

Abstract
Background
Maintaining healthy mitochondria is mandatory for muscle viability and function. An essential surveillance mechanism targeting defective and harmful mitochondria to degradation is the selective form of autophagy called mitophagy. Ambra1 is a multifaceted protein with well‐known autophagic and mitophagic functions. However, the study of its role in adult tissues has been extremely limited due to the embryonic lethality caused by full‐body Ambra1 deficiency.
Methods
To establish the role of Ambra1 as a positive regulator of mitophagy, we exploited in vivo overexpression of a mitochondria‐targeted form of Ambra1 in skeletal muscle. To dissect the consequence of Ambra1 inactivation in skeletal muscle, we generated muscle‐specific Ambra1 knockout (Ambra1 fl/fl:Mlc1f‐Cre) mice. Mitochondria‐enriched fractions were obtained from muscles of fed and starved animals to investigate the dynamics of the mitophagic flux.
Results
Our data show that Ambra1 has a critical role in the mitophagic flux of adult murine skeletal muscle and that its genetic inactivation leads to mitochondria alterations and myofibre remodelling. Ambra1 overexpression in wild‐type muscles is sufficient to enhance mitochondria clearance through the autophagy‐lysosome system. Consistently with this, Ambra1‐deficient muscles display an abnormal accumulation of the mitochondrial marker TOMM20 by +76% (n = 6–7; P < 0.05), a higher presence of myofibres with swollen mitochondria by +173% (n = 4; P < 0.05), and an alteration in the maintenance of the mitochondrial membrane potential and a 34% reduction in the mitochondrial respiratory complex I activity (n = 4; P < 0.05). Lack of Ambra1 in skeletal muscle leads to impaired mitophagic flux, without affecting the bulk autophagic process. This is due to a significantly decreased recruitment of DRP1 (n = 6–7 mice; P < 0.01) and Parkin (n = 6–7 mice; P < 0.05) to the mitochondrial compartment, when compared with controls. Ambra1‐deficient muscles also show a marked dysregulation of the endolysosome compartment, as the incidence of myofibres with lysosomal accumulation is 20 times higher than wild‐type muscles (n = 4; P < 0.05). Histologically, Ambra1‐deficient muscles of both 3‐ and 6‐month‐old animals display a significant decrease of myofibre cross‐sectional area and a 52% reduction in oxidative fibres (n = 6–7; P < 0.05), thus highlighting a role for Ambra1 in the proper structure and activity of skeletal muscle.
Conclusions
Our study indicates that Ambra1 is critical for skeletal muscle mitophagy and for the proper maintenance of functional mitochondria.
Keywords: Ambra1, Skeletal muscle, Mitophagy, Mitochondria
Introduction
Mitochondria are dynamic organelles providing energy and metabolites to eukaryotic cells, thus ensuring their metabolic needs. 1 Mitochondria also act as signalling organelles that orchestrate several cellular processes, such as programmed cell death, autophagy, cell cycle regulation, and redox balance. 2 The presence of healthy and functional mitochondria is imperative for cellular homeostasis. Therefore, cells need surveillance mechanisms able to recognize and remove dysfunctional mitochondria. 3
Macroautophagy (hereafter referred as autophagy) is a tightly regulated self‐eating process, highly conserved among species, which plays essential roles in ensuring quality control of cell components and in the maintenance of cell homeostasis. The autophagic machinery removes damaged or unnecessary organelles, protein aggregates, and pathogens through double‐membrane vesicles called autophagosomes, which are delivered to the endo‐lysosomal pathway. 4 Besides bulk autophagy, several forms of autophagy involved in the turnover of distinct cell components were recognized. Among them, mitophagy ensures the clearance of defective mitochondria during different physiological and pathological conditions, thus preventing their toxic accumulation. The mitophagic process is critical in post‐mitotic and energy‐demanding cells, such as neurons and muscle fibres, because their low regenerative capacity and the production of reactive oxygen species require high turnover rates of proteins and organelles to cope with cellular damages that may accumulate over time. 5
Skeletal muscle is a plastic tissue, able to adapt to different physiological and pathological stimuli, such as variations in nutrients, mechanical stress, physical activity, and hormones. Given its abundance within body mass, skeletal muscle plays fundamental roles in the maintenance of whole‐body metabolic homeostasis. Therefore, the fine regulation of mitochondria activity and turnover is critical for muscle function. 6 Indeed, mitophagy dysregulation was found to be involved in the aetiopathology of a broad range of disorders affecting muscles, including cancer cachexia, 7 age‐related sarcopenia, 8 insulin resistance, 9 and muscular dystrophies. 10 , 11 However, the molecular players participating in the regulation of mitophagy in muscle are still largely unknown.
Activating molecule in Beclin 1‐regulated autophagy (Ambra1) is an intrinsically disordered protein acting as a scaffold and involved in multiple cellular processes, including cell cycle, proliferation, and autophagy. 12 , 13 ,S1 During autophagy, Ambra1 interacts with Beclin 1, thus stabilizing and potentiating the activity of class‐III‐phosphoinositide‐3‐kinase (PI3K) initiation complex upon pro‐autophagic stimuli. 14 Ambra1 also plays a pivotal role in regulating mitophagy, being recruited to depolarized mitochondria where it mediates their selective clearance by interacting with the E3 ubiquitin ligase Parkin. 15 ,S2 In addition, Ambra1 facilitates the transport of mitophagic cargoes into nascent autophagosomes, thus acting as a mitophagy receptor through the interaction of microtubule‐associated protein 1 light chain 1 β (MAP 1LC3B, hereafter LC3) with the LC3‐interacting region (LIR) of Ambra1. 16 , 17 ,S3
Initial studies, exploiting a randomly mutated Ambra1 locus (Ambra1 gt/gt ) in mice, showed that Ambra1 deficiency is embryonic lethal and causes exencephaly and spina bifida, associated with autophagy impairment, unbalanced cell proliferation, and excessive apoptosis. 18 Notably, besides altered neurogenesis, Ambra1 deficiency also leads to impaired muscle development in both zebrafish and mouse embryos. 18 , 19 , 20
Because the embryonic lethality of Ambra1 gt/gt mice prevents to investigate Ambra1 function in adults, we generated a new Ambra1 f/f:Mlc1f‐Cre mouse model, in which Ambra1 depletion occurs in skeletal muscle. Here we show that Ambra1‐deficient muscles display myofibre defects, with progressive accumulation of dysfunctional mitochondria. Notably, while bulk autophagy appears unaffected, the mitophagic flux is compromised in Ambra1‐deficient muscles due to impaired recruitment of Parkin and DRP1 to depolarized mitochondria.
Methods
Generation of Ambra1 knockout mice
Whole‐body and muscle‐specific Ambra1 knockout mice were produced from Ambra1<tm1a(EUCOMM)Wtsi> embryonic stem cells (EUCOMM Consortium), which contain a targeted conditional mutation (tm1a; referenceS4) within exon 4 of Ambra1 gene in the C57BL/6N background (see Supporting Information, Figure S1a for details). Cells were aggregated with E2.5 morulas of CD1 mice, following standard procedures.S5 The nucleotide sequence of Ambra1<tm1a(EUCOMM)Wtsi> mutant allele is deposited in GenBank (#JN964599.1). Mice bearing the Ambra1 tm1a allele were crossed with FLP transgenic mice,S6 to remove lacZ and neo cassettes (tm1c allele; Figure S1a). The obtained Ambra1 fl/+ mice were backcrossed with C57BL/6N mice for one generation to get rid of the FLP transgene and then crossed with CAG‐Cre mice,S7 to generate whole‐body Ambra1 knockout (Ambra1 −/−) mice. Skeletal muscle‐specific Ambra1 (Ambra1 fl/fl:Mlc1f‐Cre) knockout mice were generated by crossing Ambra1 fl/+ mice with Mlc1f‐Cre mice.S8 Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl littermate controls were obtained by subsequent crossings (Figure S1a). All studies were carried out in mice with C57BL/6N genetic background. Genotyping was determined by PCR with specific primers, listed in Table S1.
Animals
The newly generated mice and GFP‐LC3 miceS9 were housed in controlled temperature (23°C) and light (12 h light/12 h dark cycle) conditions, with free access to water and food. Animal procedures were approved by the Animal Ethics Committee of the University of Padova and by the Italian Ministry of Health (n. 581/2017‐PR). For starvation experiments, at 8 AM, mice were transferred for 24 h to clean cages without chow but with free water access. For colchicine treatment, mice were i.p. injected with 0.4 mg/kg/day of colchicine (Sigma‐Aldrich), 24 and 12 h before sacrifice. For chloroquine treatment, mice were i.p. injected with 50 mg/kg/day of chloroquine (Sigma‐Aldrich) for three consecutive days and sacrificed 3 h after last injection.S10 For all experiments, mice were sacrificed within 7:30 and 10:00 AM.
In vivo muscle transfection
Tibialis anterior (TA) muscles were transfected by in vivo electroporation as described.S11 Briefly, animals were anaesthetized, TA was exposed, and 30–40 μg of naked plasmid DNA was injected in the central region of muscle. Spatula electrodes were placed and five 200‐ms‐long electric pulses were delivered at 100 V/cm with Nepagene NEPA21 electroporator. Muscles were collected 10–12 days after wound closure. For transfection of flexor digitorum brevis (FDB) muscle, DNA was injected in the foot without any skin incision after hyaluronidase pre‐treatment and delivered to cells by 20 electric pulses with needle electrodes.
Histology and immunofluorescence
Embryos were fixed overnight at 4°C with 4% paraformaldehyde in phosphate‐buffered saline (PBS), dehydrated in increasing alcohol solutions, cleared with xylene, and paraffine‐embedded. Seven‐micrometre‐thick sections were deparaffinized in xylene and rehydrated in decreasing alcohol solutions before staining. TA and quadriceps muscles were frozen in pre‐cooled isopentane and stored at −80°C until analysis. Ten‐micrometre‐thick cryosections were stained with haematoxylin and eosin, dehydrated, and mounted with Eukitt Quick‐hardening mounting medium (Sigma‐Aldrich). Bright‐field micrographs were captured by a Leica DM‐R microscope equipped with digital camera. Cross‐sectional area (CSA) measurements were carried out manually using haematoxylin–eosin‐stained images. Immunostainings were performed as describedS12 and hybridized overnight with primary antibodies (listed in Table S2) and with the appropriate secondary antibodies (Jackson ImmunoResearch). Nuclei were counter‐stained with Hoechst 33258 (Sigma‐Aldrich). Slides were mounted with 80% glycerol in PBS and observed with Zeiss LSM700 and Leica SP5 confocal microscopes. Confocal images were processed by FIJI SoftwareS13 and quantitative analysis was performed with Squassh software,S14 always maintaining the same parameters. Fibre type analysis and the relative CSA measurements were carried out by exploiting Myosoft automated macro for FIJI software.S15
Transmission electron microscopy
Tibialis anterior muscles from 6‐month‐old mice were fixed overnight at 4°C with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, washed with 0.1 M cacodylate buffer, post‐fixed for 2 h with 1% osmium tetroxide, and embedded in Epon812 (Electron Microscopy Sciences). Ultrathin sections were stained with uranyl acetate and lead citrate and observed with a Philips EM400 electron microscope operating at 100 kV.
Western blotting
Frozen murine samples (whole embryos, skeletal muscles, and heart) were ground in liquid nitrogen and lysed as described.S12 For subcellular fractionation of mitochondria and nuclei, freshly collected quadriceps were processed as describedS16 and lysates stored at −80°C until use. Equal amounts of proteins, quantified by BCA Protein Assay Kit (Thermo‐Fisher), were loaded in polyacrylamide Novex NuPAGE Bis‐Tris gels (Invitrogen) and electrotransferred onto PVDF membranes (Millipore). Membranes were stained with Ponceau (Sigma‐Aldrich), saturated, and incubated overnight with primary antibodies (listed in Table S2). Horseradish peroxidase‐conjugated secondary antibodies (1:2000; Bethyl Laboratories) were used, and the signal detected with SuperSignal West Pico (Thermo‐Fisher). Densitometric quantification was carried out by FIJI software.S13
Real‐time quantitative polymerase chain reaction
Whole frozen embryos or muscles were ground in liquid nitrogen. Total RNA was isolated by TRIzol (Invitrogen), quantified, and retrotranscribed with SuperScript III First‐Strand Synthesis System (Invitrogen) using random hexamers. Real‐time PCR reactions were performed with RotorGeneQ (Qiagen), using SYBR green mastermix (Qiagen). Data were analysed with the instrument's software, using Actb as a reference gene. Primers sequences are listed in Table S1.
Mitochondrial DNA copy number
Total DNA was isolated from entire quadriceps muscles by phenol:chloroform extraction and quantified. For each sample, same DNA amounts were amplified by real‐time PCR, using primers for nuclear (Tert) and mitochondrial (Nd4) DNA (Table S1). Mitochondrial DNA copy number was calculated using Tert as a reference for nuclear DNA content.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured in ex vivo FDB myofibres. Briefly, freshly dissected FDB muscles were incubated for 1 h in ice and 50 min at 37°C in the presence of 0.4% collagenase I (Sigma‐Aldrich). Fibres were mechanically dissociated and seeded in microscope slides pre‐coated with laminin (Sigma‐Aldrich). The following day, slides were incubated for 30 min at 37°C in a saline buffer (20 mM HEPES, 35 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM MgSO4, 0.4 mM KH2PO4, 5 mM glucose) supplemented with 20 nM tetramethylrhodamine methyl ester (TMRM), and myofibres were analysed with SP5 inverted confocal microscope (Leica). Sequential images of TMRM fluorescence were acquired every 5 s. When indicated, 10 μM oligomycin or 10 μM carbonyl cyanide m‐chlorophenyl hydrazone (CCCP) were added. Quantification of TMRM fluorescence was performed using FIJI software.S13
Treadmill exercise
Six‐month‐old mice were first adapted for two consecutive days to treadmill, set in horizontal position, for 15 min at a speed of 8 cm/s. Acute exercise was performed, by setting treadmill in uphill position (+5° slope) at increasing speeds (10 min at 0 cm/s, 5 min at 11 cm/s, 10 min at 18 cm/s, 15 min at 25 cm/s, and 50 min at 33 cm/s).
Respiratory chain activity
Activities of NADH:coenzyme Q1 oxidoreductase (Complex I), succinate dehydrogenase (Complex II), ubiquinol:cytochrome c oxidoreductase (Complex III), NADH:cytochrome c reductase (Complex I–III), succinate:cytochrome c reductase (Complex II–III), cytochrome c oxidase (Complex IV), and citrate synthase (CS) in skeletal muscles were determined by spectrophotometric assays as described.S17
RNA extraction and library preparation
Total RNA was extracted from TA muscles with TRIzol (Invitrogen), and its concentration measured with QuantiFluor RNA System (Promega). RNA integrity was assessed by Agilent 2100 Bioanalyzer. From each sample, 800 ng total RNA was used to prepare two pools, one from Ambra1 fl/fl TA and one from Ambra1 fl/fl:Mlc1f‐Cre TA, which provided inputs for sample preparations. Libraries for RNA‐seq were constructed with the TruSeq Stranded mRNA Kit (Illumina), according to manufacturer's instructions.
RNA‐seq analysis
Fastq files were processed with the ‘Salmon’ software v.1.3.0S18 to quantify transcript expression, using mm10.refMrna mouse transcripts as reference. Log2ratios for each transcript were calculated from transcript per million bases (TPM) values. Differentially expressed transcripts were shortlisted by setting the minimum TPM value of 3 and the minimum absolute log2ratio of 0.7. Pathway enrichment analysis was performed for selected transcripts using the PANTHER Overrepresentation Test (http://pantherdb.org/tools/compareToRefList.jsp), and the results were graphically reported using the function barplot of R v.4.1.1 software (https://www.R‐project.org/).
Statistics
Observed and expected outcomes of mouse matings (Figure S1d) were compared by χ 2 test. All comparisons were made by using two‐tailed unpaired Student's t‐test, except when indicated in figure captions, and P < 0.05 was considered as statistically significant. Data are provided as mean ± SEM. The number of biological replicates (always greater than three) is indicated in figures' captions.
Results
In vivo Ambra1‐ActA overexpression in skeletal muscle induces mitochondria clearance through the autophagy‐lysosome system
To study the contribution of Ambra1 in regulating mitophagy in adult skeletal muscle, we first exploited in vivo overexpression of a mitochondria‐targeted form of Ambra1 (Ambra1‐ActA) in TA and FDB muscles of GFP‐LC3 reporter mice, which allow to easily monitor autophagy by fluorescence microscopy. 16 We transfected muscles with a bicistronic vector that simultaneously drives the expression of FLAG‐tagged Ambra1‐ActA and mitoDsRed, thus allowing for mitochondria labelling in transfected myofibres. Fluorescence microscopy of TA cross‐sections showed more GFP‐LC3B‐positive puncta in Ambra1‐ActA‐transfected myofibres, and many of these puncta colocalized with mitochondrial signal (Figure 1A). Moreover, when acidification of lysosomes was inhibited by chloroquine administration, with a resulting block of the autophagic flux, myofibres transfected with Ambra1‐ActA showed increased mitochondrial signal co‐localizing with GFP‐LC3‐positive dots, when compared with fibres transfected with control plasmid (Figure 1A). These data suggested that Ambra1‐ActA overexpression enhances mitochondrial clearance by triggering autophagosome formation. In agreement with this, western blotting highlighted a significant chloroquine‐dependent accumulation of the mitochondrial protein cytochrome c oxidase subunit 4 (COX4) only when Ambra1‐ActA was overexpressed (Figure 1B). Interestingly, even transfected Ambra1‐ActA protein itself accumulated after autophagy inhibition, suggesting that it was subjected to the same clearance process as other autophagosome proteins (Figure 1C). In addition, Ambra1‐ActA overexpression in muscle led to increased accumulation of the lipidated form of LC3, a well‐known autophagosome marker, when chloroquine was provided (Figure 1D). Taken together, these results indicate that overexpression of a mitochondria‐targeted form of Ambra1 is sufficient to enhance mitochondrial degradation through the autophagy‐lysosome degradative system in adult muscle.
Figure 1.
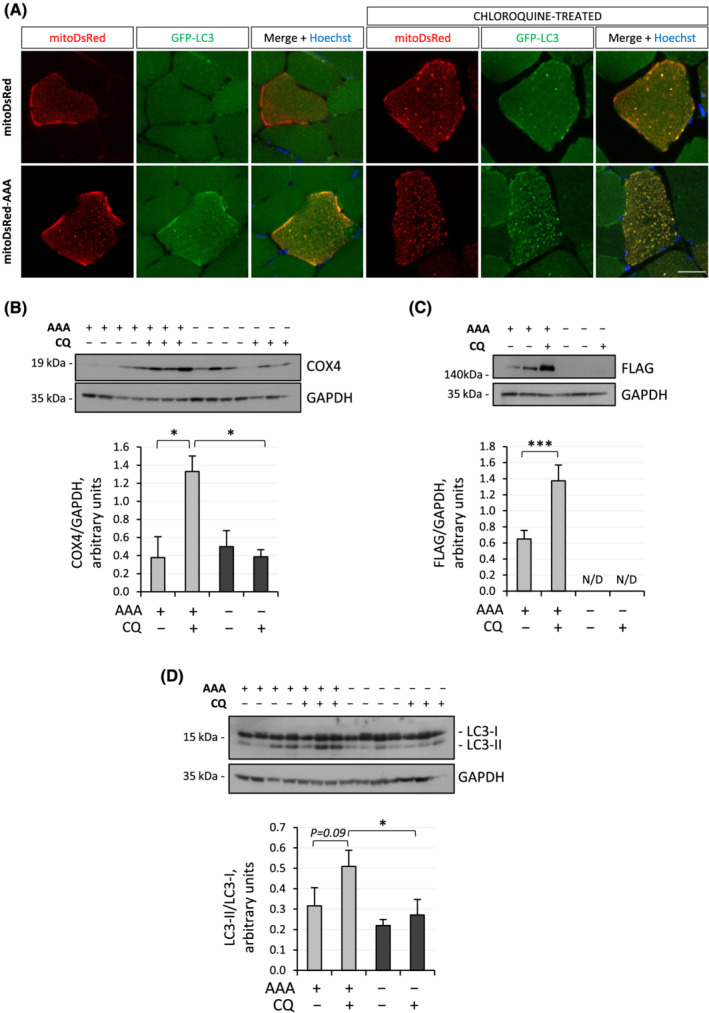
In vivo Ambra1‐ActA overexpression in skeletal muscle induces mitophagy. (A) Representative confocal immunofluorescence images for mitoDsRed (red) and GFP‐LC3 (green) in cross sections of TA muscles electroporated with control plasmid (mitoDsRed) or Ambra1‐ActA‐FLAG plasmid (mitoDsRed‐AAA), in GFP‐LC3 reporter mice treated or not with chloroquine. Nuclei were counterstained with Hoechst (blue). Scale bar, 20 μm. (B–D) Western blot analysis for COX4 (B), FLAG (C), and LC3 (D) in protein lysates of FDB muscle electroporated with Ambra1‐ActA‐FLAG plasmid (AAA, +) or control plasmid (−), in wild‐type mice treated (+) or not (−) with chloroquine (CQ). GAPDH was used as loading control. The densitometric quantifications of FLAG vs. GAPDH, COX4 vs. GAPDH, and LC3‐II vs. LC3‐I are shown below the respective western blot panels. Data are provided as mean ± SEM (n = 3–4, each condition; *, P < 0.05; ***, P < 0.001). N/D, not detectable.
Whole‐body Ambra1 knockout is embryonic lethal
To assess the consequences of in vivo Ambra1 ablation, we generated Ambra1 conditional knockout mice (Materials and Methods and Figure S1a). Global, whole‐body Ambra1 null (Ambra1 −/− ) mice were obtained by crossing Ambra1 fl/fl mice with transgenic mice expressing Cre recombinase under the control of a promoter active at the zygote stage. Western blotting and RT‐qPCR confirmed correct ablation of Ambra1 mRNA and protein in Ambra1 −/− embryos (Figure S1b,c). We found that no homozygous Ambra1 −/− pups were present in litters, whereas heterozygous Ambra1 fl/− were born at the expected Mendelian ratios (Figure S1d), confirming that Ambra1 null allele in homozygosity causes embryonic lethality at mid‐gestation. Macroscopic and histological analyses at E13.5 showed major abnormalities in Ambra1 −/− embryos, with severe disruption of brain and neural tube development (Figure S1e), thus resembling the exencephaly and spina bifida defects of Ambra1 gt/gt embryos. 18 Western blotting for autophagic markers revealed a significant increase of p62/SQSTM1 levels and a concurrent decrease of LC3‐II levels in Ambra1 −/− embryos (Figure S1f), pointing at autophagic impairment with decreased autophagosome formation and accumulation of SQSTM1‐tagged autophagic cargoes. These results support the essential role of Ambra1 for proper neural development and confirm that its global inactivation leads to embryonic lethality at mid‐gestation.
Conditional inactivation of Ambra1 in skeletal muscle leads to myofibre remodelling in postnatal life
Muscle‐specific ablation of Ambra1 was obtained by crossing Ambra1 fl/fl mice with Mlc1f‐Cre mice, which express Cre recombinase in differentiated myofibres.S8 Ambra1 fl/fl:Mlc1f‐Cre mice did not display any major difference in body weight, life expectancy, or fertility when compared with Ambra1 fl/fl littermate controls (data not shown). Western blotting confirmed depletion of Ambra1 in skeletal muscles (Figure S2a), but not in heart or other tissues (Figure 2A and data not shown). Haematoxylin–eosin staining of Ambra1‐deficient muscles did not show any overt alteration of the three‐dimensional architecture of skeletal muscles (Figure 2B). Nevertheless, morphometric analyses revealed that Ambra1 deletion led to decreased myofibre sizes in both juvenile (3‐month‐old) and adult (6‐month‐old) Ambra1 fl/fl:Mlc1f‐Cre mice, with a significant decrease of the mean myofibre CSA and of the percentage of myofibres with larger CSA, when compared with Ambra1 fl/fl control mice (Figure 2C). In addition, Ambra1 fl/fl:Mlc1f‐Cre muscles displayed a significant decrease in the percentage of type IIA myofibres, pointing at a switch from oxidative to glycolytic myofibres (Figure 2D). Analysis of the CSA distributions within different fibre types at 6 months of age showed decreased CSA in both type IIB/IIX and type IIA myofibres of Ambra1 fl/fl:Mlc1f‐Cre muscles (Figure S2b). RNA‐seq experiments on TA, aimed at the identification of differentially expressed genes between Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl muscles, highlighted changes in several gene ontology (GO) transcripts classes. Among the most enriched GO classes for ‘biological process’ (Figure 2E) and ‘cellular components’ (Figure 2F), there was significant enrichment in transcripts coding for proteins involved in sarcomere structure and function. Furthermore, these experiments revealed altered expression of genes coding for mitochondria‐related proteins in Ambra1‐depleted muscles (Figure 2F), prompting us to investigate further the mitochondrial compartment. Altogether, these data indicate that Ambra1 ablation in skeletal muscle leads to extensive remodelling both in terms of myofibre size and gene expression.
Figure 2.
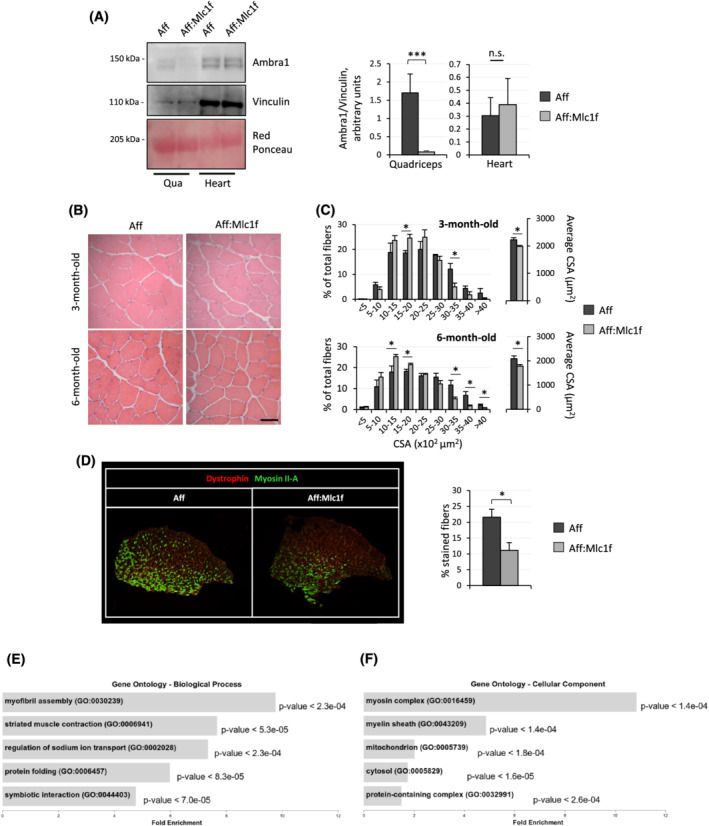
Muscle‐specific ablation of Ambra1 leads to myofibre remodelling. (A) Western blotting for Ambra1 in protein extracts of quadriceps muscle (Qua) and heart from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Vinculin was used as loading control. Densitometric quantifications of Ambra1 vs. vinculin, as determined by at least three independent experiments, are shown on the right. Data are provided as mean ± SEM (n = 4, each condition; ***, P < 0.001; n.s., not significant). (B) Haematoxylin–eosin staining of TA cross sections from 3‐ and 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Scale bar, 50 μm. (C) Morphometric analysis for cross‐sectional area (CSA) distribution among myofibres (left) and average cross‐sectional area (right), as determined by microscopy images of TA muscle cross sections as in (B). Data are shown as mean ± SEM (n = 4–9 mice, each genotype; *, P < 0.05; **, P < 0.01). (D) Merged immunofluorescence confocal images for Dystrophin (red) and Myosin IIA (green) in cross cryosections of TA muscle from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Quantification of the percentage of green‐positive fibres is shown on the right. Data are shown as mean ± SEM (n = 6–7 mice, each genotype; *, P < 0.05). (E,F) Bar plots representing the five most enriched gene ontology (GO) classes for biological process (E) and cellular component (F) categories, as determined from RNA‐seq data for differentially expressed genes in tibialis anterior muscles of 6‐month‐old Ambra1 fl/fl and Ambra1 fl/fl:Mlc1f‐Cre mice. Fold enrichment is shown in x axes; P‐value for each class is shown on the right of each bar.
Ambra1‐deficient muscles accumulate mitochondria and endo‐lysosomal vesicles
Considering that Ambra1 is a positive regulator for autophagy, we investigated bulk autophagy in Ambra1‐depleted muscles. Unexpectedly, analysis of the autophagic flux upon colchicine administration did not reveal any significant difference in LC3 lipidation and LC3‐II accumulation between Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl muscles, thus suggesting that bulk autophagy is not overtly affected by lack of Ambra1 in muscles (Figure S2c). Because we found that Ambra1‐ActA overexpression led to mitochondria degradation via the autophagy‐lysosome system (see Figure 1), we investigated the mitochondrial and endo‐lysosomal compartments in Ambra1‐deficient TA and quadriceps muscles. Western blotting (Figure 3A; Figure S2d) and quantitative immunofluorescence (Figure 3B,C) revealed that lysosome‐associated membrane protein 1 (LAMP1) and translocase of outer mitochondrial membrane 20 (TOMM20), which respectively mark endo‐lysosomes and mitochondria, were significantly increased in Ambra1 fl/fl:Mlc1f‐Cre muscles, pointing at an accumulation of mitochondria and endo‐lysosomes. This prompted us to explore whether the increased mitochondria content of Ambra1‐depleted muscles was attributable to defective degradation or to enhanced biogenesis. Quantification of gene expression and protein levels for peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) and mitochondrial transcription factor A (TFAM), two key regulators of mitochondrial biogenesis, showed that their levels were unaltered in Ambra1 fl/fl:Mlc1f‐Cre muscles (Figure S3a–c), thus ruling out a major impact of increased mitochondrial biogenesis in the observed differences. In agreement with this, mitochondrial DNA content was not significantly different between Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl muscles (Figure S3d). Accumulation of structurally abnormal mitochondria in Ambra1‐deficient muscles was confirmed by transmission electron microscopy, which showed an increased percentage of myofibres with swollen mitochondria and abnormal cristae in Ambra1 fl/fl:Mlc1f‐Cre TA sections (Figure 3D,E). Electron microscopy also confirmed increased lysosomal content of Ambra1‐deficient muscles, with markedly increased percentages of myofibres with lysosomal accumulation in Ambra1 fl/fl:Mlc1f‐Cre TA sections (Figure 3D,E). Altogether, these data indicate that Ambra1 deficiency in skeletal muscle strongly impinges on the mitochondrial compartment and the endo‐lysosomal system, with no overt defects in the bulk autophagic pathway.
Figure 3.
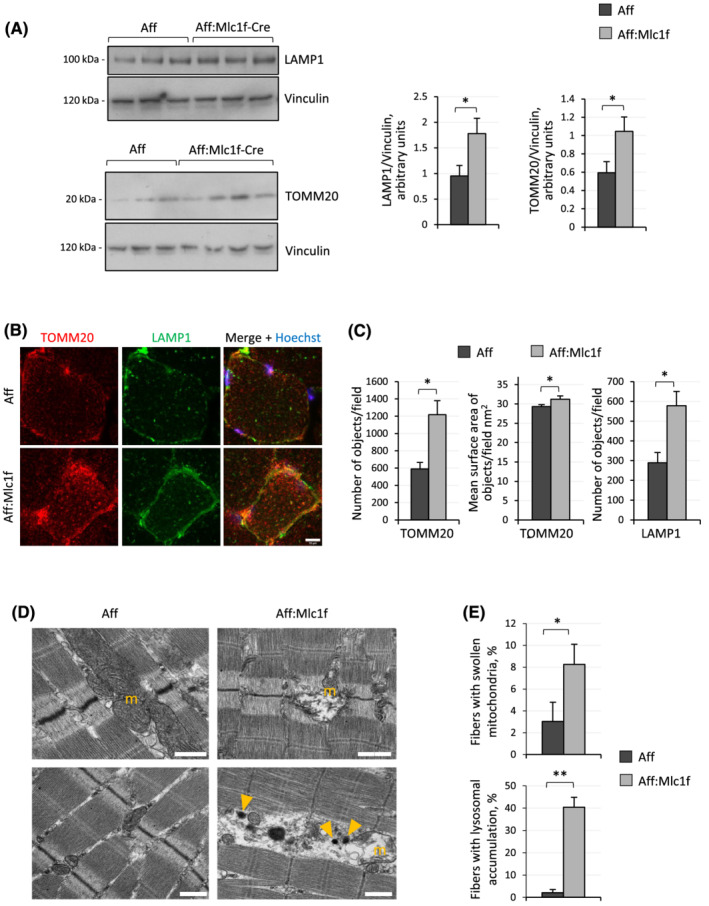
Mitochondria and endo‐lysosomal accumulation in Ambra1 null muscles. (A) Western blotting for LAMP1 and TOMM20 in protein extract of quadriceps muscle from 6‐month‐old Ambra1 f/f (Aff) and Ambra1 f/f:Mlc1f‐Cre (Aff:Mlc1f‐Cre) mice. Vinculin was used as loading control. Densitometric quantifications of LAMP1 vs. Vinculin and of TOMM20 vs. Vinculin, as determined by at least three independent experiments, are shown on the right panels. Data are provided as mean ± SEM (n = 6–7 mice, each genotype; *, P < 0.05). (B) Representative confocal immunofluorescence images for TOMM20 (red) and LAMP1 (green) in cross cryosections of quadriceps muscle from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Nuclei were counterstained with Hoechst (blue). Scale bar, 10 μm. (C) Automated quantifications of the number of objects per cell and of the mean surface of objects per cell for TOMM20 (left and middle panels) and LAMP1 (right panel) signals, as determined by confocal immunofluorescence images as in (B). Data are shown as mean ± SEM (n = 4 mice, each genotype; *, P < 0.05). (D) Representative transmission electron microscopy images of longitudinal sections of TA muscle from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Yellow arrowheads point at lysosomes. Scale bar, 1 μm. m, mitochondria. (E) Quantification of the percentage of myofibres with swollen mitochondria (upper panel) and with lysosomal accumulation (lower panel), based on electron microscopy images as in (D). Data are shown as mean ± SEM (n = 4 mice, each genotype; Mann–Whitney test; *, P < 0.05; **, P < 0.01).
Ambra1 null muscles display dysfunctional mitochondria
To understand whether defective mitochondria accumulated along postnatal life in Ambra1‐deficient muscles and how the observed ultrastructural defects impacted on mitochondrial function, we carried out further studies in mice at different ages. Transmission electron microscopy showed that mitochondria were moderately affected in 3‐month‐old Ambra1 fl/fl:Mlc1f‐Cre muscles, whereas they became markedly swollen in 6‐month‐old Ambra1 fl/fl:Mlc1f‐Cre samples (Figure 4A). Confocal TMRM analysis for mitochondrial membrane potential in myofibres isolated from FDB muscles of 6‐month‐old Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl mice revealed that Ambra1‐depleted myofibres did not hyperpolarize upon oligomycin addition, at difference from control myofibres, pointing at mitochondrial dysfunction (Figure 4B). Finally, we investigated the activity of respiratory chain complexes in mitochondria isolated from muscles of Ambra1 fl/fl:Mlc1f‐Cre and Ambra1 fl/fl mice. As the activity of the different respiratory chain complexes was not significantly affected in mice kept in standard resting conditions (Figure S3e), we subjected mice to acute treadmill exercise, to assess the impact of locomotor activity in the observed mitochondrial defects. This experiment highlighted a significant decrease of complex I activity in Ambra1‐deficient mitochondria, together with a trend towards decreased activities of complex III and complex I + III (Figure 4C). Altogether, these data indicated that lack of Ambra1 affects the structure and function of the mitochondrial compartment of skeletal muscles.
Figure 4.
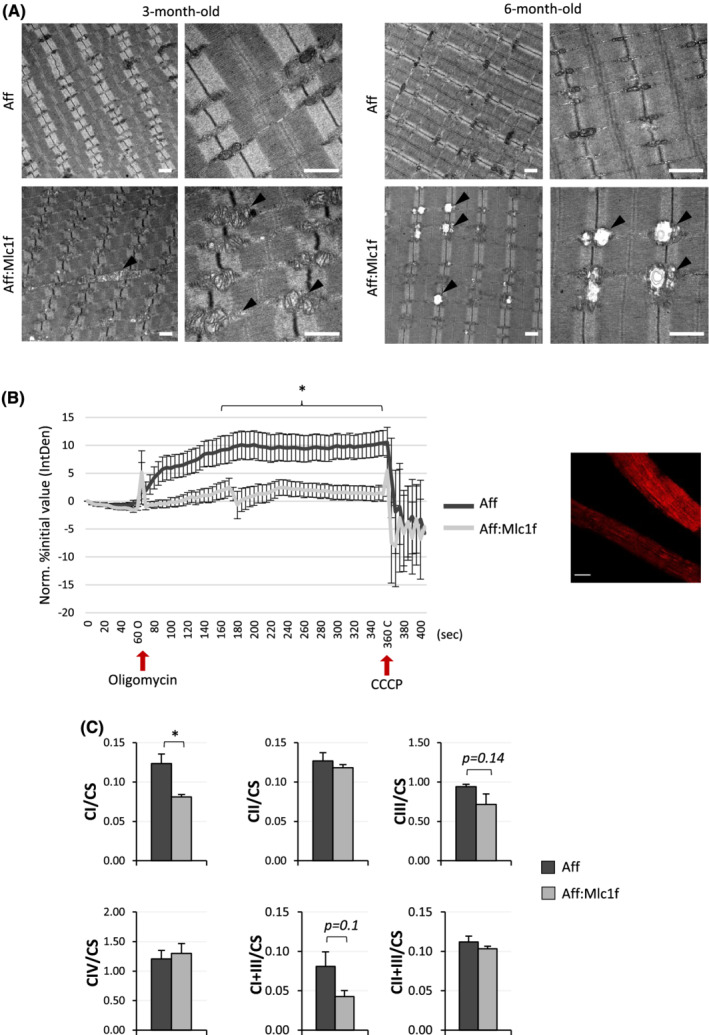
Ambra1 null muscles display dysfunctional mitochondria. (A) Representative transmission electron microscopy images of longitudinal sections of TA muscle from 3‐ and 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Black arrowheads point at some structurally abnormal mitochondria displayed by Ambra1‐deficient muscles. Scale bar, 1 μm. (B) Quantification of TMRM fluorescence in isolated FDB myofibres from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. Red arrows indicate the time points of oligomycin and CCCP addition, respectively. The graph shows the integrated density value (IntDen) normalized as the percentage of the initial value. Data are shown as mean ± SEM (n = 23–24 myofibres from four different mice, each genotype; *, P < 0.05). A representative confocal microscopy image of TMRM‐labelled myofibres from control Ambra1 fl/fl mice is shown on the right. Scale bar, 20 μm. (C) Quantification of the activity of respiratory chain complex I (CI), complex II (CII), complex III (CIII), complex IV (CIV), complex I + III (SCI + III), and complex II + III (SCII + III) in mitochondria isolated from quadriceps muscles of 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice subjected to acute treadmill exercise as described in Methods section. The activity of the different complexes was normalized on citrate synthase (CS) activity. Data are shown as mean ± SEM (n = 4 mice, each genotype; *, P < 0.05).
Ambra1 ablation in muscles affects mitophagy and leads to defective mitophagic flux
Mitochondrial dynamics and mitophagy are highly interconnected mechanisms, and the recruitment of pro‐fission factors at the mitochondrial compartment is an essential prerequisite for mitochondria turnover. 21 Therefore, we investigated key mitophagic proteins in mitochondrial fractions isolated from quadriceps muscles of 3‐ and 6‐month‐old mice. Western blot quantifications revealed that both dynamin‐related protein 1 (DRP1), a key regulator of mitochondria fission, and Parkin (PARK2), an E3 ubiquitin ligase with a central role in mitophagy, were significantly less recruited to mitochondria in muscles of both 3‐ and 6‐month‐old Ambra1 fl/fl:Mlc1f‐Cre animals when compared with Ambra1 fl/fl controls (Figure 5A,B). These findings pointed at a defective regulation of the mitophagic process in Ambra1‐deficient muscles. To investigate this aspect in further detail, we studied the mitophagic flux by quantifying the amount of lipidated LC3 recruited to muscle mitochondria upon starvation, as an inducing stimulus, and following in vivo administration of colchicine, to block the flux and fusion with lysosomes. 22 These experiments revealed that LC3‐II did not accumulate after colchicine treatment in mitochondria of Ambra1‐deficient muscles, at difference from control samples (Figure 5C). Taken together, these data indicate that Ambra1 ablation in skeletal muscles leads to defective recruitment of LC3 to mitochondria, thus reducing the rate of mitophagic flux and prompting to an abnormal accumulation of dysfunctional mitochondria.
Figure 5.
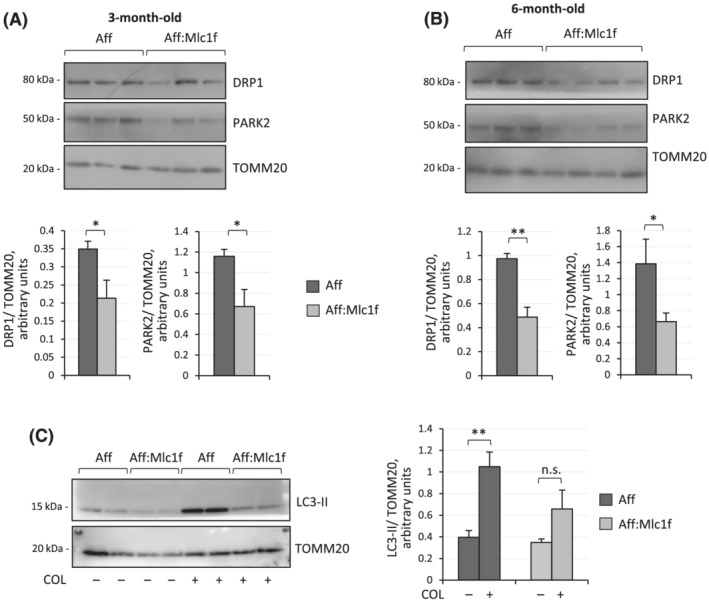
Ambra1 ablation in muscles leads to defective mitophagy. (A,B) Western blotting for DRP1 and PARK2 in mitochondria‐enriched protein extracts of quadriceps muscle from 3‐month‐old (A) and 6‐month‐old (B) Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1f) mice. TOMM20 was used as loading control. Densitometric quantifications of DRP1 vs. TOMM20 and of PARK2 vs. TOMM20, as determined by at least three independent experiments, are shown on the bottom panels. Data are shown as mean ± SEM (n = 6–7 mice, each genotype; *, P < 0.05; **, P < 0.01). (C) Western blotting for LC3 in mitochondria‐enriched protein extract of quadriceps muscle from 6‐month‐old Ambra1 fl/fl (Aff) and Ambra1 fl/fl:Mlc1f‐Cre (Aff:Mlc1) mice, starved for 24 h to induce mitophagy and treated (+) or not (−) with colchicine (COL). TOMM20 was used as loading control. The right panel shows the densitometric quantifications of LC3‐II vs. TOMM20, as determined by at least three independent experiments. Data are shown as mean ± SEM (n = 5–6 mice, each condition; **, P < 0.01; n.s., not significant).
Discussion
Both bulk and selective autophagy play fundamental roles in skeletal muscles, because myofibres are continuously subjected to mechanical and metabolic stress during their activity. In particular, mitophagy is critical for the maintenance of myofibre homeostasis, by selectively removing dysfunctional mitochondria, which otherwise would undergo a gradual accretion within cells and dramatically compromise the functionality of the entire muscle. 23 , 24 , 25
In skeletal muscle, the autophagy‐lysosome system was extensively studied mainly by focusing on transcription factors and cofactors that govern its transcriptional regulation, such as FOXOs 26 ,S11 and PGC‐1α. 27 Further, it is well known how these proteins regulate autophagy upon muscle denervation, starvation, and exercise, as well as in pathological conditions, such as muscular dystrophies, myopathies, and age‐related sarcopenia. 21 Despite this, there is a knowledge gap in the field. Indeed, there is still a limited number of studies in which proteins directly involved in mediating either bulk or selective autophagy were targeted for in vivo manipulation in mammalian skeletal muscles. Among such mediators of the autophagic process, Ambra1 emerged as a major inducer of mitophagy via direct binding to LC3. 15 , 16 In addition, recent findings highlighted an important involvement of Ambra1 in cell cycle regulation, in particular controlling the abundance of D‐type cyclins. 28 ,S1,S19 These findings, together with the identification of a c‐Myc/Ambra1/STAT3 oncogenic pathway, 29 pointed at cancer as one major field of study for Ambra1.
Although Ambra1 was described as a positive regulator of autophagy, little is known about its physiological role in a post‐mitotic tissue such as adult skeletal muscle. Previous studies showed that Ambra1 deficiency during development causes abnormal muscle structure in both zebrafish and mouse embryos, together with several neurological defects. 18 , 19 Moreover, in vitro studies showed that Ambra1 controls the interaction between TRIM32 E3‐ubiquitin ligases and ULK1 serine/threonine kinase for induction of the atrophy process in a muscle cell line. 30
Here, we investigated the role of Ambra1 in mediating autophagy and mitophagy in skeletal muscle, by exploiting different in vivo approaches in mice. First, we found that mitochondria undergo massive autophagy‐dependent degradation upon forced in vivo expression of a mitochondria‐targeted form of Ambra1 in muscle. Based on this finding, we moved forward and generated both full body and muscle‐specific knockout mouse models for Ambra1. Interestingly, our data indicate that Ambra1‐deficient muscles accumulate dysfunctional mitochondria, with disorganized cristae and impaired regulation of mitochondrial membrane potential, with no significant impact on the rate of mitochondria biogenesis. In spite of these defects, the bulk autophagic flux is not appreciably affected by Ambra1 ablation in muscle. Instead, Ambra1‐deficient muscles display a marked enhancement of LAMP1‐positive endo‐lysosomal vesicles, thus providing evidence that the degradative pathways are actually compromised, leading to the accumulation of dysfunctional mitochondria. To further investigate this aspect, we isolated mitochondria‐enriched fractions from fresh muscles and assessed the selective clearance of mitochondria by mitophagy. The data reveal that upon starvation and colchicine treatment, the lipidated form of LC3 does not accumulate in Ambra1‐deficient mitochondria, thus indicating that the mitophagy flux is compromised in muscles lacking Ambra1.
Mitochondrial dynamics and the occurrence of mitochondrial fission and fusion events play prominent roles in the clearance of dysfunctional mitochondria within cells by mitophagy. Indeed, mitochondrial fission is a mandatory step that precedes mitochondrial clearance via mitophagy. 21 In agreement with this concept, our data indicate that in the absence of Ambra1, the pro‐fission protein DRP1 and the E3 ubiquitin ligase Parkin are not properly recruited to mitochondria, even when the mitophagic flux is induced, thus accounting for the impaired mitophagy of Ambra1‐deficient muscles. Previous studies demonstrated that Ambra1 is a major interactor of Parkin and plays a critical role in recruiting Parkin to dysfunctional mitochondria. 15 , 31 Indeed, if Parkin is not properly translocated to the membrane of damaged mitochondria, it cannot be activated by the PTEN‐induced kinase 1 (PINK1). Thus, ubiquitination of mitochondrial outer membrane proteins, which in turn leads to the recruitment of autophagic cargo receptors, does not occur in such conditions. 32 , 33 Our data support a role for the Ambra1/Parkin axis in the maintenance of muscle homeostasis, by promoting the selective removal of dysfunctional mitochondria in myofibres. In agreement with this concept, a recent work showed that Parkin ablation in mice negatively impacts muscle homeostasis. Indeed, Park2 knockout mice display mild impairment in muscle contractility accompanied by defective mitochondrial function, with decreased activity of respiratory chain complexes, and deregulation of DRP1 turnover. 34 This latter evidence is of particular interest, as literature studies identified DRP1 as a substrate for Parkin‐mediate ubiquitination, 35 thus highlighting a further level of regulation between mitophagy and mitochondrial dynamics. Consistently, muscle‐specific Drp1 knockout leads to postnatal lethality in mice, with several major negative effects on muscle homeostasis. 36 In addition, inducible Drp1 ablation in skeletal muscles of adult mice still results in mitochondrial alterations, 36 further pointing at mitophagy and mitochondrial dynamics as fundamental for muscle function.
So far, few other autophagy‐related proteins were specifically inactivated in skeletal muscle in vivo. These include muscle‐specific Atg5, 37 , 38 Atg7, 39 and Vps15 40 knockout mouse models. As expected, these models display markedly compromised bulk autophagy, with a detrimental accumulation of dysfunctional organelles, leading to severe muscle atrophy with myopathy. The severe phenotypes and the overtly compromised bulk autophagy of these mouse models make it difficult to elucidate the roles of these proteins in the regulation of the selective removal of mitochondria via mitophagy. Conversely, the relatively mild phenotype of muscle‐specific Ambra1 knockout mice and the data we obtained in the present work point at Ambra1 as an autophagy protein with a main role for the mitophagic pathway in muscles, being involved in the recruitment of DRP1 and Parkin on dysfunctional mitochondria.
Interestingly, similar to our Ambra1‐depleted model, muscles lacking ATG5 show increased lysosomal size and density. 37 The endo‐lysosomal compartment represents the final district of the autophagic pathway, and thus, it is not surprising that lysosomes are altered when autophagy is impaired. On the other hand, accumulation of endo‐lysosomal vesicles, clearly displayed by Ambra1 knockout muscles, was not described before in other in vitro and in vivo Ambra1‐deficient models. Therefore, in the near future, it would be interesting to further investigate new lysosomal‐related functions of Ambra1, under both physiological and pathological conditions.
Finally, our data suggest that Ambra1 may be a valuable molecular target to selectively enhance mitophagy and improve mitochondria health in myofibres, with possible implications for the prospective treatment of muscular dystrophies and metabolic diseases in which the mitophagy pathway is affected, as well as in age‐related sarcopenia. Furthermore, they point at the search for AMBRA1 gene mutations in patients affected by myopathies with abnormal mitochondrial buildup and hinting at defective mitophagy regulation.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Table S1. List of primer sequences used for genotyping or qPCR.
Table S2. List of antibodies and concentrations used for western blotting (WB) or immunofluorescence (IF).
Figure S1. Generation and characterization of Ambra1 knockout mice.
Figure S2. Characterization of muscle‐specific Ambra1 knockout mice.
Figure S3. Investigate mitochondria in muscles lacking Ambra1.
Acknowledgements
We are thankful to Dr. Gaia Gherardi for advice and technical support with TMRM analysis, to Dr. Pasquale D'Aqunzo for the cloning of the bicistronic vector, and to Dr. Dario Bizzotto for the maintenance of mouse colonies. We gratefully thank Prof. Noboru Mizushima for providing us with GFP‐LC3 reporter mice. We also thank Prof. Leonardo Salviati and Prof. Marco Sandri for their helpful discussions. We acknowledge the light and electron microscopy facility of the Biology Department of the University of Padova and the Next Generation Sequencing facility of the Istituto di Ricerca Pediatrica (IRP, Padova). We also thank all the members of Bonaldo laboratory for daily discussions and suggestions. This work was supported by the Italian Ministry of University and Research (Grants 2015FBNB5Y and 201742SBXA), the Telethon Foundation (Grants GGP14202 and GGP19229), the Cariparo Foundation, and the University of Padova. FS was supported by an AFM‐Telethon trampoline grant (AFM‐18376).
Gambarotto L., Metti S., Chrisam M., Cerqua C., Sabatelli P., Armani A., Zanon C., Spizzotin M., Castagnaro S., Strappazzon F., Grumati P., Cescon M., Braghetta P., Trevisson E., Cecconi F., and Bonaldo P. (2022) Ambra1 deficiency impairs mitophagy in skeletal muscle, Journal of Cachexia, Sarcopenia and Muscle, 13, 2211–2224, 10.1002/jcsm.13010
References
- 1. Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 2018;20:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 2020;21:204–224. [DOI] [PubMed] [Google Scholar]
- 3. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012;148:1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizushima N. Autophagy: process and function. Genes Dev 2007;21:2861–2873. [DOI] [PubMed] [Google Scholar]
- 5. Drake J, Yan Z. Mitophagy in maintaining skeletal muscle mitochondrial proteostasis and metabolic health with ageing. J Physiol 2017;595:6391–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Ende M, Grefte S, Plas R, Meijerink J, Witkamp RF, Keijer J, et al. Mitochondrial dynamics in cancer‐induced cachexia. Biochimica et Biophysica Acta (BBA) ‐ Rev Cancer 2018;1870:137–150. [DOI] [PubMed] [Google Scholar]
- 8. Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz‐Bonilla V, Sala D, et al. Mfn2 deficiency links age‐related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 2016;35:1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Su Z, Nie Y, Huang X, Zhu Y, Feng B, Tang L, et al. Mitophagy in hepatic insulin resistance: therapeutic potential and concerns. Front Pharmacol 2019;10:1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernardi P, Bonaldo P. Mitochondrial dysfunction and defective autophagy in the pathogenesis of collagen VI muscular dystrophies. Cold Spring Harb Perspect Biol 2013;5:a011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 2012;3:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cianfanelli V, de Zio D, di Bartolomeo S, Nazio F, Strappazzon F, Cecconi F. Ambra1 at a glance. J Cell Sci 2015;128:2003–2008. [DOI] [PubMed] [Google Scholar]
- 13. Maiani E, Milletti G, Nazio F, Holdgaard SG, Bartkova J, Rizza S, et al. AMBRA1 regulates cyclin D to guard S‐phase entry and genomic integrity. Nature 2021;592:799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010;191:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, Strooper B, et al. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 2011;31:10249–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 2015;22:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strappazzon F, di Rita A, Peschiaroli A, Leoncini PP, Locatelli F, Melino G, et al. HUWE1 controls MCL1 stability to unleash AMBRA1‐induced mitophagy. Cell Death Differ 2019;27:1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fimia GM, Stoykova A, Romagnoli A, Giunta L, di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007;447:1121–1125. [DOI] [PubMed] [Google Scholar]
- 19. Skobo T, Benato F, Grumati P, Meneghetti G, Cianfanelli V, Castagnaro S, et al. Zebrafish ambra1a and ambra1b knockdown impairs skeletal muscle development. PLoS ONE 2014;9:e99210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meneghetti G, Skobo T, Chrisam M, Fontana CM, Facchinello N, Nazio F, et al. Zebrafish ambra1a and ambra1b silencing affect heart development. Zebrafish 2020;17:163–176. [DOI] [PubMed] [Google Scholar]
- 21. Romanello V, Sandri M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol Life Sci 2020;78:1305–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oost LJ, Kustermann M, Armani A, Blaauw B, Romanello V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J Cachexia Sarcopenia Muscle 2019;10:630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010;16:1313–1320. [DOI] [PubMed] [Google Scholar]
- 24. Luan P, 'Amico D D, Andreux PA, Laurila P‐P, Wohlwend M, Li H, et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med 2021;13:319. [DOI] [PubMed] [Google Scholar]
- 25. Xia Q, Huang X, Huang J, Zheng Y, March ME, Li J, et al. The role of autophagy in skeletal muscle diseases. Front Physiol 2021;12:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- 27. Vainshtein A, Desjardins EM, Armani A, Sandri M, Hood DA. PGC‐1α modulates denervation‐induced mitophagy in skeletal muscle. Skel Musc 2015;5:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. di Leo L, Bodemeyer V, Bosisio FM, Claps G, Carretta M, Rizza S, et al. Loss of Ambra1 promotes melanoma growth and invasion. Nat Commun 2021;12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nazio F, Po A, Abballe L, Ballabio C, Diomedi Camassei F, Bordi M, et al. Targeting cancer stem cells in medulloblastoma by inhibiting AMBRA1 dual function in autophagy and STAT3 signalling. Acta Neuropathol 2021;142:537–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. di Rienzo M, Piacentini M, Fimia GM. A TRIM32‐AMBRA1‐ULK1 complex initiates the autophagy response in atrophic muscle cells. Autophagy 2019;15:1674–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cianfanelli V, Nazio F, Cecconi F. Connecting autophagy: AMBRA1 and its network of regulation. Mol Cell Oncol 2015;2:e970059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Narendra D, Tanaka A, Suen D‐F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin SM, Youle RJ. PINK1‐ and Parkin‐mediated mitophagy at a glance. J Cell Sci 2012;125:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gouspillou G, Godin R, Piquereau J, Picard M, Mofarrahi M, Mathew J, et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J Physiol 2018;596:2565–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H, Song P, Du L, Tian W, Yue W, Liu M, et al. Parkin ubiquitinates Drp1 for proteasome‐dependent degradation: implication of dysregulated mitochondrial dynamics in parkinson disease. J Biol Chem 2011;286:11649–11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, et al. DRP1‐mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun 2019;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 2008;17:3897–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507–515. [DOI] [PubMed] [Google Scholar]
- 39. Raben N, Schreiner C, Baum R, Takikita S, Xu S, Xie T, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—murine Pompe disease. Autophagy 2010;6:1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nemazanyy I, Blaauw B, Paolini C, Caillaud C, Protasi F, Mueller A, et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med 2013;5:870–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of primer sequences used for genotyping or qPCR.
Table S2. List of antibodies and concentrations used for western blotting (WB) or immunofluorescence (IF).
Figure S1. Generation and characterization of Ambra1 knockout mice.
Figure S2. Characterization of muscle‐specific Ambra1 knockout mice.
Figure S3. Investigate mitochondria in muscles lacking Ambra1.