Molecular Therapy 28, 352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montini E et al. (2006) Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol 24, 687–696 [DOI] [PubMed] [Google Scholar]
- 47.Emery A and Swanstrom R (2021) HIV-1: To Splice or Not to Splice, That Is the Question. Viruses 13, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katzav S et al. (1991) Loss of the amino-terminal helix-loop-helix domain of the vav proto-oncogene activates its transforming potential. Mol Cell Biol 11, 1912–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lenasi T et al. (2008) Transcriptional Interference Antagonizes Proviral Gene Expression to Promote HIV Latency. Cell Host & Microbe 4, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y et al. (2008) Orientation-Dependent Regulation of Integrated HIV-1 Expression by Host Gene Transcriptional Readthrough. Cell Host & Microbe 4, 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazo A et al. (2007) Transcriptional interference: an unexpected layer of complexity in gene regulation. Journal of Cell Science 120, 2755–2761 [DOI] [PubMed] [Google Scholar]
- 52.Shearwin KE et al. (2005) Transcriptional interference – a crash course. Trends Genet 21, 339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guan Y et al. (2022) Comprehensive analysis of DOK family genes expression, immune characteristics, and drug sensitivity in human tumors. Journal of Advanced Research 36, 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayr C (2019) What Are 3′ UTRs Doing? Cold Spring Harb Perspect Biol 11, a034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Z et al. (2015) Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet 47, 158–163 [DOI] [PubMed] [Google Scholar]
- 56.Ojesina AI et al. (2014) Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adey A et al. (2013) The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagaraj N et al. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vandegraaff N and Engelman A (2007) Molecular mechanisms of HIV integration and therapeutic intervention. ERM 9, [DOI] [PubMed] [Google Scholar]
- 60.Engelman AN and Singh PK (2018) Cellular and molecular mechanisms of HIV-1 integration targeting. Cell Mol Life Sci 75, 2491–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang D et al. (2020) VISDB: a manually curated database of viral integration sites in the human genome. Nucleic Acids Res 48, D633–D641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janjetovic S et al. (2022) Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses 14, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anisenko AN and Gottikh MB (2019) Role of Cellular DNA Repair Systems in HIV-1 Replication. Mol Biol 53, 313–322 [DOI] [PubMed] [Google Scholar]
- 64.Álvarez EG et al. (2021) Aberrant integration of Hepatitis B virus DNA promotes major restructuring of human hepatocellular carcinoma genome architecture. Nat Commun 12, 6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu M et al. (2019) Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics 9, 1115–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cameron DL et al. (2021) VIRUSBreakend: Viral Integration Recognition Using Single Breakends. Bioinformatics 37, 3115–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin SY et al. (2021) Recurrent HBV Integration Targets as Potential Drivers in Hepatocellular Carcinoma. Cells 10, 1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho Y-C et al. (2013) Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pollack RA et al. (2017) Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 21, 494–506.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tchasovnikarova IA et al. (2015) GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stevenson EM et al. (2021) HIV-specific T cell responses reflect substantive in vivo interactions with antigen despite long-term therapy. JCI Insight 6:e142640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bruner KM et al. (2019) A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566, 120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tempera I et al. (2010) CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6, e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dias JD et al. (2022) Crosstalk between Hepatitis B Virus and the 3D Genome Structure. Viruses 14, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paris C et al. (2015) CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J Virol 89, 4770–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ong C-T and Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15, 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu X et al. (2021) HPV16-LINC00393 Integration Alters Local 3D Genome Architecture in Cervical Cancer Cells. Front Cell Infect Microbiol 11, 785169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cao C et al. (2020) HPV-CCDC106 integration alters local chromosome architecture and hijacks an enhancer by three-dimensional genome structure remodeling in cervical cancer. Journal of Genetics and Genomics 47, 437–450 [DOI] [PubMed] [Google Scholar]
- 79.Adeel MM et al. (2021) Structural Variations of the 3D Genome Architecture in Cervical Cancer Development. Frontiers in Cell and Developmental Biology 9:706375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang B et al. (2020) 3D landscape of Hepatitis B virus interactions with human chromatins. Cell Discov 6, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moreau P et al. (2018) Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat Commun 9, 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Katano H et al. (2007) Integration of HIV-1 caused STAT3-associated B cell lymphoma in an AIDS patient. Microbes and Infection 9, 1581–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Warburton A et al. (2021) Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. npj Genom. Med 6, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Niederer HA et al. (2014) HTLV-1 proviral integration sites differ between asymptomatic carriers and patients with HAM/TSP. Virology Journal 11, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zapatka M et al. (2020) The landscape of viral associations in human cancers. Nat Genet 52, 320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hai H et al. (2014) Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World Journal of Gastroenterology 20, 6236–6243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Coffin JM et al. (2021) Integration in oncogenes plays only a minor role in determining the in vivo distribution of HIV integration sites before or during suppressive antiretroviral therapy. PLOS Pathogens 17, e1009141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yeh Y-HJ et al. (2021) The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence. Viruses 13, 1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maldarelli F (2016) The role of HIV integration in viral persistence: no more whistling past the proviral graveyard. Journal of Clinical Investigation 126, 438–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bedwell GJ et al. (2021) rigrag: high-resolution mapping of genic targeting preferences during HIV-1 integration in vitro and in vivo. Nucleic Acids Res 49, 7330–7346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haworth KG et al. (2018) HIV infection results in clonal expansions containing integrations within pathogenesis-related biological pathways. JCI Insight 3, 99127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Satou Y et al. (2017) Dynamics and mechanisms of clonal expansion of HIV-1-infected cells in a humanized mouse model. Sci Rep 7, 6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patro SC et al. (2019) Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proceedings of the National Academy of Sciences 116, 25891–25899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ke W et al. (2022) miR-134–5p promotes inflammation and apoptosis of trophoblast cells via regulating FOXP2 transcription in gestational diabetes mellitus. Bioengineered 13, 319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li Q et al. (2004) A death receptor-associated anti-apoptotic protein, BRE, inhibits mitochondrial apoptotic pathway. J Biol Chem 279, 52106–52116 [DOI] [PubMed] [Google Scholar]
- 96.Li J et al. (2020) TNFAIP2 Promotes Non-Small Cell Lung Cancer Cells and Targeted by miR-145–5p. DNA Cell Biol 39, 1256–1263 [DOI] [PubMed] [Google Scholar]
- 97.Pai SI et al. (2016) The role of antagonists of the PD-1:PD-L1/PD-L2 axis in head and neck cancer treatment. Oral Oncology 61, 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pollicino T and Caminiti G (2021) HBV-Integration Studies in the Clinic: Role in the Natural History of Infection. Viruses 13, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coffin JM et al. (2019) Clones of infected cells arise early in HIV-infected individuals. JCI Insight 4, 128432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ebina H et al. (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 3, 2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Z et al. (2022) Updates on CRISPR-based gene editing in HIV-1/AIDS therapy. Virologica Sinica 37, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin D-C et al. (2014) The genomic landscape of nasopharyngeal carcinoma. Nat Genet 46, 866–871 [DOI] [PubMed] [Google Scholar]
- 103.Zheng H et al. (2016) Whole-exome sequencing identifies multiple loss-of-function mutations of NF-κB pathway regulators in nasopharyngeal carcinoma. Proc Natl Acad Sci U S A 113, 11283–11288 [DOI] [PMC free article] [PubMed] [Google Scholar]