

This cohort study aims to elucidate the genetic spectrum of uncombable hair syndrome among 107 included patients.
Key Points
Question
What is the genetic spectrum of uncombable hair syndrome (UHS)?
Findings
This cohort study of 107 index patients describes pathogenic variants in 80 individuals with a suspected diagnosis of UHS in the genes PADI3, TCHH, and TGM3. Of these individuals, 76 carried 12 different pathogenic variants in PADI3, 2 carried 3 pathogenic variants in TCHH, and 2 carried 3 pathogenic variants in TGM3; a haplotype analysis suggested a founder effect for the 4 most common pathogenic PADI3 variants.
Meaning
This study expands the genotypic spectrum of UHS and establishes that 2 pathogenic missense variants in PADI3 account for the majority of the UHS cases.
Abstract
Importance
Uncombable hair syndrome (UHS) is a rare hair shaft anomaly that manifests during infancy and is characterized by dry, frizzy, and wiry hair that cannot be combed flat. Only about 100 known cases have been reported so far.
Objective
To elucidate the genetic spectrum of UHS.
Design, Setting, and Participants
This cohort study includes 107 unrelated index patients with a suspected diagnosis of UHS and family members who were recruited worldwide from January 2013 to December 2021. Participants of all ages, races, and ethnicities were recruited at referral centers or were enrolled on their own initiative following personal contact with the authors. Genetic analyses were conducted in Germany from January 2014 to December 2021.
Main Outcomes and Measures
Clinical photographs, Sanger or whole-exome sequencing and array-based genotyping of DNA extracted from blood or saliva samples, and 3-dimensional protein modeling. Descriptive statistics, such as frequency counts, were used to describe the distribution of identified pathogenic variants and genotypes.
Results
The genetic characteristics of patients with UHS were established in 80 of 107 (74.8%) index patients (82 [76.6%] female) who carried biallelic pathogenic variants in PADI3, TGM3, or TCHH (ie, genes that encode functionally related hair shaft proteins). Molecular genetic findings from 11 of these 80 individuals were previously published. In 76 (71.0%) individuals, the UHS phenotype were associated with pathogenic variants in PADI3. The 2 most commonly observed PADI3 variants account for 73 (48.0%) and 57 (37.5%) of the 152 variant PADI3 alleles in total, respectively. Two individuals carried pathogenic variants in TGM3, and 2 others carried pathogenic variants in TCHH. Haplotype analyses suggested a founder effect for the 4 most commonly observed pathogenic variants in the PADI3 gene.
Conclusions and Relevance
This cohort study extends and gives an overview of the genetic variant spectrum of UHS based on molecular genetic analyses of the largest worldwide collective of affected individuals, to our knowledge. Formerly, a diagnosis of UHS could only be made by physical examination of the patient and confirmed by microscopical examination of the hair shaft. The discovery of pathogenic variants in PADI3, TCHH, and TGM3 may open a new avenue for clinicians and affected individuals by introducing molecular diagnostics for UHS.
Introduction
Uncombable hair syndrome (UHS 1-3; OMIM 191480, 617251, 617252, respectively) is a rare, autosomal recessive disorder characterized by an abnormal hair shaft.1,2,3 Dermatologists also refer to UHS as spun glass hair syndrome or pili trianguli et canaliculi. Children with UHS present with dry, frizzy, wiry, and often light-colored, shiny hair that stands out from the scalp and cannot be combed flat. Uncombable hair syndrome usually manifests in infancy or early childhood, frequently improves with age, and in some cases, by adulthood the UHS phenotype becomes barely recognizable. Nevertheless, UHS often leaves a lasting imprint on the childhood years of the affected owing to pejorative responses to their unusual appearance. Although additional clinical features have been reported, such as ectodermal dysplasia, retinitis pigmentosa, juvenile cataract, and polydactyly, UHS typically manifests as an isolated hair anomaly. No medical therapy is yet available.
In 2016, we published, to our knowledge, the first investigation into the molecular genetics of UHS.1 This investigation included 18 individuals with UHS (13 females and 5 males) and identified biallelic variants in the genes PADI3 (OMIM 606755), TCHH (OMIM 190370), and TGM3 (OMIM 600238) in 11 individuals. PADI3, TCHH, and TGM3 encode proteins whose sequential interactions play a role in the formation of the hair shaft.4,5 PADI3 is responsible for the deimination of the positively charged l-arginine residues of proteins into neutral citrulline residues in the presence of calcium ions.5 One of the main substrates of PADI3 is the structural hair shaft protein trichohyalin (TCHH). Deimination by PADI3 reduces the overall charge of TCHH and thus enables its subsequent interaction with the intermediate keratin filaments. The citrullinated-TCHH and keratin filaments are then cross-linked by the TGM3 enzyme, a process that is necessary for the stabilization and hardening of the hair shaft.4,5,6,7 Disturbance of this cascade leads to a deficiency in the shaping and mechanical strengthening of the hair shaft, thus resulting in the UHS phenotype.1
Prior to our 2016 study,1 the assignment of a clinical diagnosis of UHS was mainly based on signs detected during a clinical examination, with confirmation via shrinking-tube technique light microscopy or scanning electron microscopy of hair shaft cross sections.8 In at least 50% of examined hair samples, scanning electron microscopy reveals longitudinal grooves along the entire length of the hair shaft and a triangular or heart-shaped outline in the cross-section, as opposed to the normal circular shape.9 As a direct consequence of our study,1 we now base the assignment or confirmation of a clinical diagnosis of UHS on molecular genetic diagnostics.
At the time of our 2016 report,1 around 100 cases of UHS had been reported. Subsequent to our report, we were contacted by a large number of clinicians and members of the public from across the world with information on further possible UHS cases. Several of the individuals from the general public reported having had a history of UHS symptoms during childhood, without the assignment of a clinical diagnosis. As a result of these contacts, EDTA blood samples, saliva, or DNA were sent to our laboratory from 89 unrelated index patients. For 51 of these patients, samples were also received from affected and/or unaffected family members. We obtained DNA to screen an affected sibling or an affected parent in 7 and 2 families, respectively. Herein, we report the results of the molecular genetic analyses.
Methods
Participants
All study procedures were performed in accordance with the principles of the Declaration of Helsinki. Ethical approval was obtained from the ethics committee of the Medical Faculty of the University of Bonn, and all participants, or legal guardians in the case of minors, provided written informed consent prior to blood sampling. The countries of origin of the respective individuals who were genetically elucidated are presented in relation to the identified pathogenic variants to provide an overview of the diversity of the UHS-related genetic variation spectrum worldwide. Note that this information is not equivalent to race or ethnicity. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Genetic Analyses
Sanger sequencing was performed using the BigDye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems) and an ABI 3100 genetic analyzer (Applied Biosystems). Exome sequencing was performed at the Cologne Center for Genomics in Cologne, Germany. Data generation and filtering procedures are described in detail in the eMethods in the Supplement. Haplotype analysis was performed for the 4 most commonly observed pathogenic variants in PADI3 using single-nucleotide polymorphism (SNP) chip data as described in detail in the eMethods in the Supplement.
Results
In addition to the 18 individuals reported in our 2016 study,1 genetic screening of an additional 89 unrelated individuals with suspected UHS (69 females and 20 males) led to the identification of pathogenic variants in 69 of them. The affected individuals presented with the typical dry, frizzy, and shiny hair that was difficult to be combed flat (Figure 1 and eFigure 1 in the Supplement). Sixty-seven of these individuals harbored recurrent and/or novel pathogenic variants in PADI3 in either a homozygous or a compound heterozygous state, and 2 individuals carried previously unreported compound heterozygous pathogenic variants in either TGM3 or TCHH. From the newly elucidated 67 individuals with pathogenic PADI3 variants, 56 carried 1 or 2 of the 4 variants reported in our 2016 study.1 In the remaining 11 individuals, the following 8 previously unreported pathogenic PADI3 (NM_016233.2) variants were identified: c.60_66delinsTGCTTGG (p.Gly22Trp); c.505C>T (p.Gln169*); c.556C>T (p.Arg186Trp); c.652G>A (p.Gly218Ser); c.1114A>G (p.Arg372Gly); c.1115G>T (p.Arg372Met); c. 1183del (p.Glu395Asnfs*7); and c.1868C>T (p.Pro623Leu) (Figure 2). This expands the UHS-associated variant spectrum of PADI3 to a total of 12 pathogenic variants identified in individuals from 20 countries (eTable 1 in the Supplement). Of these, 9 lead to single amino acid substitutions, 2 are nonsense variants, and 1 is a frameshift variant leading to a premature stop codon.
Figure 1. Clinical Manifestation of Uncombable Hair Syndrome (UHS).

The typical manifestation of UHS with shiny, frizzy, and dry hair can be observed in 8 individuals carrying biallelic pathogenic variants in the PADI3 gene. See eFigure 1 in the Supplement for more individuals (n = 22) with their respective genotypes.
Figure 2. Additional Pathogenic Variants in PADI3.
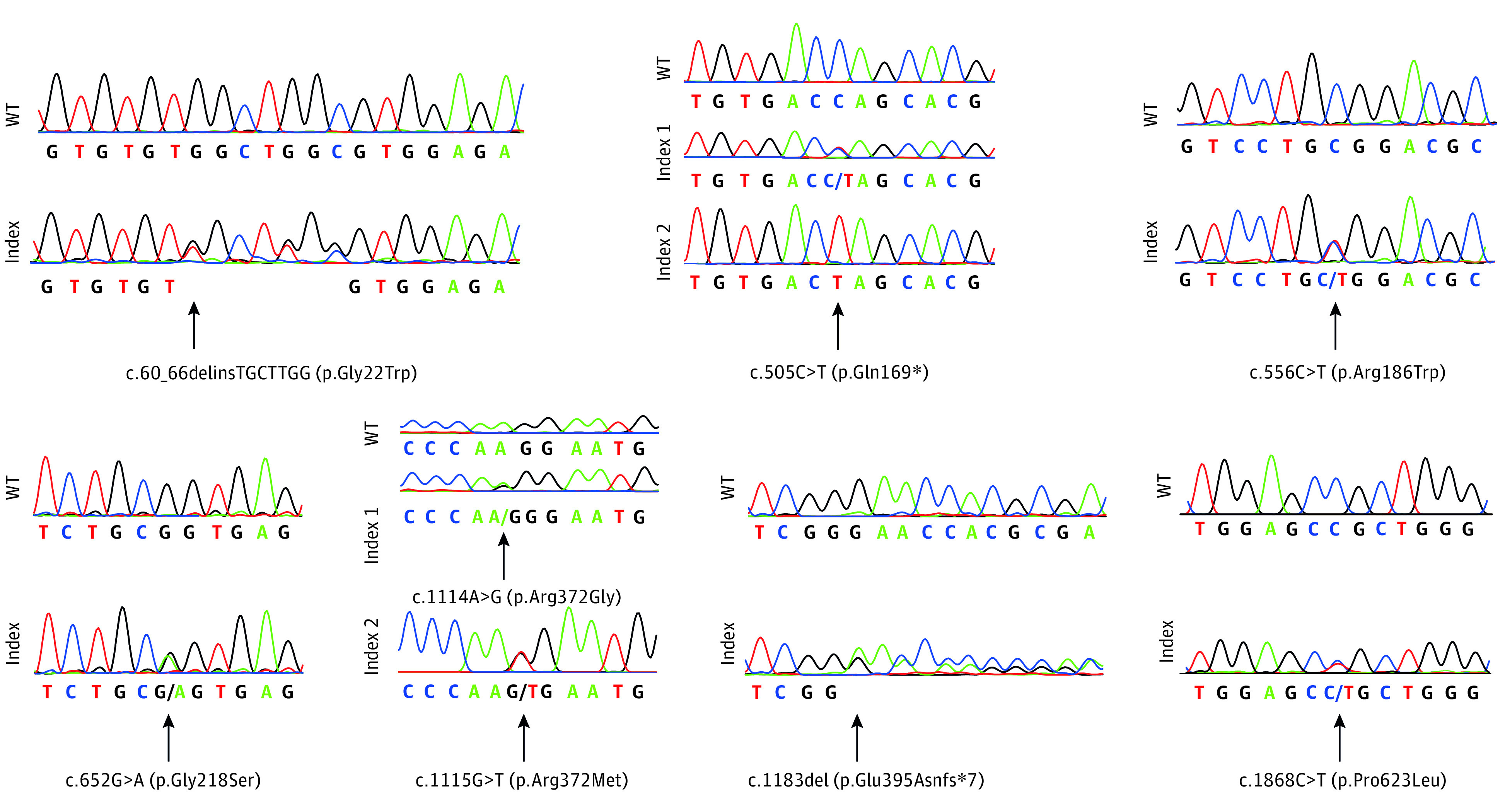
Electropherograms showing the newly discovered pathogenic variants c.60_66delinsTGCTTGG (p.Gly22Trp), c.505C>T (p.Gln169*) in heterozygosity and homozygous state, c.556C>T (p.Arg186Trp), c.652G>A (p.Gly218Ser), c.1114A>G (p.Arg372Gly), c.1115G>T (p.Arg372Met), c.1183del (p.Glu395Asnfs*7), and c.1868C>T (p.Pro623Leu) in comparison with the respective wild-type (WT) sequences. The 2 variants c.1114A>G (p.Arg372Gly) and c.1115G>T (p.Arg372Met) are located adjacently and therefore depicted below the same wild-type sequence. Of note, variant c.60_66delinsTGCTTGG (p.Gly22Trp) may also represent a rare haplotype composed of 3 single nucleotide variants, namely rs1360902614, rs1221559173, and rs1344213588, which have been observed in the same single individual (minor allele frequency = 6.5 × 10−6) in gnomAD (Genome Aggregation Database).
To examine the possible consequences of the newly discovered PADI3 variants on 3-dimensional protein structure and function, protein modeling was performed (Figure 3 and eFigure 2 in the Supplement). Interestingly, 2 of the newly discovered missense variants (p.Arg372Gly and p.Arg372Met) affect the same amino acid residue Arg372, which is involved in substrate binding via the formation of hydrogen bonds with the substrate arginine (Figure 3B). Modeling revealed that substitution of this residue by glycine or methionine results in the loss of these hydrogen bonds (Figure 3B). This is presumably associated with weaker substrate binding and lower enzyme activity.
Figure 3. Three-Dimensional Protein Modeling of Newly Discovered PADI3 Variants.
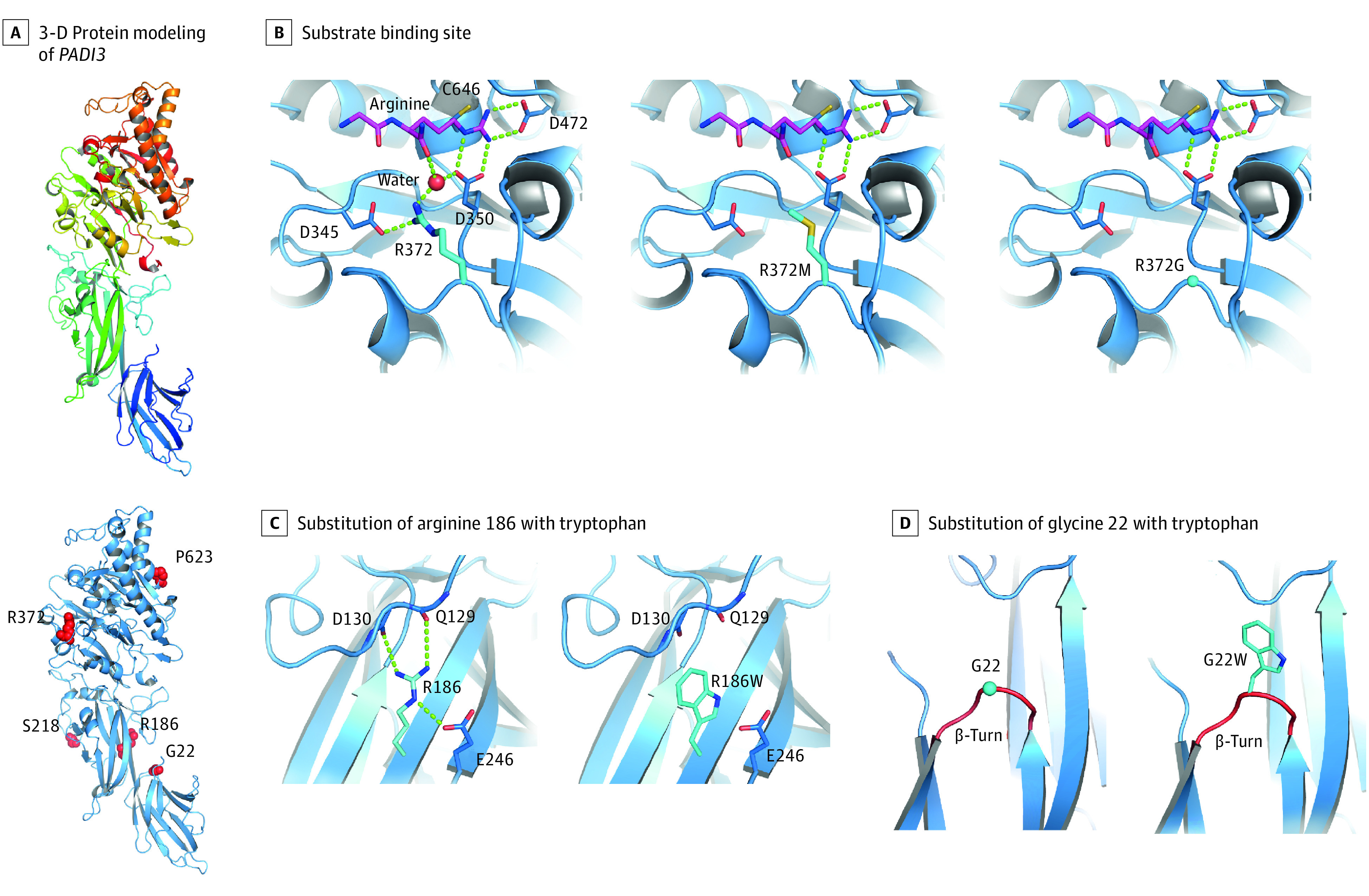
An overview of the entire 3-dimensional model of the wild-type PADI3 gene (A, upper panel) and the locations of the newly identified missense substitutions (A, bottom panel). Arginine 372 is located at the substrate binding site of PADI3 and is involved in the binding of l-arginine residues of target proteins via formation of hydrogen bonds (B, left); its substitution by a methionine (B, middle) or glycine (B, right) is expected to lead to the loss of these hydrogen bonds, thus a weaker substrate binding and lower enzyme activity. Substitution of arginine 186 in the wild-type PADI3 gene (C, left) with a tryptophan (C, right), as well as substitution of glycine 22 (D, left) with a tryptophan (D, right), are expected to destabilize PADI3 owing to loss of stabilizing hydrogen bonds (C) and insertion of a bulky residue in a β-turn loop reducing the flexibility at this site (D).
The modeling also demonstrated that 2 other missense variants have probable consequences on the stability of PADI3. More specifically, substitution of arginine 186 to tryptophan results in the loss of stabilizing hydrogen bonds (Figure 3C). Substitution of glycine 22 to tryptophan results in the insertion of a bulky residue in a β-turn (Figure 3D). β-Turn loops require a small residue allowing for flexibility. Thus, in all β-turn loops, glycine is the most frequent residue in this position. The replacement of glycine by any other amino acid residue will destabilize the protein.10 The most common previously reported pathogenic missense variants in PADI3 were also remodeled and showed that the residues Leu112, Ala294, and Pro605 are located in the hydrophobic core of the enzyme (eFigure 3 in the Supplement). The substitution of these residues by amino acids with larger side chains causes sterical clashes (eFigure 3 in the Supplement) in the hydrophobic core of the protein, and thus most likely destabilizes PADI3.
For TCHH (NM_007113.3), our 2016 study1 identified 1 individual with the pathogenic homozygous stop variant c.991C>T (p.Gln331*). In the expanded cohort, there was 1 individual identified with compound heterozygosity for 2 novel TCHH frameshift variants, each of which leads to a premature stop codon: c.2435del (p.Pro812Argfs*67) and c.4245del (p.Lys1415Asnfs*9) (Figure 4A).
Figure 4. Newly Discovered Pathogenic Variants in TCHH and TGM3.

Electropherograms showing the newly discovered pathogenic frameshift variants c.2435del (p.Pro812Argfs*67) and c.4245del (p.Lys1415Asnfs*9) in TCHH (A) and the compound heterozygous pathogenic missense variants c.1966C>T (p.Arg656Trp) and c.1993C>T (p.Arg665Trp) in TGM3 (B) in comparison with the respective wild-type (WT) sequences. Protein modeling suggests that mutation p.Arg665Trp leads to the loss of 2 hydrogen bonds destabilizing the contact of barrel 2 domain to the core domain (C). Domain organization of TGM3 and location of residues 656 and 655 is indicated (C, left). Arg655 forms hydrogen bonds to the side chain of Asp156 and the backbone carbonyl of Lys24 (C, right upper panel). The mutation of Arg655 to Trp abolishes these hydrogen bonds (C, right lower panel).
Notably, 2 individuals in the present cohort carried only 1 heterozygous loss-of-function variant in TCHH, c.991C>T (p.Gln331*) and the novel frameshift variant c.4005_4008del (p.Asp1335Glufs*88), respectively. No second pathogenic variant in the coding sequence was detected for either individual. In these 2 individuals, a plausible hypothesis is the existence of a structural variant on the other allele, the detection of which may be challenged by short-read exomes or Sanger sequencing. Until this is investigated further, the genetic cause of these 2 cases remains unclear.
In our 2016 study,1 1 individual was identified with the pathogenic homozygous stop variant c.1351C>T (p.Gln451*) in TGM3. In the expanded cohort, 1 individual was identified with 2 rare pathogenic missense variants in TGM3 (NM_003245.3) in a compound heterozygous state: c.1966C>T (p.Arg656Trp) (rs151074346 with a minor allele frequency in the Genome Aggregation Database [gnomAD] = 5.00 × 10−4) and c.1993C>T (p.Arg665Trp) (rs150949349 with a minor allele frequency in gnomAD = 3.16 × 10−3) (Figure 4B). Protein modeling suggests that the Arg665Trp variation destabilizes the contact of the C-terminal barrel 2 domain with the core domain. Arg665 forms 2 hydrogen bonds with the side chain of Asp156 and the backbone carbonyl of Lys24. Both hydrogen bonds are lost by mutation to Trp (Figure 4C). The Arg656Trp substitution will most likely not affect protein stability but will decrease the surface charge of the barrel 2 domain.
The total present cohort included 107 unrelated index patients with UHS. For 80 (74.8%) of these patients, the genetic associations have been established, while for 27 (25.2%), the pathogenic variants/associated genes remain unknown (Figure 5A). Briefly, in 76 (71.0%) individuals, the UHS phenotype was associated with biallelic variants in PADI3, while TGM3 and TCHH together (2 individuals each) were associated with only 3.8% of the UHS cases.
Figure 5. Genetic Spectrum of UHS and Common Haplotypes Surrounding the Most Common PADI3 Pathogenic Variants.
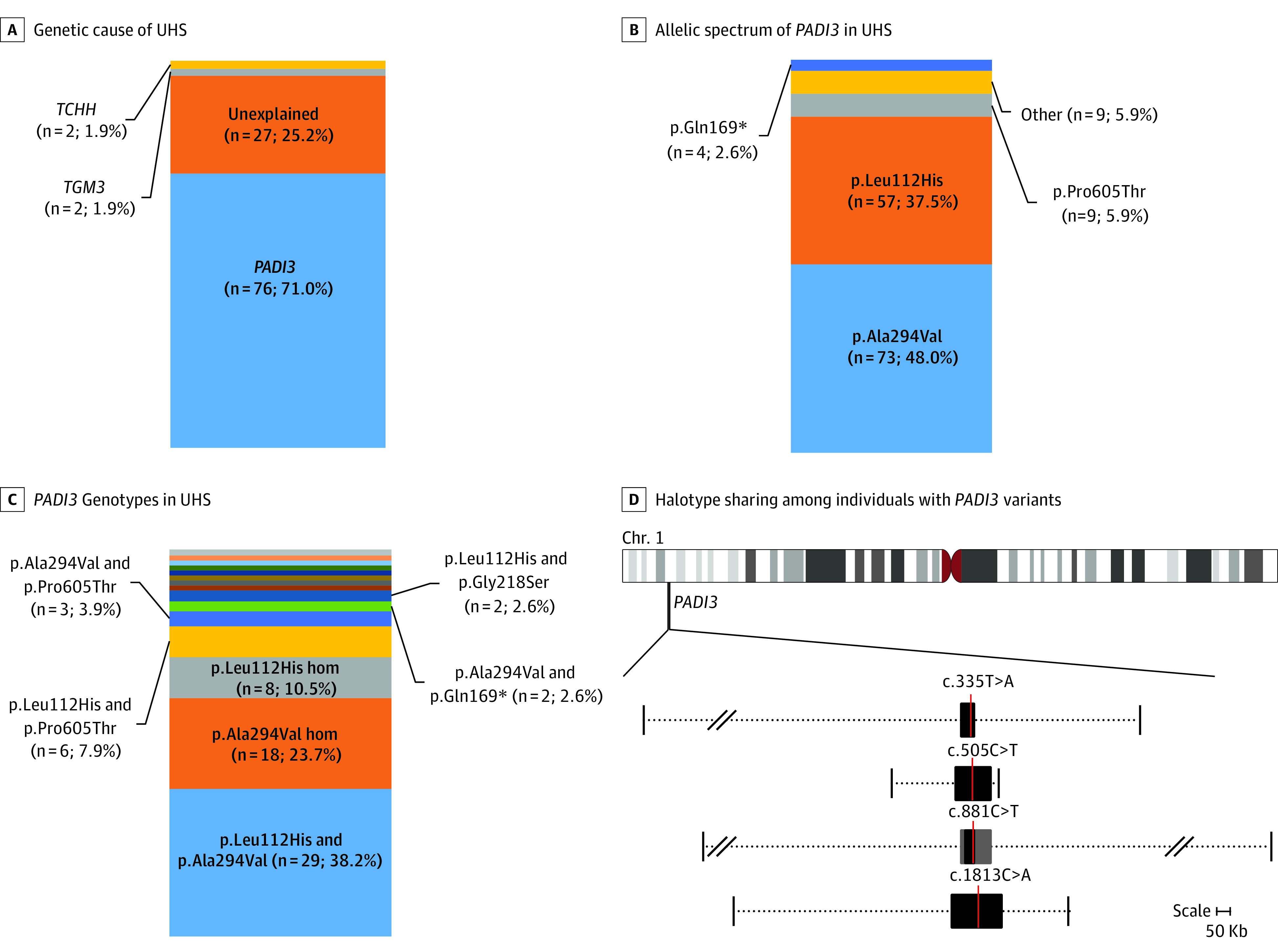
A, Stacked percentage bar plots depict the respective proportion of affected index cases that could be explained by pathogenic variants in either 1 of the 3 known uncombable hair syndrome (UHS)-associated genes and those not yet genetically elucidated within the total cohort. B, Individual pathogenic variants within all mutated PADI3 alleles (other includes p.Gly22Trp [n = 1], p.Arg186Trp [n = 1], p.Gly218Ser [n = 2], p.Arg372Gly [n = 1], p.Arg372Met [n = 1], p.Glu395Asnfs*7 [n = 1], p.Lys578* [n = 1], and p.Pro623Leu [n = 1]). C, Pathogenic genotypes within all PADI3-associated UHS cases: p.Gln169* homozygous; p.Leu112His and p.Glu395Asnfs*7; p.Leu112His and Arg186Trp; p.Leu112His and p.Arg372Met; p.Leu112His and p.Gly22Trp; p.Ala294Val and p.Arg372Gly; p.Ala294Val and p.Pro623Leu; and p.Ala294Val and p.Lys578* were singleton observations represented with different color codes without text, and each constitutes approximately 1% of the bar plot. D, The minimally shared regions across all haplotypes carrying a pathogenic variant are denoted with black boxes, and the location of the respective variations are depicted with a red line. The gray boxes surrounding c.881C>T denote a common haplotype shared over 90% of all analyzed haplotypes. The variable extent of haplotype sharing across all analyzed samples is denoted by the dotted lines, whereby the vertical lines on either end of the dotted lines mark the furthest extent of haplotype sharing between at least 2 samples upstream and downstream of the respective pathogenic variants. Slash marks show omitted whole sizes owing to space restraints. See eFigure 5 and eTables 2 through 5 in the Supplement for a depiction of haplotype sharing on individual level.
The most recurrent pathogenic PADI3 variants were c.881C>T (p.Ala294Val) and c.335T>A (p.Leu112His) (73 [48.0%] and 57 [37.5%] of 152 pathogenic PADI3 alleles identified in total, respectively; Figure 5B). Of the 76 individuals with PADI3 pathogenic variants, 75 carried either 1 or both of these variants. On the genotype level, 29 of 76 (38.2%) individuals, whose UHS was associated with PADI3, had a combination of these 2 pathogenic variants; 18 (23.7%) carried c.881C>T (p.Ala294Val) in a homozygous state, 8 (10.5%) carried c.335T>A (p.Leu112His) in a homozygous state, and 20 (26.3%) carried either one of these variants in combination with another pathogenic variant (Figure 5C). The third most commonly observed recurrent pathogenic variant, c.1813C>A (p.Pro605Thr) (9 of 152 [5.9%] pathogenic PADI3 alleles identified in total; Figure 5B), was observed in a heterozygous state in 9 of the 76 (11.8%) individuals with a PADI3 variant (Figure 5C). The newly identified stop variant c.505C>T (p.Gln169*) was the fourth most common pathogenic variant (4 of 152 [2.6%] variant alleles; Figure 5B). This was carried by 3 individuals, with 1 showing a homozygous state (Figure 5C). One missense variant, c.652G>A (p.Gly218Ser), was carried by 2 individuals, and the remaining pathogenic variants were all single heterozygous observations (Figure 5C).
Interestingly, the cohort included 3 families, which include affected parent-child duos who appear to have an autosomal dominant mode of inheritance. For 2 of these families, DNA was also received from the affected parent, and it was confirmed that they each carried 2 pathogenic PADI3 variants (eFigure 4 in the Supplement). More specifically, in 1 family both the affected mother and son carried p.Leu112His and p.Arg294Val (eFigure 4A in the Supplement). In the other family, the affected mother and daughter carried p.Leu112His in homozygosity. For the latter case, DNA was also available from the unaffected father, and he was found to be a heterozygous carrier of p.Leu112His (eFigure 4B in the Supplement). These results revealed that also in these families, a recessive inheritance was present rather than dominant.
A haplotype analysis for the 4 most common pathogenic PADI3 variants was performed using genotype data of 40 individuals from 34 families to investigate whether these variants derive from a single mutational event (common ancestor) or have arisen independently multiple times. Shared haplotype blocks of variable sizes were detected in all of the investigated individuals for each of the 4 pathogenic variants with common or individual haplotype break points (Figure 5D and eTables 2-5 and eFigure 5 in the Supplement). The minimally shared haplotype block across all haplotypes carrying c.335T>A (p.Leu112His) was 47.744 base pairs (bp) long and tagged by 19 SNPs, 11 of which were intergenic (Figure 5D). The individual shortest and longest common haplotype blocks detected were 64.919 and 1.606.067 bp long, respectively, and 2 families from Germany shared the latter with the same haplotype break points (eTable 3 and eFigure 5 in the Supplement). Haplotype sharing between at least 2 individuals was observed up to 466 and 169 SNPs upstream and downstream of the pathogenic variant, respectively. Notably, several common haplotype breakpoints were observed across multiple individuals both upstream and downstream of the site of the pathogenic variation (eTable 3 and eFigure 5 in the Supplement).
The minimally shared haplotype block across all haplotypes carrying c.881C>T (p.Ala294Val) was 34.820 bp long and tagged by 11 SNPs, 10 of which were intragenic (Figure 5D). Notably, a larger block of 99.064 bp long was shared between 31 of the 34 (91.1%) investigated haplotypes carrying this variant. The individual shortest and longest common haplotype blocks detected were 51.669 and 1.674.521 bp long, respectively. Haplotype sharing between at least 2 individuals was observed up to 378 and 306 SNPs upstream and downstream of the pathogenic variant, respectively, and similar to c.335T>A (p.Leu112His); common breakpoints were observed on either side of the pathogenic variant (eTable 4 and eFigure 5 in the Supplement).
For the third most common pathogenic variant c.1813C>A (p.Pro605Thr), 4 haplotypes were analyzed (eTable 5 in the Supplement). The minimally shared haplotype block across them was 162.538 bp long (Figure 5D), exceeding PADI3 and extending over the neighboring genes PADI1 and PADI4. Haplotype sharing between at least 2 individuals was observed up to 230 and 88 SNPs upstream and downstream of the pathogenic variant, respectively. The minimally shared haplotype block across all haplotypes carrying c.505C>T (p.Gln169*) was 116.238 bp long (Figure 5D) and also extended over the neighboring genes PADI1 and PADI4. Haplotype sharing between at least 2 individuals was observed up to 144 and 34 SNPs upstream and downstream of the pathogenic variant, respectively (eTable 2 in the Supplement). Altogether, these results support the founder mutation hypothesis for each of the 4 analyzed pathogenic variants.
Discussion
Taken together, the present results show that in the majority of the present cohort, UHS was associated with pathogenic variants in the gene PADI3, including 2 recurrent variants that were detected in around 99% of all PADI3 variant carriers. The present study led to discovery of 8 novel pathogenic variants in PADI3. Of these, 6 were single observations and 2 were observed in 3 and 2 individuals, respectively. Therefore, we anticipate that other very rare pathogenic variants in PADI3 exist and that these are likely to be discovered in the future.
Remarkably, in 3 of the newly referred families, the examination of exome sequencing data revealed pathogenic variants in either LIPH (2 families) or LPAR6 (1 family) genes (ie, genes underlying hypotrichosis simplex with woolly hair). This suggests that the woolly hair phenotype (with or without hypotrichosis) may be mistaken for UHS by clinicians or affected individuals and, therefore, we suggest that woolly hair should be considered in the differential diagnosis of UHS.
In the present cohort, all of the individuals with pathogenic variants in PADI3, TGM3, or TCHH showed an autosomal recessive mode of inheritance. In the literature prior to the genetic elucidation of UHS, some pedigrees were reported who appeared to have an autosomal dominant inheritance with a respective affected parent. In this cohort, we performed genetic screening in 2 such families with an apparent autosomal dominant mode of inheritance. The results indeed revealed a pseudodominance because the affected parents also happened to be carriers of biallelic PADI3 variants even though the child had inherited the phenotype in an autosomal recessive fashion. In fact, this was not surprising because the allele frequency of some of the pathogenic PADI3 variants is not very low in the population (eTable 1 in the Supplement). Therefore, we suggest that at least some of those pedigrees from the literature with a presumed autosomal dominant mode of inheritance may in fact also have an autosomal recessive inheritance of pathogenic PADI3 variants similar to the 2 families reported herein. However, given that the genetic associations of around one-fourth of the cohort remains unclear, another plausible hypothesis is that pathogenic variants in other genes may also be associated with UHS and that some of these genes may have an autosomal dominant pattern of inheritance. Nonetheless, based on the exome data of the genetically unclarified cases, we can assume that the UHS phenotype is unlikely to be related to a fourth major gene that is recurrently affected by pathogenic coding variants. We therefore speculate that a large heterogeneity may exist with several other UHS-associated genes, each of which may explain the phenotype in only a single or very few families, as is the case for TCHH and TGM3.
Limitations
It cannot be excluded that some of the unexplained cases may be related to variants that affect the splicing or expression of known UHS-associated genes and have remained undetected by the sequencing approaches used here. In fact, it is well known that large structural variants such as deletions/duplications and complex rearrangements are difficult to detect via short-read sequencing approaches. Similarly, deep introns and regulatory regions such as promoters are mostly not covered by whole-exome or Sanger sequencing. Accordingly, we plan in the near future to use a targeted long-read sequencing approach for screening of the entire coding and noncoding regions in and around the known UHS-associated genes in all of the unexplained cases to address this concern.
Conclusions
This cohort study expands the pathogenic variant spectrum of UHS and found that PADI3 was most commonly associated with this rare hair phenotype. The present results also suggest that the 4 most commonly observed pathogenic PADI3 variants are far more likely to have descended from a respective common ancestor rather than having had occurred independently multiple times.
eMethods. Whole-exome sequencing and haplotype analysis
eFigure 1. Clinical manifestation of UHS in additional individuals and the respective underlying genotypes
eFigure 2. 3-Dimensional protein modeling of the pathogenic stop and frameshift variants identified in PADI3
eFigure 3. 3-Dimensional protein modeling of the three most common PADI3 variants
eFigure 4. Genetic screening of two pedigrees suggesting an autosomal dominant inheritance
eFigure 5. The variable extent of sharing across haplotypes carrying the respective PADI3 pathogenic variants
eTable 1. Pathogenic PADI3 variants underlying UHS
eTable 2. The variable extent of sharing across individual haplotypes carrying c.505C>T (p.Gln169*)
eTable 3. The variable extent of sharing across individual haplotypes carrying c.335T>A (p.Leu112His)
eTable 4. The variable extent of sharing across individual haplotypes carrying c.881C>T (p.Ala294Val)
eTable 5. The variable extent of sharing across individual haplotypes carrying c.1813C>A (p.Pro605Thr)
eReferences
References
- 1.Ü Basmanav FB, Cau L, Tafazzoli A, et al. Mutations in three genes encoding proteins involved in hair shaft formation cause uncombable hair syndrome. Am J Hum Genet. 2016;99(6):1292-1304. doi: 10.1016/j.ajhg.2016.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calderon P, Otberg N, Shapiro J. Uncombable hair syndrome. J Am Acad Dermatol. 2009;61(3):512-515. doi: 10.1016/j.jaad.2009.01.006 [DOI] [PubMed] [Google Scholar]
- 3.Navarini AA, Kaufmann F, Kaech A, Trüeb RM, Weibel L. Picture of the month: uncombable hair (pili trianguli et canaliculi). Arch Pediatr Adolesc Med. 2010;164(12):1165-1166. doi: 10.1001/archpediatrics.2010.228-a [DOI] [PubMed] [Google Scholar]
- 4.Tarcsa E, Marekov LN, Andreoli J, et al. The fate of trichohyalin: sequential post-translational modifications by peptidyl-arginine deiminase and transglutaminases. J Biol Chem. 1997;272(44):27893-27901. doi: 10.1074/jbc.272.44.27893 [DOI] [PubMed] [Google Scholar]
- 5.Méchin MC, Takahara H, Simon M. Deimination and peptidylarginine deiminases in skin physiology and diseases. Int J Mol Sci. 2020;21(2):566. doi: 10.3390/ijms21020566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinert PM, Parry DA, Marekov LN. Trichohyalin mechanically strengthens the hair follicle: multiple cross-bridging roles in the inner root shealth. J Biol Chem. 2003;278(42):41409-41419. doi: 10.1074/jbc.M302037200 [DOI] [PubMed] [Google Scholar]
- 7.Westgate GE, Botchkareva NV, Tobin DJ. The biology of hair diversity. Int J Cosmet Sci. 2013;35(4):329-336. doi: 10.1111/ics.12041 [DOI] [PubMed] [Google Scholar]
- 8.Kiliç A, Oğuz D, Can A, Akil H, Gürbüz Köz O. A case of uncombable hair syndrome: light microscopy, trichoscopy and scanning electron microscopy. Acta Dermatovenerol Croat. 2013;21(3):209-211. [PubMed] [Google Scholar]
- 9.Hicks J, Metry DW, Barrish J, Levy M. Uncombable hair (cheveux incoiffables, pili trianguli et canaliculi) syndrome: brief review and role of scanning electron microscopy in diagnosis. Ultrastruct Pathol. 2001;25(2):99-103. doi: 10.1080/01913120117514 [DOI] [PubMed] [Google Scholar]
- 10.Shapovalov M, Vucetic S, Dunbrack RL Jr. A new clustering and nomenclature for beta turns derived from high-resolution protein structures. PLoS Comput Biol. 2019;15(3):e1006844. doi: 10.1371/journal.pcbi.1006844 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods. Whole-exome sequencing and haplotype analysis
eFigure 1. Clinical manifestation of UHS in additional individuals and the respective underlying genotypes
eFigure 2. 3-Dimensional protein modeling of the pathogenic stop and frameshift variants identified in PADI3
eFigure 3. 3-Dimensional protein modeling of the three most common PADI3 variants
eFigure 4. Genetic screening of two pedigrees suggesting an autosomal dominant inheritance
eFigure 5. The variable extent of sharing across haplotypes carrying the respective PADI3 pathogenic variants
eTable 1. Pathogenic PADI3 variants underlying UHS
eTable 2. The variable extent of sharing across individual haplotypes carrying c.505C>T (p.Gln169*)
eTable 3. The variable extent of sharing across individual haplotypes carrying c.335T>A (p.Leu112His)
eTable 4. The variable extent of sharing across individual haplotypes carrying c.881C>T (p.Ala294Val)
eTable 5. The variable extent of sharing across individual haplotypes carrying c.1813C>A (p.Pro605Thr)
eReferences