

Summary:
Cell size is tightly controlled in healthy tissues, but it is unclear how deviations in cell size affect cell physiology. To address this, we measured how cell proteome changes with cell size. Size-dependent protein concentration changes are widespread and predicted by subcellular localization, size-dependent mRNA concentrations, and protein turnover. As proliferating cells grow larger, concentration changes typically associated with cellular senescence are increasingly pronounced, suggesting that large size may be a cause rather than just a consequence of cell senescence. Consistent with this hypothesis, larger cells are prone to replicative, DNA damage-, and CDK4/6i-induced senescence. Size-dependent changes to the proteome, including those associated with senescence, are not observed when an increase in cell size is accompanied by an increase in ploidy. Together, our findings show how cell size could impact many aspects of cell physiology through remodeling the proteome and provide a rationale for cell size control and polyploidization.
Graphical Abstract
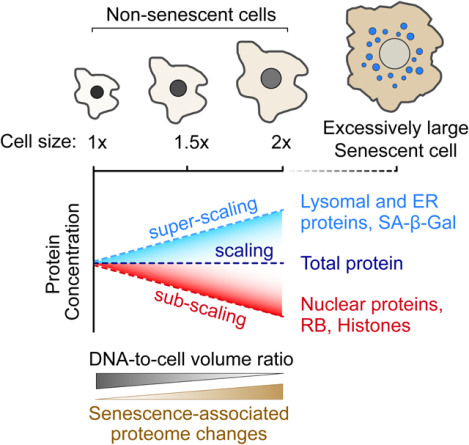
eTOC Blurb
Lanz et al. used mass spectrometry to measure how proteome composition changes with cell size (i.e., DNA-to-volume ratio). They found that different cellular components scale differently with size. Large cell size elicited senescence-like proteome changes and contributed to irreversible cell cycle exit upon replicative aging and exposure to DNA damage.
Introduction
Cells have dedicated mechanisms to control their size, which is one of the most prominent characteristics of distinct cell types (Ginzberg et al., 2015; Lloyd, 2013; Zatulovskiy and Skotheim, 2020). The link between the characteristic cell size and cell function is more obvious at the extremes of the cell size range. Red blood cells and lymphocytes need to be small to squeeze through tight spaces, while macrophages must be larger to engulf a wide range of targets. However, in the middle of the cell size range, including epithelial cells and fibroblasts, the link between cell size and function is unclear. One possibility is that these cells control their size to enhance proliferation, as their rapid turnover is a key part of their physiological function (Lengefeld et al., 2021; Miettinen and Bjorklund, 2016). Yet, even if these cells are optimized for growth and proliferation, as has been indicated (Lengefeld et al., 2021; Neurohr et al., 2019), it is unclear why there would be an optimal cell size. As cells get larger, it has long been assumed that most proteins and RNA remain at constant concentrations (Berenson et al., 2019; Crissman and Steinkamp, 1973; Kempe et al., 2015; Lin and Amir, 2018; Padovan-Merhar et al., 2015; Son et al., 2015; Williamson and Scopes, 1961; Zlotek-Zlotkiewicz et al., 2015), and organelle volumes, such as the nucleus, increase in direct proportion to cell size (Cantwell and Nurse, 2019; Chan and Marshall, 2010; Marshall, 2020). If protein and RNA concentrations remain constant, larger cells should be capable of proportionally more biosynthesis. However, this is not the case. There is a limit to the size range of efficient biosynthesis (Zhurinsky et al., 2010), and excessively large cells exhibit loss of mitochondrial potential (Miettinen and Bjorklund, 2016), dilution of their cytoplasm (Neurohr et al., 2019), and reduced proliferation (Demidenko and Blagosklonny, 2008). Moreover, recent work has demonstrated the remarkable effect even small variations in cell size can have on hematopoietic stem cell proliferation (Lengefeld et al., 2021).
One possible explanation for why there is an optimal cell size for biosynthesis would be if many key cellular proteins did not remain at constant concentration as cells grow. Then, the further cells are from their target size, the more concentrations of these proteins would change, and the more growth and metabolism would deviate from the optimum. Intriguingly, investigations of the mechanisms cells use to control their size identified a class of proteins whose concentrations change with cell size. In budding yeast, human, and plant cells, key cell cycle inhibitors are not synthesized in proportion to cell size so that they are diluted by cell growth, a behavior defined as sub-scaling (Figure 1A) (D’Ario et al., 2021; Schmoller et al., 2015; Zatulovskiy et al., 2020). Larger cells therefore have lower concentrations of cell cycle inhibitors, which promotes their division. In fission yeast, division is in part promoted by a size-dependent increase in the concentration of a cell cycle activator (Keifenheim et al., 2017), a behavior defined as super-scaling (Figure 1A) (Chen et al., 2020). If this phenomenon of size-dependent protein concentration changes were widespread across the proteome it would provide an explanation for why there is an optimal cell size. Namely, the further a cell would be from its target size, the further its intracellular protein concentrations would be from their optimum. To explore this possibility, we first measured how the proteome changes as a function of the natural cell size variation in proliferating human cells.
Figure 1. Cell size shapes the human proteome.
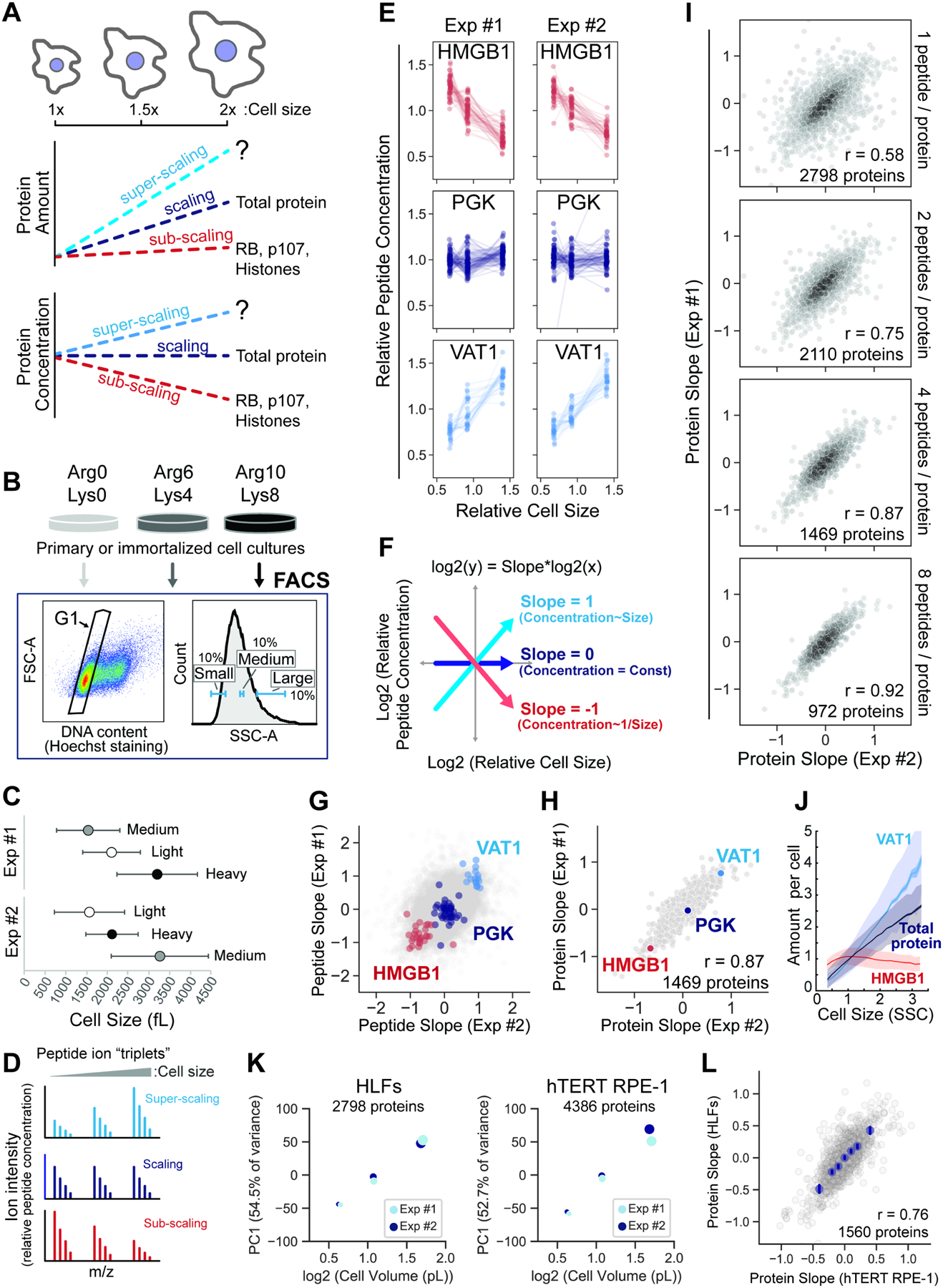
(A) Schematic illustration of the potential scaling relationships between protein amount, concentration, and cell size. (B) Metabolically labeled HLFs cells were gated by G1 DNA content and sorted into three size bins based on the side scatter parameter (SSC) as a proxy for cell size using FACS. (C) The attainment of differentially sized G1 cells was confirmed using a Coulter counter. Central dots represent the mean volume for each size bin and error bars represent the standard deviation. SILAC labeling orientation was swapped for replicate experiments. (D) Ion intensities within SILAC “triplets” represent relative peptide concentrations. (E) SILAC channel intensities are plotted for three proteins that exemplify different size scaling behaviors. Each dotted line represents an independent peptide measurement. (F) Derivation of a slope value that describes the scaling behavior of each peptide triplet. Proteins with a slope of 0 maintain a constant cellular concentration regardless of cell volume. A slope value of 1 corresponds to an increase in concentration that is proportional to the increase in volume and a slope of −1 corresponds to dilution (1/volume). (G) Peptide and (H) Protein slope values from two replicate experiments. Only proteins with at least 4 independent peptide measurements in both experiments are shown. (I) Correlation of protein slope values from two replicate experiments. A threshold for the minimum number of peptide measurements per protein is indicated in each panel. (J) Immunofluorescence intensity measured as a function of SSC (cell size) using flow cytometry. Total protein amount is inferred from the measurement of carboxyfluorescein succinimidyl ester (CFSE) dye. The data were binned by cell size and plotted as mean protein amounts per cell for each size bin (solid lines). Dark shaded area shows standard error of the mean for each bin, and light shaded area shows the standard deviation. A representative plot is shown for n=3 biological replicates for each experiment. 100,000 cells were analyzed for each sample. (K) Principal component analysis of the relative concentration of each protein in small-, medium-, and large-sized cells. The 1st principal component is plotted against cell volume for two cell lines, HLFs (fibroblast) and hTERT RPE-1 (epithelial). Dot size represents mean cell volume. (L) Similarity of the cell size-dependent concentration changes between primary HLF and immortalized hTERT RPE-1 cells. Protein Slope values for each cell type are the mean of two biological replicate experiments. Only proteins with at least 3 independent peptide measurements in each biological replicate are depicted (Table S1).
Results
Measuring cell size-dependent changes to the human cell proteome
To measure the scaling behavior of the human proteome (Figure 1A), we developed a method based on triple-SILAC (Stable Isotope Labeling by Amino acids In Cell Culture) proteomics (described in Figure S1), which enables the simultaneous measurement of thousands of individual proteins. Asynchronously proliferating SILAC-labeled primary Human Lung Fibroblasts (HLFs) and hTERT-immortalized RPE-1 epithelial cells were gated for G1 DNA content and sorted into three size bins (small, medium, and large) using fluorescence-activated cell sorting (FACS) (Figure 1B). SILAC labeling orientation was swapped for replicate experiments, and the attainment of differentially sized G1 cells was confirmed using a Coulter counter (Figure 1C and S2A). The SILAC channel intensities within a peptide “triplet” represent relative peptide concentrations (Figure 1D and S1A), so we used the behavior of multiple independent peptide triplets to determine the size scaling relationship for individual proteins. HMGB1, PGK, and VAT1 are proteins we chose to exemplify sub-scaling, scaling, and super-scaling behavior, respectively (Figure 1E). Rather than measure SILAC ratios, as is typically done, we calculated a slope value to describe the size scaling behavior of each peptide triplet (Figure 1F, S1D–G). Peptide triplets with positive and negative slopes represent peptides (Figure 1G), and therefore proteins (Figure 1H), whose concentrations are super- and sub-scaling, respectively, whereas triplets with a slope value near 0 maintain an approximately constant concentration as a function of G1 cell size.
We observed a continuum of size scaling behaviors across the proteome, which spanned slope values from −1 to 1, indicating that a 2-fold increase in cell size can lead to a 2-fold increase or decrease in concentration for a given protein (Figure 1H; Table S1). Several chromatin-associated High Mobility Group proteins (HMGs), including HMGB1, are diluted in larger cells and so exhibit sub-scaling behavior like what has been previously described for RB, Whi5, and histones (Claude et al., 2020; Swaffer et al., 2021a; Zatulovskiy et al., 2020). We also identified a diverse set of super-scaling proteins, such as VAT1, whose concentration increases as a function of cell size. Setting a requirement for multiple independent measurements per protein significantly improved the correspondence between replicate experiments and yielded a high-confidence set of ~1,500 proteins, each having at least 4 distinct peptide measurements (Figure 1I; Table S1). We validated candidate super- and sub-scaling behaviors for a subset of proteins using immunofluorescence combined with flow cytometry (Figure 1J and S3). In both primary lung fibroblasts (HLF) and immortalized epithelial cells (hTERT RPE-1), changes to the proteome were directly proportional to changes in G1 cell volume, indicating that similar concentration changes take place when cell size increases from small to medium as when it increases from medium to large (Figure 1K and S4). Importantly, the size-dependency of the proteome is not specific to a single human cell type, as we found a striking degree of similarity in the proteome scaling of both cell lines we measured (Figure 1L).
We also confirmed that the process of cell sorting did not affect our proteome measurements (Figure S5A) and that isolation of different sized cells using SSC (Side Scatter) or the total protein dye CFSE (carboxyfluorescein succinimidyl ester) as proxies for cell size (Berenson et al., 2019; Tzur et al., 2011) yielded similar results (Figure S5B–D; Table S2). Since larger G1 cells have on average spent more time in G1, it is possible that the protein concentration changes we observe reflect time in G1 rather than cell size. To control for this possibility, we selected different sized G1 cells that were synchronously released from a Thymidine-Nocodazole cell cycle arrest. We found a highly similar size-scaling relationship in our synchronous and asynchronous experiments (Figure S6; Table S3), indicating that cell size, not time in the G1 cell cycle phase, is driving changes to the proteome.
Size scaling of protein concentrations correlate with subcellular localization
To examine how cell size may influence different aspects of cellular biology, we asked if different groups of related or interacting proteins exhibited similar size scaling behavior. Indeed, this was the case. For example, all 17 detected histone variants sub-scaled and had slope values near −1 (Figure 2A), indicating that histones are maintained in proportion to DNA (Wisniewski et al., 2014) and are therefore diluted by cell growth in G1 (Claude et al., 2020; Swaffer et al., 2021a). Moreover, 5 of the 6 detected cathepsin proteases were strongly super-scaling and nearly doubled in concentration from the smallest to the largest cell size bin. Interestingly, we found that all individual protein subunits of the ribosome and proteasome showed small but highly consistent sub- and super-scaling behaviors respectively (Figure 2A) and that, more generally, proteins that are partners in a complex scale similarly (Figure 2B; Table S4). We also re-confirmed the sub-scaling behavior of the cell cycle inhibitor RB (Zatulovskiy et al., 2020) (Figure S5E and S7E), which was the 20th most sub-scaling protein (of a total of 4,387 proteins) identified in our RPE-1 dataset.
Figure 2. Differential size scaling of organelle protein content.
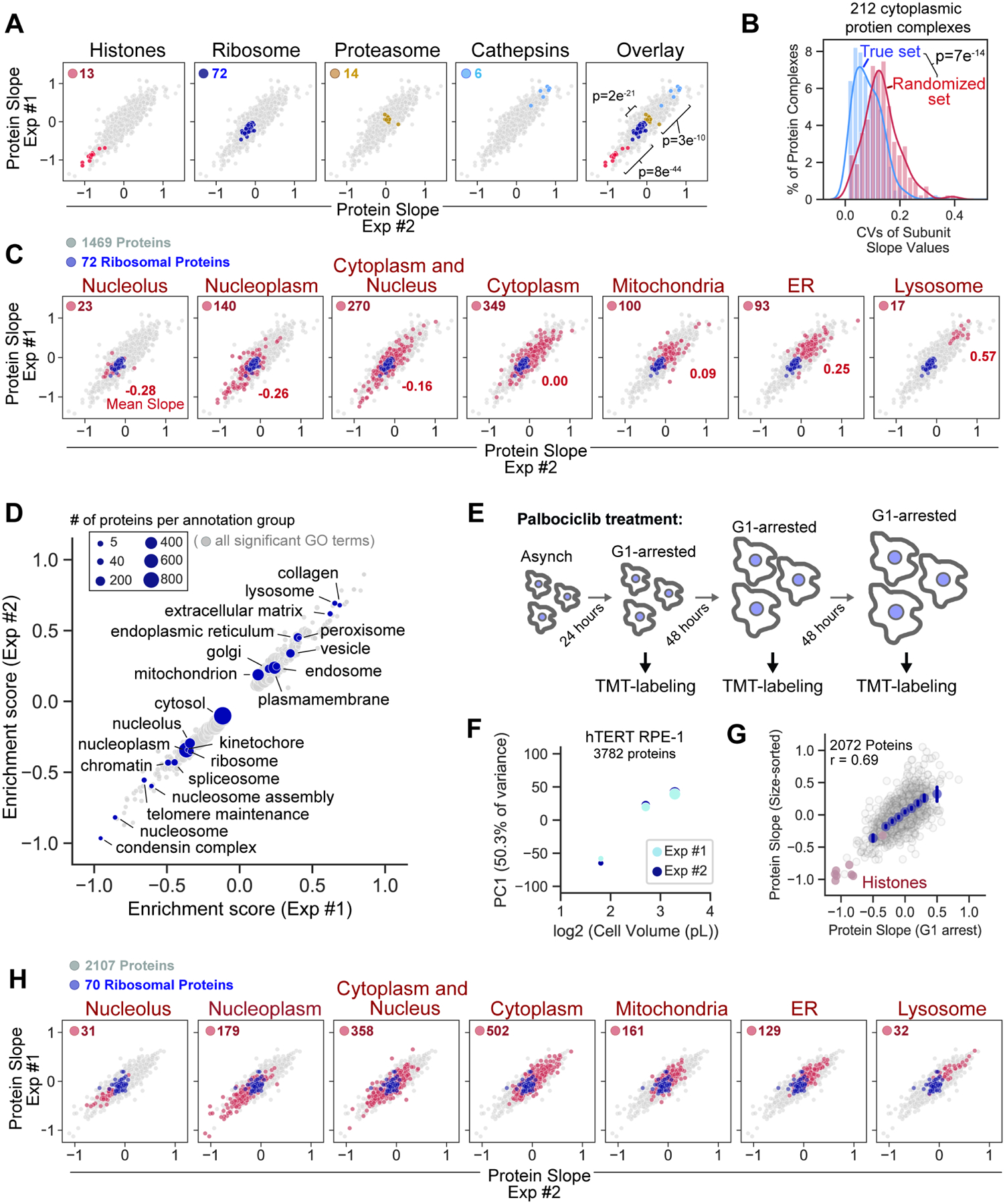
(A) Scaling behavior of protein groups. Significance is determined by t-test between adjacent protein groups. (B) Variance of subunit Protein Slope values for 212 annotated protein complexes (Table S4). Only proteins with cytoplasmic annotation are considered. Significance is determined by t-test between the proteins grouped by complex and a randomized control set. (C) Scaling behavior of proteins based on subcellular localization (see methods). The number of highlighted proteins and their average slope are indicated for each panel. Ribosomal proteins are plotted in dark blue. (D) 2D annotation enrichment analysis using the Protein Slope values calculated in HLF cells. Table S5 contains a complete list of enrichment scores for significantly super- or sub-scaling GO terms. (E) Asynchronous RPE-1 cells were arrested in G1 with 1μM Palbociclib and (F) the relative change in the concentrations of individual proteins determined using TMT quantitation (Table S6). Dot size represents mean cell size. (G) Correlation of slope values derived from size-sorted (asynchronous) and G1-arrested (Palbociclib treated) RPE-1 cells. For G1 arrested cells, slope values for individual proteins were determined using the TMT reporter ions (MS3-level) and calculated as described in Figure 1. X-axis bins are shown in dark blue. Error bars represent the 95% confidence interval and r denotes the Pearson correlation coefficient. (H) Size-scaling of organelle protein content in cells arrested in G1 with 1μM Palbociclib, as plotted in (C).
We next assessed how size-scaling behavior relates to subcellular location. To do this, we first annotated the proteome based on a strict association with a single, major cellular compartment. Subcellular localization was a strong predictor of size scaling behavior, with ER- and lysosome-resident proteins becoming increasingly concentrated with size, and nucleoplasmic/nucleolar proteins becoming more dilute (Figure 2C, S7A, and S7B). We confirmed lysosome super-scaling by measuring the lysosome-labelling dye Lysotracker and the lysosomal protein LAMP1 (Figure S7C). While we did not observe a difference in scaling behavior of luminal- and membrane-associated organelle proteins, we were able to identify multiple protein domains, sequence features, and post-translation modifications that are preferentially associated with super- or sub- scaling proteins (Figure S8A). A less stringent 2D annotation enrichment revealed a range of intermediate scaling behaviors across multiple different cellular compartments and annotation groups (Figure 2D and S8B; Table S5). Importantly, we confirmed that large G1 cells exposed to generated by long-term treatment with the CDK4/6 inhibitor Palbociclib, which arrests cells in G1 but does not inhibit cell growth (Figure 2E) (Neurohr et al., 2019; Tan et al., 2021), recapitulated the proteome changes we observed in size-sorted G1 cells (Figure 2F–H; Table S6). In further support of these findings, a proteomic analysis revealed similar size-scaling relationships when the relative protein concentration was plotted against the mean cell size for each cancer cell line in the NCI60 collection (Cheng et al., 2021). Taken together, these analyses refute the commonly held assumption that the protein content of cellular components scales in uniform proportion to cell volume. While total protein concentration may be mostly constant within a cell’s natural size range, this is not necessarily true for individual proteins.
A linear model based on diverse mechanisms predicts protein size scaling
After identifying size-dependent changes in the concentrations of individual proteins, we sought to investigate the underlying mechanisms. We first tested whether size-dependent changes in a protein’s concentration are explained by changes in the concentration of its mRNA transcript. To do this, we performed RNA-seq on size-sorted G1 cells and calculated mRNA slope values analogous to the protein slopes described in Figure 1. We found that although there was significant correlation between the protein and RNA slope values, there was large variability in protein scaling slopes for any given RNA slope (Figure 3A–C, and S9A; Table S7). Overall, changes in mRNA concentrations explained only a minority of the size-dependent variation in protein concentration. We therefore explored additional post-transcriptional variables using linear models. We found that a protein’s turnover (Zecha et al., 2018) and codon affinity (Frenkel-Morgenstern et al., 2012) both significantly correlated with the Protein Slope (Figure S9B and S9C; Table S8). Iterative incorporation of each parameter (mRNA Slope, protein turnover, and codon affinity score) significantly improved the model with minimal collinearity (Figure 3D and S10). Since none of these parameters explained the striking difference in size scaling highlighted in Figure 2C, we incorporated an additional variable to account for a protein’s subcellular localization (Figure 3D and S10). Using the correlation between biological replicates as a benchmark for the maximum size-dependent variation any model could predict, we find that our composite model predicts ~60% of this maximum size-dependent variance. Taken together, this analysis indicates that the size-dependency of a protein’s concentration is regulated both pre- and post-transcriptionally.
Figure 3. Diverse mechanisms control proteome size-dependency.
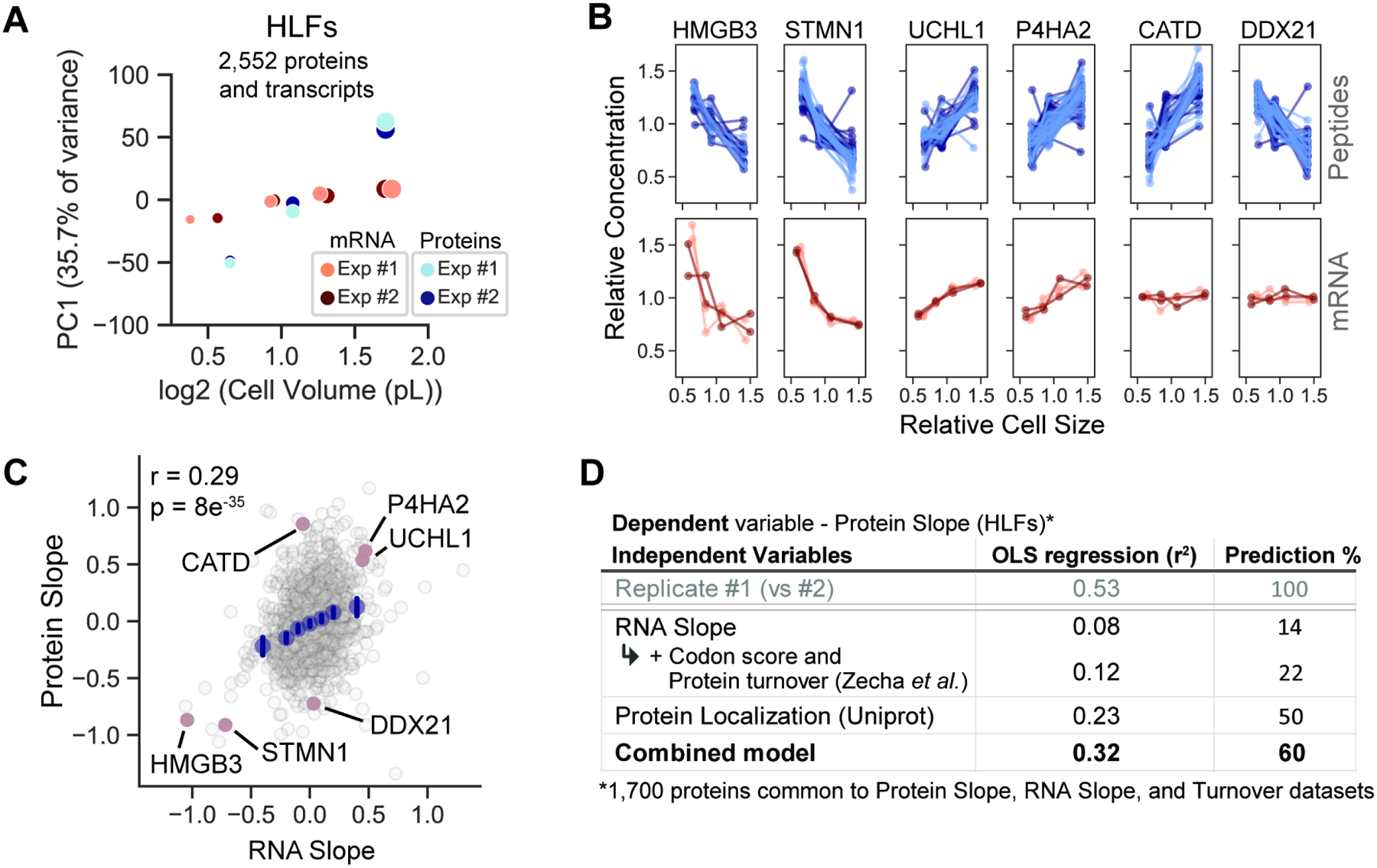
(A) Principal component analysis of the relative concentration of each protein and its corresponding transcript in small-, medium-, and large-sized cells. The 1st principal component is plotted against cell volume. Size dependency of the transcriptome was determined by sequencing the mRNA of size-sorted G1 cells. Dot size represents mean cell volume. (B) Size-dependent concentration changes for a representative set of proteins and their corresponding mRNA transcripts. For proteins, each connected line represents a unique peptide measurement from two biological replicate experiments (light and dark blue). For RNA, technical replicates are denoted in the same color, while biological replicates are denoted in different colors (4 replicates in total). (C) Correlation of size-dependent proteome and transcriptome changes. Examples in (B) are highlighted are in red. RNA Slope is calculated in a manner analogous to the Protein Slope using RPKM values (Table S7). (D) Ordinary least squares regression model predicts the size scaling behavior of 1,700 individual proteins based on their subcellular localization and additional features. Prediction % denotes the amount of the variance predicted by the model divided by the variance of a replicate experiment (r2model/r2replicates), the benchmark for how much a model could predict. See Figure S10 for a full description of the model.
Large size promotes senescence in primary human fibroblasts
Curiously, increasing cell size in proliferating cells is accompanied by proteomic changes normally associated with cell senescence, including the upregulation of beta-galactosidase, lysosomal proteins, and metalloproteases, and the downregulation of Ki67, HMGB proteins, and LaminB (Hernandez-Segura et al., 2018) (Figure 4A and S7D). Senescence-associated secretory phenotype (SASP) proteins (Ruscetti et al., 2018) were also super scaling (Figure 4B and S11). While large cell size is associated with senescence (Hernandez-Segura et al., 2018), it has generally been thought that large size results from a senescent cell’s inability to divide while at the same time maintaining cell growth. However, our experiments indicate that in proliferating (non-senescent) cells, increasing cell size itself results in proteome changes that gradually approach those found in senescent cells, which supports the hypothesis that cell size per se promotes senescence (Lengefeld et al., 2021; Neurohr et al., 2019). This is consistent with earlier reports that continued cell growth and hypertrophy are required to induce senescence in cell cycle arrested cells (Demidenko and Blagosklonny, 2008).
Figure 4. Senescence-associated changes to the proteome are increasingly pronounced in large proliferating cells.
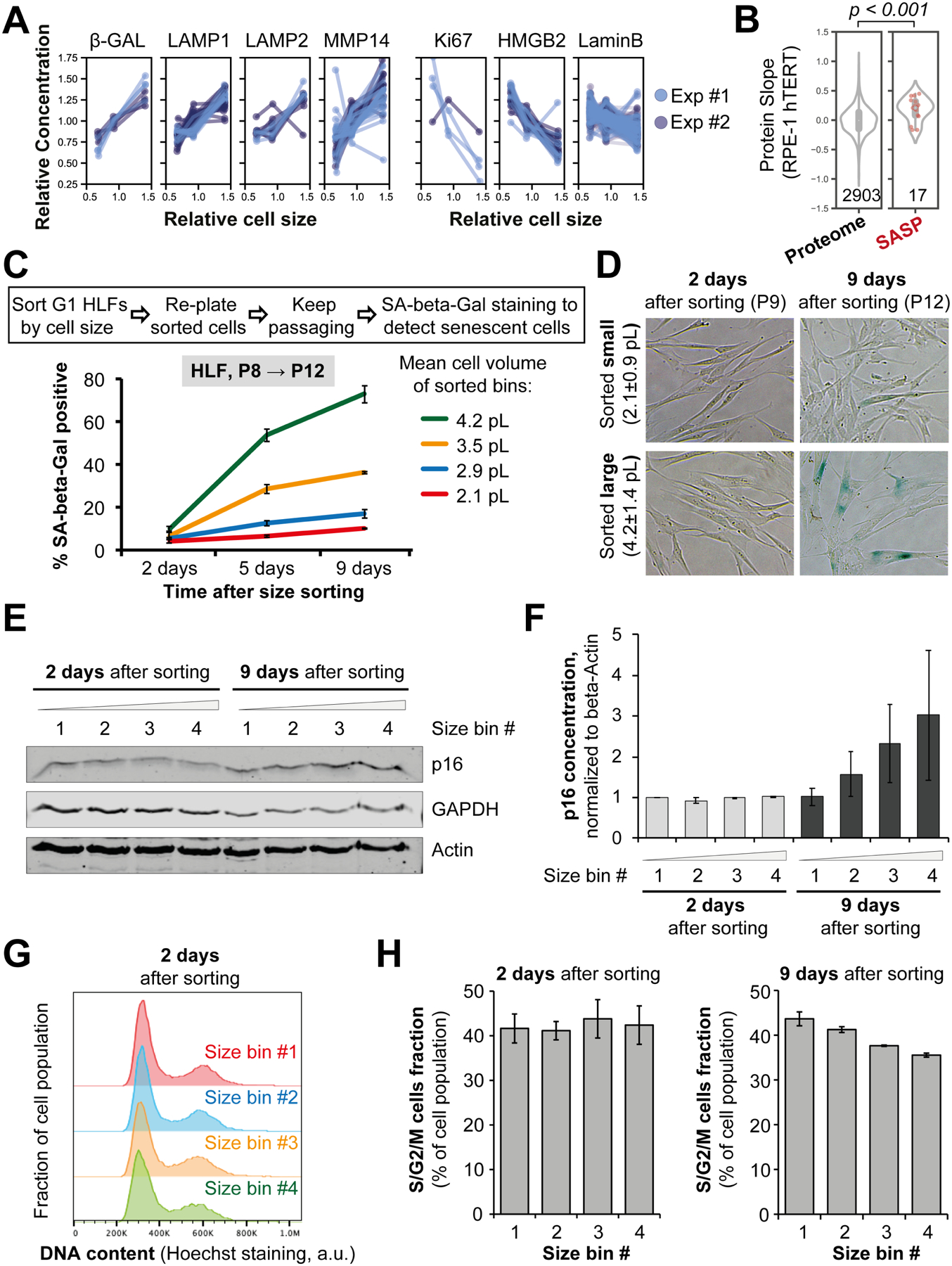
(A, B) Examples of protein concentration changes typically associated with senescence that we observe in proliferating cells. This includes size-dependent intracellular proteins (A) and SASP (senescence-associated secretory phenotype) proteins (B). (C-F) Replicative senescence of different-sized primary HLF cells. Asynchronous HLFs were gated for G1 DNA content and sorted into four bins by size using FACS, then replated and stained for the senescence marker SA-beta-Gal at the indicated time points (D). Percentage of blue-stained SA-beta-Gal positive cells (C) was calculated for each time point and plotted for each sorted size bin as mean ± standard error. Cell sizes for each bin are shown as mean ± SD in (C). P denotes the cell passage number. (E) A representative immunoblot showing the dynamics of the senescence marker p16 in different-sized HLF cells at the indicated time after size sorting. (F) Quantification of the immunoblots measuring p16 in different-sized HLF cells. Data are shown as mean ± standard error, n=3. The antibodies against senescence markers were validated using immunoblotting and immunofluorescent staining of early- and late-passage primary HLFs (Figure S13). (G) Cell cycle distributions of HLF cells 2 days after sorting by G1 cell size and re-plating. Size bins correspond to the bins in panel (C). (H) Percentage of cells in S/G2/M phases of the cell cycle for each of the four sorted cell size bins, 2 and 9 days after sorting.
To test if large cell size contributes to senescence, we used primary human lung fibroblasts (HLF) that naturally senesce after 10–15 passages. We sorted HLFs into 4 cell size bins at passage #8, re-plated the cells, and then cultured them for an additional 4 passages. After these 4 additional passages, cells that were larger at the time of sorting had higher concentrations of the senescence marker p16 and had higher senescence-associated beta-galactosidase (SA-beta-Gal) staining (Figure 4C–F). This is despite the fact large and small cells did not differ in the mRNA concentrations of senescence markers shortly after sorting and were equally proliferative (Figure 4G, 4H, and S12A–D). There was also no correlation between cell size and telomere length (Figure S12E). Moreover, when the cells were induced to grow large through G1 cell cycle arrest with the CDK4/6 inhibitor Palbociclib (Figure 5A), the expression of senescence markers p21 and p16, as well as the percentage of SA-beta-Gal-positive cells, progressively increased with longer Palbociclib treatments (Figure 5B–H). Importantly, co-treatment with the mTOR inhibitor Rapamycin, which slows down cell growth, reduced the expression of senescence markers in Palbociclib-arrested cells (Figure 5C–H). This finding supports our hypothesis that large cell size promotes replicative senescence in primary cells.
Figure 5. Large cell size promotes senescence.
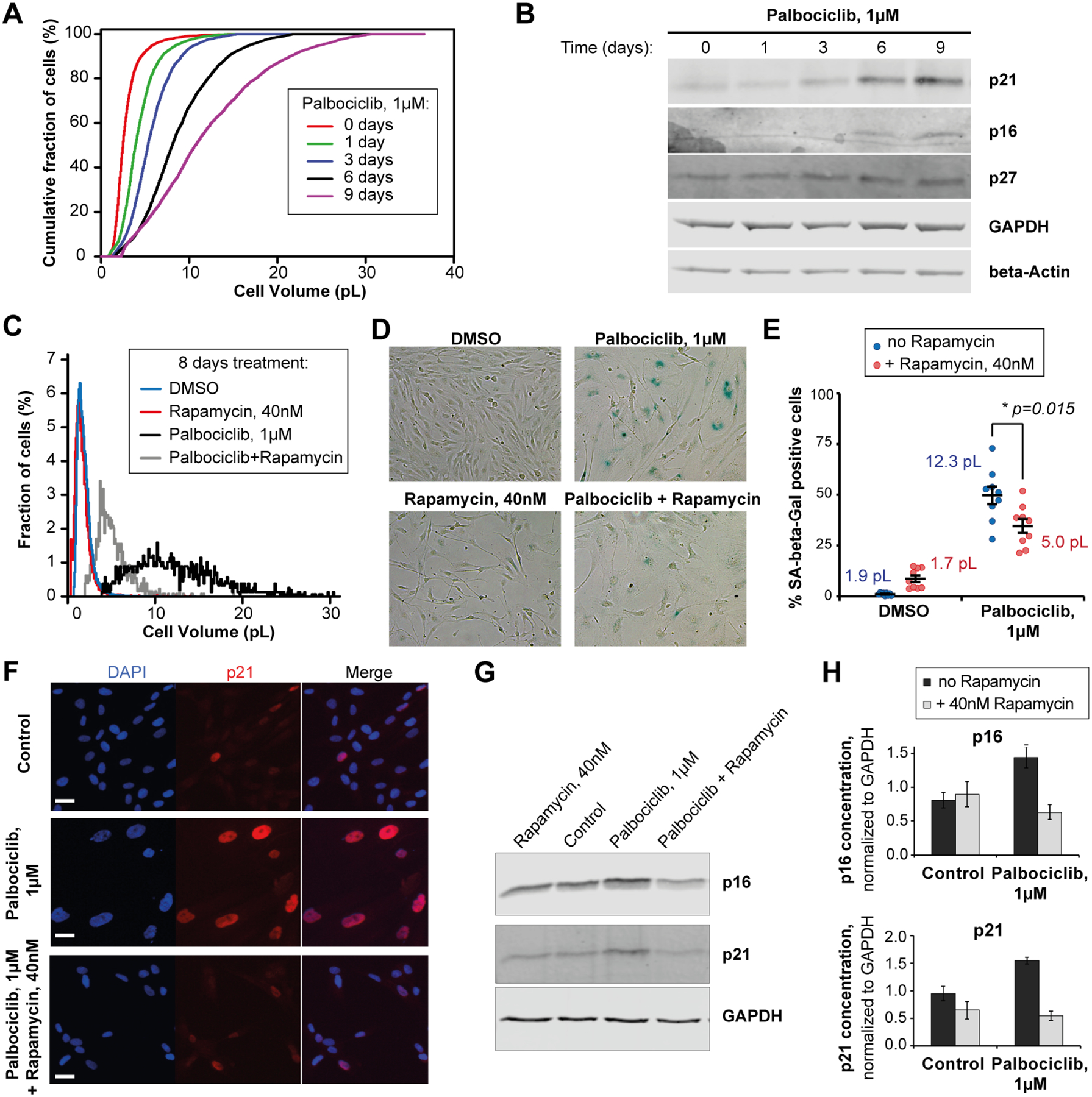
(A) Cumulative cell size distributions of HLF cells arrested in G1 phase for 0 to 9 days with 1μM of the CDK4/6 inhibitor Palbociclib. (B) A representative immunoblot showing the accumulation of senescence markers p16 and p21 in the HLF cells that increase in size upon Palbociclib arrest. (C, D) Cell size distributions (C) and SA-beta-Gal staining images (D) of the RPE-1 cells treated for 8 days with DMSO or Palbociclib, in the presence or absence of Rapamycin to determine the effect of cell size reduction on senescence dynamics. (E) Effect of Rapamycin, which reduces cell growth, on the percentage (±standard error) of SA-beta-Gal positive cells in RPE-1 cultures treated with Palbociclib for 8 days. Values shown next to each condition indicate the mean cell sizes after 8 days of treatment. SA-beta-Gal quantification for every data point in included 700–1200 cells quantified from 9 different fields of view. Values shown next to each condition indicate the mean cell sizes after 8 days of treatment. (F-H) Effects of Rapamycin on the expression of senescence markers p21 and p16 in HLF cells that were treated with Palbociclib for 8 days to increase cell size. (F) Immunofluorescent staining against p21 in HLF cells. Scale bar = 10μm. (G) A representative immunoblot against p21 and p16. (H) Quantification of p21 and p16 immunoblots. Data are shown as mean ± standard error, n = 4 biological replicates.
Large cell size promotes drug-induced senescence in telomerase-immortalized cells
Having shown that increased cell size is associated with increased rate of replicative senescence in primary cells, we next sought to examine how cell size contributes to a drug-induced senescence in the cells that do not senesce naturally. To do this, we treated size-sorted telomerase-immortalized human retinal pigment epithelium cells (hTERT RPE-1) with a low dose of a DNA damaging agent (10 ng/ml Doxorubicin), which triggers the DNA damage response. In some ways this is similar to the telomere shortening occurring in primary fibroblasts. We note the proteome’s size-dependency in untreated G1 RPE-1 cells is very similar to that of HLFs (Figure 1L and S7). Consistent with the previous result, larger cells increasingly induced the senescence marker SA-beta-Gal upon Doxorubicin treatment (Figure 6A and 6B). Moreover, co-treatment with the mTOR inhibitor Rapamycin, which reduces cell size, significantly attenuates the SA-beta-Gal staining induced by prolonged treatment with Doxorubicin or the Cdk4/6 inhibitor Palbociclib (Figure S14), a finding consistent with previous work (Demidenko et al., 2009). We note that, unlike in our experiments using primary fibroblasts, the concentrations of p16 did not increase with cell size in RPE-1 cells (Table S1), which is consistent with the observations of others (Cheng et al., 2021).
Figure 6. Larger cells are more prone to DNA damage-induced senescence.
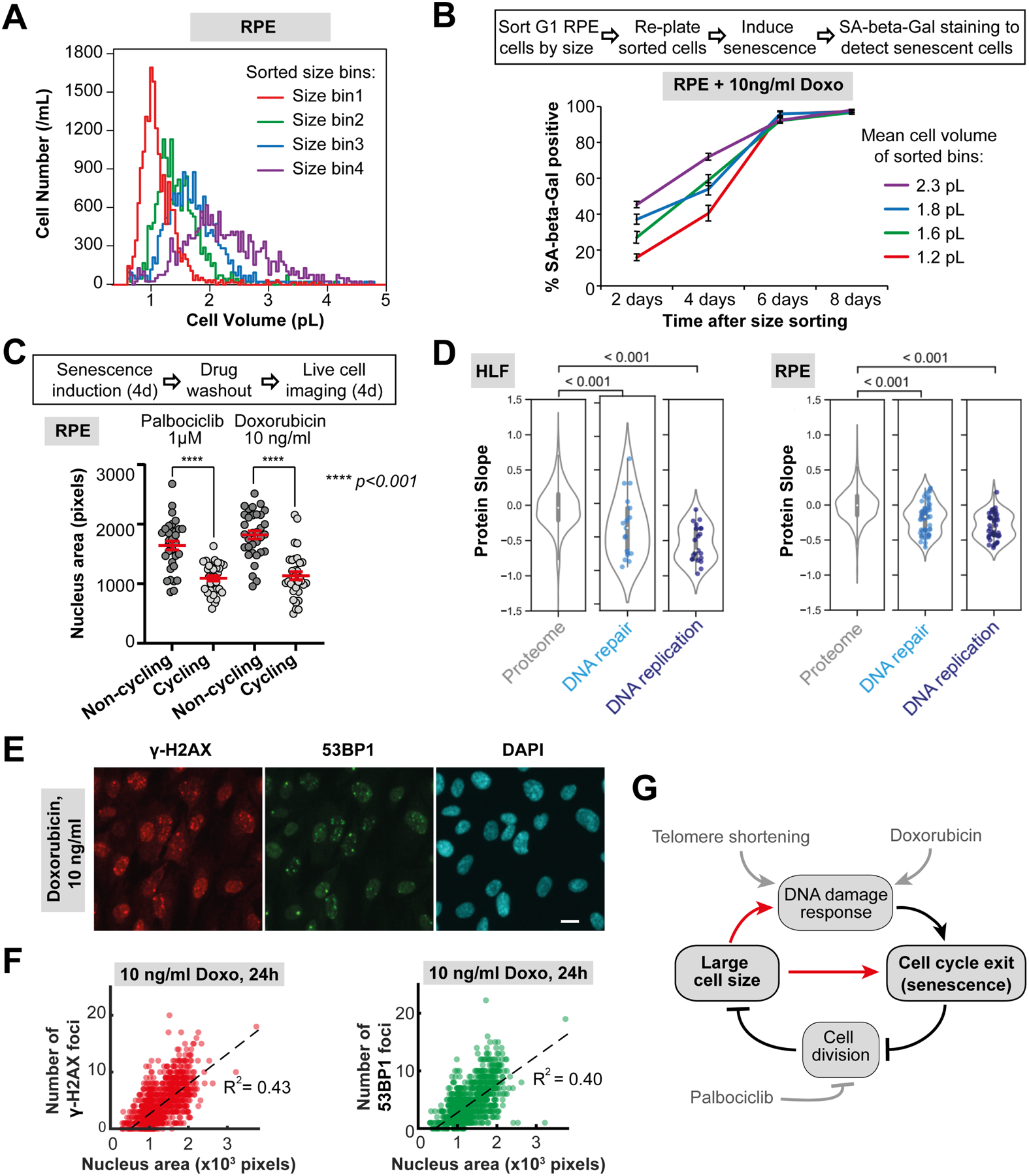
(A) Asynchronous RPE-1 cells were gated for G1 DNA content and sorted into four bins by size using FACS. (B) Sorted cells were replated, cultured in the presence of the DNA damaging agent Doxorubicin (10 ng/ml), and then stained for SA-beta-Gal at the indicated time points to determine the DNA damage-induced senescence dynamics. (C) Large cell size inhibits cell cycle re-entry after G1 arrest or DNA damage to promote senescence. RPE-1 cells were treated with Palbociclib or a low dose of Doxorubicin for 4 days. Then, the drugs were washed out, and the cells were imaged for 4 days to identify cells that re-enter the cell cycle, and cells that remain arrested in a senescent state. Nuclear area was used as a proxy for cell size and cell cycle re-entry was determined using the fluorescent cell cycle phase reporters Cdt1-mKO2 (G1 reporter) and Geminin-mAG (S/G2/M reporter) (Sakaue-Sawano et al., 2008). N = 33 cells for each data point. (D) Protein slope values for all proteins annotated as DNA repair (GO:0006281) and DNA replication (GO:0006260) that were present in our dataset. (E) Immunofluorescence staining of RPE-1 cells treated with 10 ng/ml Doxorubicin for 24 hours against γ-H2AX (red) and 53BP1 (green), with DAPI staining shown in cyan. Scale bar = 10μm. (F) Number of γ-H2AX and 53BP1 loci in RPE-1 cells treated with 10 ng/ml Doxorubicin plotted against the nucleus area, which serves as a proxy for cell size. N = 1265 cells. All the experiments were performed in n = 3 biological replicates. (G) Model of relationships between DNA-related stress, large cell size, and senescence.
So far, we have only assayed senescence using indirect markers rather than the durable cell cycle arrest that defines a senescent cell (Sharpless and Sherr, 2015). We therefore sought to more directly test whether large cell size inhibits cell cycle re-entry following the withdrawal of two treatments previously shown to induce senescence (Neurohr et al., 2019). We treated hTERT RPE-1 cells with Palbociclib or a low-dose of Doxorubicin for 4 days to induce senescence in a fraction of the cell population. We then washed out the drugs and imaged these enlarged cells for 4 additional days to determine which cells re-entered the cell cycle and which cells remained durably arrested (Figure 6C). Importantly, Palbociclib or Doxorubicin treatment arrests cells without stopping cell growth, and thus generates a population of cells with a range of large cell sizes that exhibit senescent features (Neurohr et al., 2019). Since all these cells are exposed to drug for the same duration, we can isolate cell size as a determining factor for durable cell cycle arrest. We found that the cells that were larger at the time of drug washout are more likely to remain arrested than smaller cells (Figure 6C). This supports the hypothesis that larger size makes cells more prone to senescence, consistent with findings for excessively large budding yeast (Neurohr et al., 2019). Large cell size may help explain the durable cell cycle arrest in response to transient exposure to a DNA damaging agent. This is because the excessive cell growth that occurs while cells delay cell cycle progression to repair DNA can inhibit cell cycle re-entry.
Larger cells have more DNA damage after Doxorubicin treatment
Our proteomics data show that most DNA repair and replication factors subscale with cell size (Figure 6D). To test if large cells are more prone to DNA damage than small cells, we treated the cells with a low concentration (10ng/ml) of Doxorubicin for 24 hours to induce DNA damage, and then quantified the numbers of γ-H2AX and 53BP1 loci that mark unrepaired DNA breaks in cell nuclei (Figure 6E, 6F, and S15). We found that the number of those loci per nucleus increases with the nucleus area (Figure 6F). The total amount of γ-H2AX marker per cell also increases with cells size within the G1 population of cells (Figure S15F–H). This supports the idea that larger cells have more DNA damage, which may promote senescence. Crucially, we are not stating that large cell size is the only driver of senescence, but rather proposing that the large size characteristic of senescent cells may itself further promote this state by inhibiting cell cycle progression and maintaining cell cycle arrest (Figure 6G). Consistent with this hypothesis, mutations to the Rb-family proteins that reduce cell size prevent senescence of mouse embryonic fibroblasts and hematopoietic stem cells (Dannenberg et al., 2000; Lengefeld et al., 2021; Sage et al., 2000).
Cell volume-to-ploidy ratio drives size-dependent proteome changes
Our data so far are consistent with the hypothesis that large cell size inhibits cell division, and that this may be due to widespread changes in the concentrations of individual proteins as cells grow larger. The further cells are from their target size, the more protein concentrations deviate from their optimum. However, in apparent contradiction to this model, there are many examples of large animal cells that proceed through the cell cycle and maintain highly efficient cell growth (Lee et al., 2009; Mu et al., 2020). In many cases, such large cells are polyploid and therefore do not have an aberrant cell size-to-ploidy ratio. We therefore sought to test whether protein size-scaling is determined by cell size per se or by the cell size-to-ploidy ratio. To do this, we sought to compare the proteomes of diploid, tetraploid, and octoploid (2N, 4N, 8N) G1-phase cells (Figure 7A and 7B). To obtain G1 cells with different ploidies, we induced endoreduplication in RPE-1 cells using a moderate dose of the Aurora kinase B inhibitor barasertib (75nM, 48 hours) (Mu et al., 2020) and sorted 2N, 4N, and 8N G1 cells from a mixed population using FACS (Figure 7A). While ploidy increases are accompanied by a corresponding increase in cell size (Figure 7A), proteomic analysis showed that the ploidy-sorted cells do not exhibit the same proteome changes as size-sorted cells with 2N ploidy (Figure 7C, 7D, and 7E; Table S9). Taken together, these results indicate that the proteome’s size-dependency is largely due to changes in the cell size-to-ploidy ratio. Thus, despite being different sizes, cells of different ploidy maintain similar protein concentrations, which may be why the excessively large size of polyploid cells does not impede their cell cycle progression.
Figure 7. Cell volume-to-ploidy ratio drives size-dependent proteome changes.
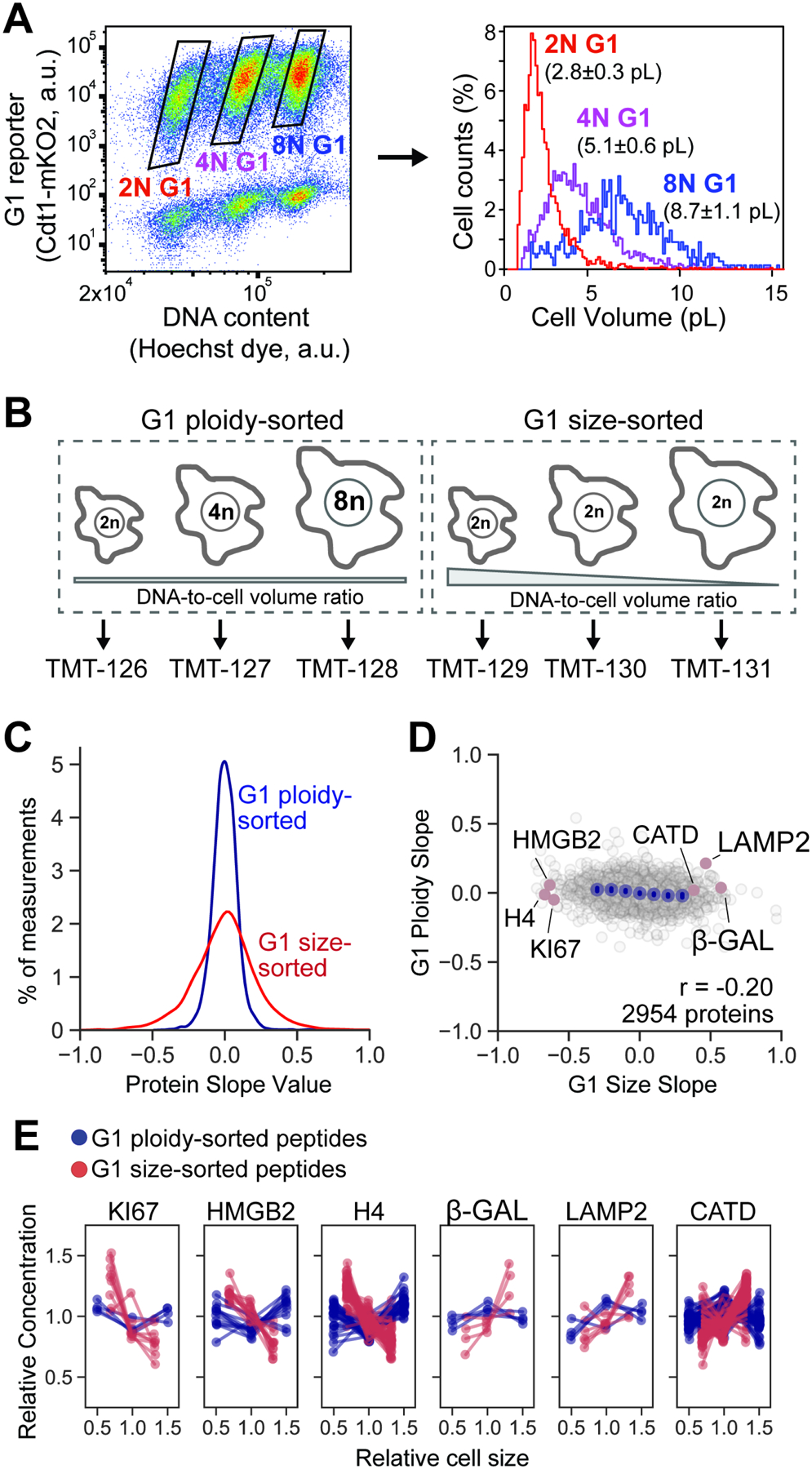
(A) hTERT RPE-1 cells expressing fluorescent cell cycle reporters (Cdt1-mKO2, Geminin-mAG) were treated with an Aurora kinase inhibitor barasertib (75nM, 48 hours) to partially inhibit cytokinesis. Cells were then sorted based on ploidy and G1 cell cycle phase. The attainment of differentially sized G1 cells was confirmed using a Coulter counter. The histogram shows a representative example of size distributions for sorted cells, and the numbers next to it represent mean cell size ± standard error for n=3 biological replicates. (B) Ploidy-sorted and Size-sorted G1 cells were isolated by FACS and their proteomes measured using TMTsixplex. Ploidy-sorted Protein Slope values were calculated by plotting the relative protein concentration against the mean cell size in the 2N, 4N, and 8N bins to obtain a slope value (Table S9). (C) Distributions of ploidy-sorted and size-sorted Protein Slope values. Despite large increases in cell size from 2N to 8N, concentration changes were minimal. (D) Slightly negative correlation between ploidy-sorted and size-sorted Protein Slope values are consistent with a small increase in DNA-to-cell volume ratio in polyploid G1 cells. (E) DNA-to-cell volume-dependent concentration changes for a representative set of proteins. For each protein panel, dotted lines represent unique peptide measurements.
Discussion
As cells grow larger, the total protein amount increases in proportion to cell volume so that total protein concentration remains constant (Figure 1A). While it was long thought that the concentrations of individual gene products also remained constant as cells grew larger (Padovan-Merhar et al., 2015; Zhurinsky et al., 2010), this paradigm had not previously been tested on a proteomic scale. In contradiction to the previous paradigm, we find that the concentrations of most individual proteins change with cell size. Some proteins sub-scaled with cell size and were diluted in larger cells, while others super-scaled with cell size so that their concentrations increased as cells grew larger. The measurement accuracy and comprehensiveness of our proteomic approach now brings to light the widespread and diverse nature of cell size scaling.
Transcriptional and post-transcriptional size-scaling mechanisms.
To begin to determine the mechanisms governing size scaling across the proteome, we built a series of linear models. We began our analysis by looking at scaling relationships of mRNA, which in budding yeast was shown to exhibit super- and sub-scaling behavior (Chen et al., 2020; Swaffer et al., 2021a). Our linear models predicted the size scaling slope of individual proteins from their relative mRNA concentration, protein half-life, codon affinity, and subcellular localization. Taken together, our models indicate that both transcriptional and post-transcriptional size-scaling mechanisms govern protein size scaling. One possible mechanism linking protein stability and size scaling could be through changes in cellular biosynthesis. In the large cell size regime, where growth efficiency (growth rate per unit mass) begins to slow, proteins that are stable would become more prevalent than those that are actively degraded (Neurohr et al., 2019). This is because the concentration of unstable proteins reflects a balance of their synthesis rate per unit mass, which likely scales in proportion to the growth efficiency, and their degradation rate, which is likely to be independent of cell size. While our linear models represent a good starting point, further investigation of specific proteins with diverse size scaling properties will be required to identify the underlying molecular mechanisms.
Differential size scaling in the proteome content of organelles.
The characteristic that best predicts a protein’s scaling behavior is its subcellular localization. Using microscopy, others have previously measured fluorescently tagged markers of several different organelles (Neumann and Nurse, 2007; Rafelski et al., 2012). The consensus from these experiments is that organelle volume typically scales in proportion to cell size. Though this is inconsistent with our measurements of organelle protein content, several factors could explain this discrepancy. First, aggregate organelle protein concentration may not reflect the scaling behavior of any single “marker” protein, but instead reflect an average across several proteins that is weighted by their abundance. Second, our proteomic method may be more precise than previous imaging-based analyses. For example, we find statistically significant ~1.15x differences in the scaling of organelle protein content within a ~2-fold range of cell size (e.g., Cytoplasm compared to Mitochondria or Nucleoplasm; see Figure S7). It is unclear whether this type of resolution can be achieved using fluorescence microscopy. Finally, it is possible that organelle volume may not scale with its protein content, so individual proteins within the organelle may become increasingly concentrated or diluted as cells grow. This last possibility is particularly intriguing since it would likely result in a size-dependence of organelle function, which has been observed in mitochondria (Miettinen and Bjorklund, 2016).
Causal relationship between large cell size and senescence.
Our results also shed light on the phenomenon of cellular senescence (Hernandez-Segura et al., 2018; Kumari and Jat, 2021; Sharpless and Sherr, 2015). While the senescent cell state has been associated with large cell size, this was mostly thought to be a passive consequence of continuing biosynthesis after cell cycle arrest. However, consistent with recent reports (Lengefeld et al., 2021; Neurohr et al., 2019), we demonstrate that large cell size also inhibits entry into the cell cycle (Figure 6C), implying that the relationship between cell size and senescence can also be inverted. Moreover, as proliferating cells grow larger, we detect senescence-like changes in several common “markers” of senescence (Hernandez-Segura et al., 2018; Kumari and Jat, 2021; Sharpless and Sherr, 2015). It could be that these senescence-associated changes to the proteome collectively contribute to the exit of larger cells from the cell cycle. Alternatively, it may be that only some (or few) senescence-associated changes are consequential for cell cycle arrest while other “markers” of senescence are merely markers of large cell size.
One potential reason large cells exit the cell cycle is that they accumulate more DNA damage than smaller cells (Figure 6). There are at least two plausible hypotheses for why we observe more DNA damage in larger cells that are not mutually exclusive. The repair of damaged DNA could be compromised in larger cells, or larger cells may be more prone to accumulate DNA damage in the first place. The fact that many DNA repair proteins sub-scale with cell size supports the first hypothesis. However, the fact that DNA replication factors also subscale with cell size supports the hypothesis that larger cells might accumulate more DNA damage than smaller cells during S phase. The sub-scaling nature of many DNA replication factors may explain the detrimental effect of long-term CDK4/6 inhibition on cells subsequently released into S phase (Crozier et al., 2022), since long-term CDK4/6 inhibition results in large cell size (Figure 2F) and concomitant changes to the proteome (Figure 2G).
Polyploidy as a mechanism to generate large non-senescent cells.
Our observation here that most protein concentrations change as cells grow provides a rationale for why many cells control their size. If the proteome content that supports optimal cell physiology is only found near the target cell size, then the further a cell deviates from its target size, the further protein concentrations will be from their functional and growth-supporting optimum. While a small change in the concentration of a single protein may not significantly affect cell physiology, the cumulative effect of thousands of small protein concentration changes could account for the drastic drop in the efficiency of biosynthesis in large cells (Demidenko and Blagosklonny, 2008; Lengefeld et al., 2021; Liu et al., 2022; Neurohr et al., 2019). The inability to maintain biosynthesis efficiency at large cell sizes is likely due to the decreasing amount of DNA in larger cells, which becomes a limiting factor for transcription (Berry et al., 2022; Sun et al., 2020; Swaffer et al., 2021b; Zhurinsky et al., 2010). Consistent with DNA being a limiting factor, we find that proteome concentrations mostly depend on the cell size-to-ploidy ratio rather than cell size per se. Interestingly, growth efficiency can be maintained over a 100-fold range if cell ploidy is scaled proportionally to cell size (Mu et al., 2020). Thus, our findings highlight the utility of polyploidization as an elegant mechanism for organisms to generate large, non-senescent cells capable of efficient and balanced protein synthesis.
Limitations of the study
Although we successfully measured the concentrations of about 3000 proteins across different cell sizes, there are still thousands of proteins that we were unable to consistently detect using proteomic mass spectrometry. As a result, our datasets are enriched for structural proteins and metabolic enzymes because of their high abundance in the cells. We therefore still lack information about many signaling and regulatory proteins, which are generally less abundant. In addition, we do not capture the size-dependency of post-translational modifications that modulate protein activities. Importantly, this work and a contemporaneous study from the Bjorklund lab (Cheng et al., 2021) suggest that the proteome’s size-dependency will be similar across many of the common lineages of cultured mammalian cells. However, we expect the relationship between cell size and proteome content will be more complex in the context of a tissue. Thus, it is unclear to what extent our measurements in vitro will reflect the how cell size impacts cell physiology in vivo. Furthermore, because cultured mammalian cells proliferate in a non-native environment, single-celled organisms may represent a simpler and more physiological system to study the relationship between cell size and the proteome.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jan Skotheim (skotheim@stanford.edu).
Materials availability
All cell lines used in this study are available from the lead contact upon request without restriction.
Data and code availability
Proteomic and RNA-seq data have been deposited to PRIDE and GEO, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy and western blotting data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Primary human fetal lung fibroblasts from Cell Applications (Cat#506–05f) and hTERT-immortalized RPE-1 (RRID: CVCL_4388).
Method Details
Cell Culture
Recently isolated primary fetal human lung fibroblasts (HLF) were purchased from Cell Applications, telomerase-immortalized retinal pigment epithelium (hTERT RPE-1, here also referred to as RPE-1 for brevity) cells were obtained from the Stearns laboratory at Stanford. All cells were cultured at 37°C with 5% CO2 in Dulbecco’s modification of Eagle’s medium (DMEM) with L-glutamine, 4.5 g/l glucose and sodium pyruvate (Corning), supplemented with 10% heat-inactivated fetal bovine serum (FBS, Corning) and 1% penicillin/streptomycin.
Fluorescence-activated cell sorting (FACS)
Fluorescence-activated cell sorting was used to sort live cells by their size and cell cycle phase. To do this, the cells were harvested from dishes by trypsinization, stained with 20 μM Hoechst 33342 DNA dye in PBS for 15 minutes at 37°C, and then sorted on a BD FACSAria Fusion flow cytometer. Consecutive SSC-A over FSC-A, and FSC-H over FSC-A gates were used to isolate single cells. Then, G1 cells were gated by DNA content (Hoechst staining). Finally, we collected the 10% smallest and 10% largest cells, as well as another 10% of the cells near the average size using the gating based on SSC-A signal. During sorting, all cell samples and collection tubes were kept at 4°C. To determine the cell size distributions of the collected samples, aliquots were taken from each sorted size bin and measured on a Z2 Coulter counter (Beckman). Sorted cells were used for mRNA or protein isolation, or re-plated for assessing senescence dynamics.
Stable Isotope Labeling In Cell culture (SILAC)
Cells for SILAC were cultured in special Lysine/Arginine-free DMEM for SILAC (Thermo Scientific) with 10% dialyzed heat-inactivated FBS (HyClone) and penicillin/streptomycin. These cultures were supplemented with “light”, “intermediate”, or “heavy” versions of Lysine (0.8mM) and Arginine (0.4mM) (Cambridge Isotope Laboratories) (Deng et al., 2019). The “light” (Agr0 Lys0) version of the media contained L-Arginine and L-Lysine built with normal 12C and 14N isotopes; the “intermediate” (Arg6 Lys4) version had L-Arginine containing six 13C atoms and L-Lysine containing four deuterium atoms; the “heavy” (Arg10 Lys8) version had L-Arginine containing six 13C and four 15N atoms and L-Lysine containing six 13C and two 15N atoms. Proline (200 mg/l) was added to the media to prevent conversion of isotope-coded Arginine into Proline in cells. We confirmed that cell proliferation is not impaired in our SILAC medium (Figure S2B). To ensure complete labelling, the cells were cultured in SILAC media for 5 passages (approximately 10 doublings) prior to the experiments.
LC-MS/MS sample preparation - SILAC
See Table S10 for a complete list of proteomic experiments. Small, medium, and large cells sorted by FACS were pelleted by centrifugation at 500xg for 10 minutes and lysed for 40 minutes on ice in RIPA lysis buffer (Abcam) containing a protease and phosphatase inhibitor cocktail. SILAC-labeled different sized cells were mixed prior to lysis in order to minimize handling error in protein extraction, proteolytic digestion, and peptide desalting. Cell lysates were cleared by centrifugation at 15000xg for 30 minutes at 4°C. The mixed lysates were then denatured in 1% SDS, reduced with 10mM DTT, alkylated with 5mM iodoacetamide, and then precipitated with three volumes of a solution containing 50% acetone and 50% ethanol. Proteins were re-solubilized in 2 M urea, 50 mM Tris-HCl, pH 8.0, and 150 mM NaCl, and then digested with TPCK-treated trypsin (50:1) overnight at 37°C. Trifluoroacetic acid and formic acid were added to the digested peptides for a final concentration of 0.2%. Peptides were desalted with a Sep-Pak 50mg C18 column (Waters). The C18 column was conditioned with 5 column volumes of 80% acetonitrile and 0.1% acetic acid and washed with 5 column volumes of 0.1% trifluoroacetic acid. After samples were loaded, the column was washed with 5 column volumes of 0.1% acetic acid followed by elution with 4 column volumes of 80% acetonitrile and 0.1% acetic acid. The elution was dried in a Concentrator at 45°C.
LC-MS/MS sample preparation - TMT
Lysis, denaturation, reduction, and precipitation for SILAC analysis was the same for TMT analysis (working solution of Iodoacetamide was dissolved in HEPES rather than Tris buffer). Our method for TMT labeling was adapted from Zecha et al. (Zecha et al., 2019) and the Thermo TMT10plex™ Isobaric Label Reagent Set Protocol. In brief, acetone precipitated samples were resuspended in 100μm TEAB and digested O/N with TPCK trypsin (50:1) in the absence of Tris or Urea. After digestion, peptide concentration was ~1μg/ul in 100μM TEAB for all samples. 20μg of peptide was labeled using 100μg of Thermo TMT10plex™ in a reaction volume of 25μl for 1 hour. The labeling reaction was quenched with 8μL of 5% hydroxylamine for 15 minutes. Labeled peptides were pooled, acidified to a pH of ~2 using drops of 10% formic acid, and desalted with a Sep-Pak 50mg C18 column as described above.
HILIC fractionation - SILAC
Desalted peptide samples were reconstituted in 80% acetonitrile and 1% formic acid and fractionated by hydrophilic interaction liquid chromatography (HILIC) with a TSK gel Amide-80 column (2 mm × 150 mm, 5 μm; Tosoh Bioscience). 90 second fractions were collected between 10 and 25 minutes of the gradient. Three solvents were used for the gradient: buffer A (90% acetonitrile), buffer B (80% acetonitrile and 0.005% trifluoroacetic acid), and buffer C (0.025% trifluoroacetic acid). The gradient used consists of a 100% buffer A at time = 0 min; 88% of buffer B and 12% of buffer C at time = 5 min; 60% of buffer B and 40% of buffer C at time = 30 min; and 5% of buffer B and 95 % of buffer C from time = 35 to 45 min in a flow of 150 μl/min. HILIC fractions were dried in a SpeedVac and reconstituted in 0.1% trifluoroacetic acid. A total of 10 fractions were collected and pooled back into 5 fractions (1–6, 2–7, 3–8, 4–9, 5–10).
High-pH reverse phase fractionation - TMT
TMT-labeled peptides (Experiment from Figure S5D) were fractionated using a Pierce™ High pH Reversed-Phase Peptide Fractionation The eight default fractions were either injected separately or pooled back into 4 fractions (1–5, 2–6, 3–7, 4–8). Dried peptides were reconstituted in 0.1% formic acid.
LC-MS/MS data acquisition - SILAC
Peptide samples were analyzed using a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with Dionex Ultimate 3000 LC systems (Thermo Fisher Scientific, San Jose, CA). Peptides were separated by capillary reverse phase chromatography on a 25 cm reversed phase column (100 μm inner diameter, packed in-house with ReproSil-Pur C18-AQ 3.0 m resin (Dr. Maisch GmbH)). Liquid chromatography was performed using a two-step linear gradient with 4–25 % buffer B (0.1% (v/v) formic acid in acetonitrile) for 90 min followed by 25–40 % buffer B for 10 min. Data was acquired in top 20 data dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer with a resolution of 120,000 (FWHM) and m/z scan range of 340–1500. Selected precursor ions were subjected to fragmentation using higher-energy collisional dissociation (HCD) with quadrupole isolation, isolation window of 1.6 m/z, and normalized collision energy of 30%. HCD fragments were analyzed in the Orbitrap mass analyzer with a resolution of 15,000 (FWHM). Fragmented ions were dynamically excluded from further selection for a period of 15 seconds. The AGC target was set to 400,000 and 50,000 for full FTMS scans and FTMS2 scans, respectively. The maximum injection time was set to 50 ms for full FTMS scans and dynamic for FTMS2 scans.
LC-MS/MS data acquisition - TMT
Desalted TMT-labeled peptides were analyzed on a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a Thermo EASY-nLC 1200 LC system (Thermo Fisher Scientific, San Jose, CA). Peptides were separated by capillary reverse phase chromatography on a 25 cm column (75 μm inner diameter, packed with 1.6 μm C18 resin, AUR2–25075C18A, Ionopticks, Victoria Australia). Electrospray Ionization voltage was set to 1550 volts. Peptides resulting from on-bead digestion were resuspended in 10 μL of 0.1% formic acid. 2 μL was introduced into the Fusion Lumos mass spectrometer using a two-step linear gradient with 6–33% buffer B (0.1% (v/v) formic acid in 80% acetonitrile) for 145 min followed by 33–45% buffer B for 15 min at a flow rate of 300 nL/min. Column temperature was maintained at 40°C throughout the procedure. Xcalibur software (Thermo Fisher Scientific) was used for the data acquisition and the instrument was operated in data-dependent mode. Survey scans were acquired in the Orbitrap mass analyzer over the range of 380 to 1800 m/z with a mass resolution of 70,000 (at m/z 200). Ions were selected for fragmentation from the 10 most abundant ions with a charge state of either 2, 3 or 4 and within an isolation window of 2.0 m/z. Selected ions were fragmented by Higher-energy Collisional Dissociation (HCD) with normalized collision energies of 27 and the tandem mass spectra was acquired in the Orbitrap mass analyzer with a mass resolution of 17,500 (at m/z 200). Repeated sequencing of peptides was kept to a minimum by dynamic exclusion of the sequenced peptides for 30 seconds. For MS/MS, the AGC target was set to 1e5 and max injection time was set to 120ms. Relative changes in peptide concentration were determined at the MS3-level by isolating and fragmenting the 4 most dominant MS2 ion peaks.
Spectral searches - TMT and SILAC
All raw files were searched using the Andromeda engine (Cox et al., 2011) embedded in MaxQuant (v1.6.7.0) (Cox and Mann, 2008). See Table S11 for a complete summary of the search parameters used for the SILAC, TMT, and LFQ experiments. In brief, 3 label SILAC search was conducted using Maxquant’s default Arg6/10 and Lys4/8. For TMT searches, a Reporter ion MS3 search was conducted using 10plex TMT isobaric labels. For both TMT and SILAC searches, variable modifications included oxidation (M) and protein N-terminal acetylation. Carbamidomthyl (C) was a fixed modification. The number of modifications per peptide was capped at five. Digestion was set to tryptic (proline-blocked). Peptides were “Re-quantified”, and maxquant’s match-between-runs feature was not enabled. Database search was conducted using the UniProt proteome - Human_UP000005640_9606. Minimum peptide length was 7 amino acids. FDR was determined using a reverse decoy proteome (Elias and Gygi, 2007).
RNA extraction and sequencing
To compare the transcriptomes of different-sized G1 cells, primary HLFs were arrested in the G1 phase of the cell cycle by a 24-hour treatment with 1μM of the Cdk4/6 inhibitor Palbociclib and then sorted into size bins using FACS. To extract RNA, each sample of sorted HLF cells was split into two technical replicates each of which contained 200,000 HLF cells, which were then mixed with 100,000 D. melanogaster S2 cells as a spike-in. Each sample was then pelleted and RNA was extracted using Direct-zol™ RNA Microprep Kit (Zymo Research). mRNA was enriched using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, #E7490). The NEBNext Ultra II RNA Library Prep Kit for Illumina® (NEB, #E7775) was then used to prepare libraries for paired-end (2×150 bp) Illumina sequencing (Novogene). Two independent biological replicates of each sample were collected and for each biological replicate, two technical replicates (i.e., separate lysis, library prep, and sequencing) were processed. Approximately 40 million reads were sequenced per replicate.
RNAseq data processing
RNA samples contained a mixture of H. sapiens and D. melanogaster spike-in. A combined H. sapiens and D. melanogaster genome file was created using the hg38 and dm6 versions of the respective genomes and a combined transcriptome annotation was created using the H. sapiens gene models from the v29 version of the GENCODE annotation (Frankish et al., 2019) and the BDGP6 D. melanogaster gene models from ENSEMBL release 90 (Yates et al., 2020). For the purposes of RNA-seq data quality evaluation, genome browser track generation, and calculating the hg38-to-dm6 ratio, reads were aligned against the combined genomes and combined annotated set of splice junctions using the STAR aligner (version 2.5.3a; settings: --limitSjdbInsertNsj 10000000 --outFilterMultimapNmax 50 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 10 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --sjdbScore 1 --twopassMode Basic --twopass1readsN −1) (Dobin et al., 2013). Read mapping statistics and genome browser tracks were generated using custom Python scripts. For quantification purposes, reads were aligned as 2×50mers in transcriptome space against an index generated from the combined annotations described above using Bowtie (version 1.0.1; settings: -e 200 -a -X 1000) (Langmead et al., 2009). Alignments were then quantified using eXpress (version 1.5.1) (Roberts and Pachter, 2013) before effective read count values and TPM (Transcripts Per Million transcripts) were then separated for each genome and renormalized TPMs were calculated with respect to only the H. sapiens transcripts.
Flow cytometry
For flow cytometry analysis, cells were grown on dishes to ~50% confluence and harvested by trypsinization. The cells were then fixed with 3% formaldehyde for 10 minutes at 37°C, and then permeabilized with 90% methanol for 30 minutes on ice. Fixed and permeabilized cells were washed once with PBS, blocked with 3% BSA in PBS for 30 minutes at 37°C, and then stained with primary antibodies for 2 hours at 37°C. We used the following primary antibodies: p16 (Abcam, ab108349), HMGB1 (Abcam, ab79823), HMGN2 (CST, 9437S), RPLP0 (Sigma, SAB1402899), Actin (Sigma-Aldrich, A2103), UCHL1 (CST, 13179S), VAT1 (Santa Cruz, sc-515705), LAMP1 (CST, 9091), alpha-Tubulin (Abcam, ab6160), γ-H2AX (Sigma Millipore, 05–636-I). After the primary antibodies, the cells were washed twice with a wash buffer (1% BSA in PBS + 0.05% Tween® 20), stained with the fluorophore-conjugated secondary antibodies Alexa Fluor 488 goat anti-mouse (Life Technologies A11029), Alexa Fluor 594 goat anti-rabbit (Life Technologies A11037), and Alexa Fluor 405 goat anti-rat (Abcam ab175673) at 1:1000 dilution for 1 hour at 37°C, and then washed twice again. After this treatment, the cells were resuspended in PBS containing 3 μM DAPI for DNA staining, incubated for 10 minutes at room temperature, and then analyzed on a Attune NxT flow cytometer (Thermo Fisher). To compensate for the nonspecific background staining with antibodies, we measured the fluorescence of cells stained with nonspecific Isotype Control antibodies. We then performed a linear regression of this nonspecific background signal with the cell size, and subtracted the background fluorescence corresponding to the cell’s size from the actual fluorescence signal measured for each cell. For total protein staining, live cells were resuspended in PBS, then the CellTrace CFSE dye (CarboxyFluorescein Succinimidyl Ester, Thermo Fisher) was added at 5 μM concentration, incubated for 30 minutes at 37°C. The dye was then washed out with FBS-containing DMEM, and the cells were pelleted and resuspended in PBS for the flow cytometry or for the fixation and antibody staining. For the Lysotracker staining, the cells were harvested and re-suspended in growth media at a concentration of 106 cells/mL. Then, Lysotracker Red DND-99 (Thermo Fisher) was added at a concentration of 75nM and incubated at 37°C for 30 min. Cells were spun down and re-suspended for analysis or additional staining. For plotting the flow cytometry data, all protein amounts and cell size values were normalized to their means. To characterize the degree of size-dependency of protein amounts, we fit a line to the flow cytometry data after normalizing these data to mean values. We performed at least three biological replicates for each experiment that measured 100,000 cells.
To compare the proliferation efficiency of different-sized cells shortly after FACS sorting, the cells were left to settle on culture dishes for 2 days, then cultured in the presence of 1μM EdU for 24 hours to label all the cells that underwent replication within this time period. Cells were then stained using a Click-iT™ EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Molecular Probes), following the manufacturer’s protocol, and analyzed by flow cytometry. To compare the telomere lengths of different-sized cells, passage #8 HLF cells were stained using a Flow-FISH Telomere PNA Kit (Agilent DAKO) according to the manufacturer’s protocol and analyzed by flow cytometry.
Senescence-associated beta-galactosidase (SA-beta-Gal) staining
To detect senescent cells, the RPE-1 or HLF cells on a dish were stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology) following the manufacturer’s protocol. Briefly, live cells on a dish were washed once with PBS and fixed with 1x Fixation solution for 10 minutes at room temperature, then rinsed twice with PBS, and stained with β-Galactosidase Staining Solution for 48 hours at 37°C in a dry incubator (no CO2). The cells were then imaged on an EVOS M5000 imaging system to obtain a colored brightfield image. The obtained images were quantified manually, by a blinded investigator, to determine the percentage of senescent cells, i.e., the cells that have a pronounced blue staining.
Live cell fluorescence microscopy
In preparation for imaging, cells were seeded on 35-mm glass-bottom dishes (MatTek) at low density and incubated overnight at 37°C and 5% CO2. Then, the cells were moved to a Zeiss Axio Observer Z1 microscope equipped with an incubation chamber and imaged for 96 hours (Schwarz et al., 2018). Brightfield and fluorescence images were collected from three dishes at multiple positions every 20 minutes using an automated stage controlled by the Micro-Manager software. We used a Zyla 5.5 sCMOS camera, which has a large field of view allowing us to track motile cells within a field of view for long durations, and an A-plan 10x/0.25NA Ph1 objective. To distinguish G0/G1 and S/G2/M cells in time lapse imaging experiments, we used RPE-1 cells expressing the fluorescent cell cycle reporters mKO2-hCdt1 (G1), and mAG-hGeminin (S/G2) (Sakaue-Sawano et al., 2008). These reporters were introduced into RPE-1 cells using a lentivirus vector, and the positive, fluorescent population of cells was isolated using fluorescence-activated cell sorting.
Immunofluorescent staining for microscopy
Cells were seeded on a 35-mm collagen-coated glass-bottom dish (MatTek) one day before starting the cell treatments. For the staining, cells were fixed with 4% formaldehyde for 10 minutes at room temperature, permeabilized with 0.2% Triton™ X-100 (Sigma-Aldrich) for 15 minutes at 4°C, and then blocked with 3% BSA in PBS. Then, the cells were incubated with primary antibodies overnight at 4°C, washed twice with PBS, and then incubated with conjugated Alexa Fluor 647 goat-anti-mouse (Invitrogen, A32728) and Alexa Fluor 488 goat-anti-rabbit (Invitrogen, A32731) secondary antibodies at 1:1000 for 1 hour at room temperature. The cells were washed twice with PBS, incubated with 300 nM DAPI for 5 minutes at room temperature, and then imaged using a Zeiss Axio Observer Z1 microscope with an A-plan 10x/0.25NA objective. γ-H2AX and 53BP1 loci were quantified from microscopy images using standard tools implemented in FIJI software. The primary antibodies used for immunofluorescent staining were rabbit anti-53BP1 (Novus Biologicals, NB100304, 1:2000 dilution), mouse anti-γ-H2AX (Sigma Millipore, 05–636-I, 1:200), rabbit anti-p16 (Abcam, ab108349, 1:250), rabbit anti-p21 (Abcam, ab109199, 1:200), mouse anti-Lamin B1 (CST #68591, 1:200).
Immunoblotting
For immunoblotting, cells were lysed in the RIPA lysis buffer on ice. Proteins from lysates were separated on 8% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were then blocked with SuperBlock™ (TBS) Blocking Buffer (Thermo Fisher Scientific) and incubated overnight at 4°C with primary antibodies in 5% non-fat dry milk dissolved in TBST buffer. The primary antibodies were detected using the fluorescently labeled secondary antibodies Alexa Fluor 680 Goat anti-Mouse IgG (Life Technologies, A21058) and Alexa Fluor 790 Goat anti-Rabbit IgG (Life Technologies, A11369). Membranes were imaged on a LI-COR Odyssey CLx and analyzed with LI-COR Image Studio software. The primary antibodies used for immunoblotting were rabbit anti-p16 (Proteintech, 10883–1-AP, 1:300 dilution), rabbit anti-p21 (Abcam, ab109199, 1:1000), rabbit anti-beta-Actin (Sigma-Aldrich, A2103, 1:3000), mouse anti-p21 (Santa Cruz, sc6246, 1:300), mouse anti-GAPDH (Invitrogen, MA5–15738, 1:3000).
Quantification and Statistical Analysis
Peptide Quantitation – SILAC
Our SILAC analysis pipeline uses the peptide feature information in MaxQuant’s “evidence.txt” output file. Each row of the “evidence.txt” file represents an independent peptide triplet measurement. Contaminant and decoy peptide identifications were discarded. Peptides without signal in any of the three SILAC channels were also excluded. Peptide triplets (each row in the “evidence.txt” table) are assigned to a protein based on MaxQuant’s “Leading razor protein” designation. For each peptide triplet, the fraction of ion intensity in each SILAC channel was calculated by dividing the “Intensity L/M/H” column by the “Intensity” column. SILAC channels were normalized by adjusting the fraction of ion intensity in each channel by the median for all measured peptides (see Figure S1B and S1C). After normalization, the relative signal difference between the SILAC channels for each peptide triplet was plotted against the normalized cell size for each of the bins of isolated G1 cells.
For each peptide, we calculated its slope as follows (mean squared error filtering):
Y1,2,3 = Relative signal in each SILAC channel (order based on labeling orientation)
Avg. size = (mean volume of small bin + mean volume of medium bin + mean volume of large bin) / 3
x1 = (Mean volume of small size bin) / Avg. size
x2 = (Mean volume of medium size bin) / Avg. size
x3 = (Mean volume of large size bin) / Avg. size
Based on the expectation that our experimental conditions would not result in large, non-linear changes in protein expression, we exclude peptide triplets whose three data points did not loosely fit a linear regression line. Linear regressions on the ~50,000 triplets/experiment were performed using np.polyfit in Python. Regressions with a mean squared error > 0.075 were excluded. Because this filtering step significantly improved the overall data quality (Figure S1F and S1G), we concluded that our filtering method mostly excludes peptide triplets contaminated by analytical interference or near the noise floor.
Individual peptide measurements were consolidated into a protein level measurement using Python’s groupby.median. Peptides with the same amino acid sequence that were identified as different charge states or in different fractions were considered independent measurements. We summarize the size scaling behavior of individual proteins as a slope value derived from a regression (similar to what is described above for individual peptides), and each protein slope value is based on the behavior of all detected peptides.
For a given protein, we calculate its cell size-dependent slope as follows:
yi = Relative signal in the ith SILAC channel (median of all corresponding peptides in this channel)
xi = same normalized cell size xi as for the peptide slope calculations above
The protein slope value was determined from a linear fit to the log-transformed data using the equation:
Variables were log-transformed so that a slope of 1 corresponds to an increase in protein concentration that is proportional to the increase in volume and a slope of −1 corresponds to 1/volume dilution. Pearson r and p values for correlation analyses were calculated using scipy’s pearsonr module in Python.
Protein annotations
Protein annotations in Figure 2 were sourced from UniProt columns named “Subcellular location [CC]” or “Protein names” (UniProt, 2019). For Figure 2D, protein localization was strictly parsed so that each annotated protein belongs to only one of the designated groups. Proteins with 2 or more of the depicted annotations were ignored (except for the “Cytoplasm / Nucleus” category, which required a nuclear and cytoplasmic annotation).
2D annotation enrichment
2D annotation enrichment, like in Figure 2D, was performed as described previously (Cox and Mann, 2012). In brief, all annotation groups with significant enrichment in either experiment are displayed on the plot (Benjamini-Hochberg FDR value at 2%). The position of each annotation group on the plot is determined by the enrichment score (S). The enrichment score is calculated from the rank ordered distribution of Protein Slope values:
Where Rgroup and Rremaing proteins are the average ranks for the proteins within an annotation group and all remaining proteins in the experiment, respectively, and n is the total number of proteins.
Peptide quantitation - TMT
Our TMT analysis pipeline uses the peptide feature information in MaxQuant’s “evidence.txt” output file. Each row of the “evidence.txt” file represents an independent MS3 TMT measurement. Contaminant and decoy peptide identifications were discarded. Peptides without signal in any of the TMT channels were also excluded. TMT peptide measurements were assigned to protein based on MaxQuant’s “Leading razor protein” designation. For each peptide triplet, the fraction of ion intensity in each TMT channel was calculated by dividing the “Reporter ion intensity” column by the sum of all reporter ion intensities. TMT channels were normalized by adjusting the fraction of ion intensity in each channel by the median for all measured peptides (similar to the SILAC normalization in Figure S1B and S1C). After normalization, the relative signal difference between the TMT channels for each peptide triplet was plotted against the normalized cell size for each of the bins of isolated G1 cells. Slope values in Figure 2F and 2G were derived in a manner analogous to the Slope values calculated in the SILAC experiments. Pearson r and p values for correlation analyses were calculated using SciPy’s pearsonr module in Python.
Principle component analysis
PCA analysis was performed in Python using the sklearn package. Protein-level measurements were first obtained in manner similar to the slope calculation. A data frame was created that contained individual proteins as rows with columns corresponding to the relative protein concentration in each SILAC/TMT channel (obtained from the median of all peptide measurements for a given protein). These protein-level measurements were subjected to MSE-filtering method (Figure S1F and S1G) used to calculate Protein Slope values. For the comparison of mRNA and Protein datasets, the columns corresponding to the relative change in transcript concentration (determined by RPKM value) for each biological replicate experiment were added to the data frame use to for the analysis in Figure 1K.
Protein complex analysis
Protein complex annotations were derived from the CORUM database (“Core Complexes” with 3512 entries). We calculated the Coefficient of Variation (CV) between the Protein Slope values of subunits in annotated protein complexes. To do so, all slope values were normalized to a value between 0 and 1. Slope values were then grouped for each protein complex. Only proteins with a minimum of 5 independent peptide measurements (between two replicate experiments) and protein complexes with at least three quantified subunits were considered in the analysis. To generate a randomized dataset, quantified proteins (i.e., proteins with Protein Slope values) within each protein complex were replaced with randomly selected proteins using the random function from the NumPy library. CV is calculated for both real and randomized datasets and compared with ttest_ind (T-test) from scipy.stats library. Only cytoplasmic proteins and protein complexes were considered to avoid biases from the differential scaling observed in different subcellular compartments.
OLS linear regression model
Multiple linear regression analysis was performed using the statsmodels module in Python. The prediction of size scaling behavior was based on the 1,700 proteins shared between the protein turnover (HeLa cells) (Zecha et al., 2018), RNA Slope, and Protein Slope datasets (at least 2 peptides / protein). Independent variables for codon affinity score, RNA Slope, and protein turnover (T50%) were each independently standardized by subtracting all values by the dataset’s mean and then dividing by the dataset’s standard deviation. Because codons with an A or T residue in the 3rd position have lower affinity for tRNA and are thus less efficiently translated (Frenkel-Morgenstern et al., 2012), the codon affinity score we used to predict translation efficiency is the fraction of codons in a given mRNA transcript that contain an A or T residue in the 3rd position. The subcellular localization variable was based on UniProt’s “Subcellular location [CC]” annotations and entered as a binary value for each compartment (1 if a protein possessed an annotation and 0 if it did not). Only subcellular compartments that provided nonredundant predictive power were ultimately included in the model. A constant value was added to the regression equation using the add_constant function in statsmodels. We set the benchmark for predictive accuracy (Prediction %) as the correlation between biological replicate experiments (Protein Slope from Exp #1 vs Exp #2). See Figure S10 for more details on the model and its coefficients.
Supplementary Material
Table S1. Slope values for size-sorted G1 cells (HLF and hTERT RPE-1), related to Figure 1
Table S2. Slope values for size-sorted G1 cells using SSC and Total protein dye, related to Figure 1 and S5
Table S4. Slope variance for proteins in protein complexes, related to Figure 2B
Table S5. 2D gene annotation enrichments for HLF and hTERT RPE-1, related to Figure 2D
Table S7. RNA and Protein Slope values for size-sorted G1 cells, related to Figure 3
Table S8. 2D gene annotation enrichments for linear model parameters, related to Figure 3D
Table S9. Slope values for size-sorted and ploidy-sorted G1 cells, related to Figure 7
Table S10. Proteomic Experiments list and quantitation channel information, related to STAR Methods
Table S11. MaxQuant spectral search parameter files, related to STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-p16 | Abcam | Cat#ab108349; RRID: AB_10858268 |
| rabbit anti-p16 | Proteintech | Cat#10883–1-AP; RRID: AB_2078303 |
| rabbit anti-HMGB1 | Abcam | Cat#ab79823; RRID: AB_1603373 |
| rabbit anti-HMGN2 | Cell Signaling Technology | Cat#9437S; RRID: AB_10949505 |
| mouse anti-RPLP0 | Sigma | Cat#SAB1402899; RRID: AB_10738968 |
| rabbit anti-UCHL1 | Cell Signaling Technology | Cat#13179S; RRID: AB_2798141 |
| mouse anti-VAT1 | Santa Cruz Biotechnology | Cat#sc-515705 |
| rabbit anti-LAMP1 | Cell Signaling Technology | Cat#9091; RRID: AB_2687579 |
| mouse anti-γ-H2AX | Sigma Millipore | Cat#05–636-I; RRID: AB_2755003 |
| rabbit anti-p21 | Abcam | Cat#ab109199; RRID: AB_10861551 |
| mouse anti-p21 | Santa Cruz Biotechnology | Cat#sc-6246; RRID: AB_628073 |
| rabbit anti-53BP1 | Novus Biologicals | Cat#NB100304; RRID: AB_10003037 |
| mouse anti-Lamin B1 | Cell Signaling Technology | Cat#68591; RRID: AB_2799751 |
| rat anti-Tubulin | Abcam | Cat#ab6160; RRID: AB_305328 |
| rabbit anti-Actin | Sigma-Aldrich | Cat#A2103; RRID: AB_476694 |
| mouse anti-GAPDH | Thermo Fisher Scientific | Cat#MA5–15738; RRID: AB_10977387 |
| Alexa Fluor 488 goat anti-mouse IgG | Thermo Fisher Scientific | Cat#A11029; RRID: AB_2534088 |
| Alexa Fluor 594 goat anti-rabbit IgG | Thermo Fisher Scientific | Cat#A11037; RRID: AB_2534095 |
| Alexa Fluor 405 goat anti-rat IgG | Abcam | Cat#ab175673; RRID: AB_2893021 |
| Alexa Fluor 647 goat-anti-mouse IgG | Thermo Fisher Scientific | Cat#A32728; RRID: AB_2633277 |
| Alexa Fluor 488 goat-anti-rabbit IgG | Thermo Fisher Scientific | Cat#A32731; RRID: AB_2633280 |
| Alexa Fluor 680 goat anti-Mouse IgG | Thermo Fisher Scientific | Cat#A21058; RRID: AB_2535724 |
| Alexa Fluor 790 goat anti-Rabbit IgG | Thermo Fisher Scientific | Cat#A11369; RRID: AB_2534142 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| TPCK-treated Trypsin | Worthington | Cat#LS003740 |
| Trypsin GOLD TMT10plex Mass Tags | Promega Thermo Scientific | Cat#V5280 Cat#90110 |
| L-LYSINE:2HCL - Unlabeled | Cambridge Isotope Lab | Cat#ULM-8766-PK |
| L-ARGININE:HCL - Unlabeled | Cambridge Isotope Lab | Cat#ULM-8347-PK |
| L-LYSINE:2HCL (4,4,5,5-D4, 96–98%) | Cambridge Isotope Lab | Cat#DLM-2640–0.5 |
| L-ARGININE:HCL (13C6, 99%) | Cambridge Isotope Lab | Cat#CNLM-2265-H-0.25 |
| L-LYSINE:2HCL (13C6, 99%; 15N2, 99%) | Cambridge Isotope Lab | Cat#CNLM-291-H-1 |
| L-ARGININE:HCL (13C6, 99%; 15N4, 99%) | Cambridge Isotope Lab | Cat#CNLM-539-H-1 |
| DMEM for SILAC | Thermo Fisher Scientific | Cat#A33822 |
| HyClone Dialyzed heat-inactivated FBS | Cytiva | Cat#SH3007903HI |
| Palbociclib | Sigma-Aldrich | Cat#PZ0383 |
| Rapamycin | Thermo Fisher Scientific | Cat#AAJ62473MF |
| Doxorubicin | Thermo Fisher Scientific | Cat#D419325MG |
| Barasertib | Selleck Chemicals | Cat#AZD1152-HQPA |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#H1399 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dilactate) | Thermo Fisher Scientific | Cat#D3571 |
| Lysotracker Red DND-99 | Thermo Fisher Scientific | Cat#L7528 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat#23225 |
| Sep-Pak 50mg C18 Cartridges | Waters | Cat##054955 |
| High pH Revered-phased Peptide Fractionation Kit | Thermo Scientific | Cat#84868 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | Cat#9860 |
| Direct-zol™ RNA Microprep Kit | Zymo Research | Cat#R2063 |
| Click-iT™ EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat#C10424 |
| CellTrace™ CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34554 |
| Deposited data | ||
| All Proteomics raw files and spectral searches | This paper | PRIDE:PXD033332 |
| All RNAseq raw files | This paper | GEO: GSE208348 |
| Experimental models: Cell lines | ||
| hTERT RPE1 | Stearns Lab (Stanford) | RRID: CVCL_4388 |
| Primary human fetal lung fibroblasts | Cell Applications | Cat#506–05f |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| FlowJo | BD Biosciences, https://www.flowjo.com/solutions/flowjo | RRID: SCR_008520 |
| FIJI | https://imagej.net/software/fiji/ | RRID: SCR_002285 |
| MaxQuant | Mann Lab | N/A |
| Python - Jupyter Notebooks | Anaconda | N/A |
| Other | ||
Highlights.
Cell size (i.e., DNA-to-cell volume ratio) globally affects the cell proteome composition
Proteins located in a particular organelle scale similarly with cell size
Increase in cell size causes senescence-like proteome changes in non-senescent cells
Larger cells are more prone to replicative and drug-induced senescence
Acknowledgements:
We thank Shicong Xie for assistance with Python-related data analysis and Marcus Smolka for use of his HPLC for HILIC pre-fractionation. We thank Robert Brooks, Charles Sherr, Christine Jacobs-Wagner, Marcus Smolka, Jette Lengefeld, and members of the Skotheim lab for helpful feedback on the manuscript. We also thank Frank McCarty and Stanford SUMS for assistance in mass spec related data acquisition. Cell sorting was performed on an instrument in the Shared FACS Facility purchased by Parker Institute for Cancer Immunotherapy.
Funding:
National Institutes of Health R35 grant GM134858 (JMS)
National Institutes of Health F32 grant GM137522 (MCL)
Chan Zuckerberg Biohub collaborative postdoctoral fellowship (MCL)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
Authors declare that they have no competing interests.
References
- Berenson DF, Zatulovskiy E, Xie S, and Skotheim JM (2019). Constitutive expression of a fluorescent protein reports the size of live human cells. Mol Biol Cell 30, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry S, Muller M, Rai A, and Pelkmans L (2022). Feedback from nuclear RNA on transcription promotes robust RNA concentration homeostasis in human cells. Cell Syst. [DOI] [PubMed] [Google Scholar]
- Cantwell H, and Nurse P (2019). Unravelling nuclear size control. Curr Genet 65, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YH, and Marshall WF (2010). Scaling properties of cell and organelle size. Organogenesis 6, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao G, Zahumensky J, Honey S, and Futcher B (2020). Differential Scaling of Gene Expression with Cell Size May Explain Size Control in Budding Yeast. Mol Cell 78, 359–370 e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Chen J, Kong Y, Tan C, Kafri R, and Björklund M (2021). Size-scaling promotes senescence-like changes in proteome and organelle content. bioRxiv, 2021.2008.2005.455193. [Google Scholar]
- Claude K-L, Bureik D, Adarska P, Singh A, and Schmoller KM (2020). Transcription coordinates histone amounts and genome content. bioRxiv, 2020.2008.2028.272492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2012). 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 13 Suppl 16, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Crissman HA, and Steinkamp JA (1973). Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. J Cell Biol 59, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier L, Foy R, Mouery BL, Whitaker RH, Corno A, Spanos C, Ly T, Gowen Cook J, and Saurin AT (2022). CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J 41, e108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ario M, Tavares R, Schiessl K, Desvoyes B, Gutierrez C, Howard M, and Sablowski R (2021). Cell size controlled in plants using DNA content as an internal scale. Science 372, 1176–1181. [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, van Rossum A, Schuijff L, and te Riele H (2000). Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev 14, 3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko ZN, and Blagosklonny MV (2008). Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 7, 3355–3361. [DOI] [PubMed] [Google Scholar]
- Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, and Blagosklonny MV (2009). Rapamycin decelerates cellular senescence. Cell Cycle 8, 1888–1895. [DOI] [PubMed] [Google Scholar]
- Deng J, Erdjument-Bromage H, and Neubert TA (2019). Quantitative Comparison of Proteomes Using SILAC. Curr Protoc Protein Sci 95, e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel-Morgenstern M, Danon T, Christian T, Igarashi T, Cohen L, Hou YM, and Jensen LJ (2012). Genes adopt non-optimal codon usage to generate cell cycle-dependent oscillations in protein levels. Mol Syst Biol 8, 572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginzberg MB, Kafri R, and Kirschner M (2015). Cell biology. On being the right (cell) size. Science 348, 1245075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A, Nehme J, and Demaria M (2018). Hallmarks of Cellular Senescence. Trends Cell Biol 28, 436–453. [DOI] [PubMed] [Google Scholar]
- Keifenheim D, Sun XM, D’Souza E, Ohira MJ, Magner M, Mayhew MB, Marguerat S, and Rhind N (2017). Size-Dependent Expression of the Mitotic Activator Cdc25 Suggests a Mechanism of Size Control in Fission Yeast. Curr Biol 27, 1491–1497 e1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempe H, Schwabe A, Cremazy F, Verschure PJ, and Bruggeman FJ (2015). The volumes and transcript counts of single cells reveal concentration homeostasis and capture biological noise. Mol Biol Cell 26, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R, and Jat P (2021). Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol 9, 645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HO, Davidson JM, and Duronio RJ (2009). Endoreplication: polyploidy with purpose. Genes Dev 23, 2461–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengefeld J, Cheng CW, Maretich P, Blair M, Hagen H, McReynolds MR, Sullivan E, Majors K, Roberts C, Kang JH, et al. (2021). Cell size is a determinant of stem cell potential during aging. Sci Adv 7, eabk0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, and Amir A (2018). Homeostasis of protein and mRNA concentrations in growing cells. Nat Commun 9, 4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yan J, and Kirschner MW (2022). Beyond G1/S regulation: how cell size homeostasis is tightly controlled throughout the cell cycle? bioRxiv, 2022.2002.2003.478996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd AC (2013). The regulation of cell size. Cell 154, 1194–1205. [DOI] [PubMed] [Google Scholar]
- Marshall WF (2020). Scaling of Subcellular Structures. Annu Rev Cell Dev Biol 36, 219–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen TP, and Bjorklund M (2016). Cellular Allometry of Mitochondrial Functionality Establishes the Optimal Cell Size. Dev Cell 39, 370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu L, Kang JH, Olcum S, Payer KR, Calistri NL, Kimmerling RJ, Manalis SR, and Miettinen TP (2020). Mass measurements during lymphocytic leukemia cell polyploidization decouple cell cycle- and cell size-dependent growth. Proc Natl Acad Sci U S A 117, 15659–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FR, and Nurse P (2007). Nuclear size control in fission yeast. J Cell Biol 179, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, et al. (2019). Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell 176, 1083–1097 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovan-Merhar O, Nair GP, Biaesch AG, Mayer A, Scarfone S, Foley SW, Wu AR, Churchman LS, Singh A, and Raj A (2015). Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol Cell 58, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafelski SM, Viana MP, Zhang Y, Chan YH, Thorn KS, Yam P, Fung JC, Li H, Costa Lda F, and Marshall WF (2012). Mitochondrial network size scaling in budding yeast. Science 338, 822–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, and Pachter L (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J, et al. (2018). NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, and Jacks T (2000). Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Koivomagi M, and Skotheim JM (2015). Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature 526, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz C, Johnson A, Koivomagi M, Zatulovskiy E, Kravitz CJ, Doncic A, and Skotheim JM (2018). A Precise Cdk Activity Threshold Determines Passage through the Restriction Point. Mol Cell 69, 253–264 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, and Sherr CJ (2015). Forging a signature of in vivo senescence. Nat Rev Cancer 15, 397–408. [DOI] [PubMed] [Google Scholar]
- Son S, Kang JH, Oh S, Kirschner MW, Mitchison TJ, and Manalis S (2015). Resonant microchannel volume and mass measurements show that suspended cells swell during mitosis. J Cell Biol 211, 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XM, Bowman A, Priestman M, Bertaux F, Martinez-Segura A, Tang W, Whilding C, Dormann D, Shahrezaei V, and Marguerat S (2020). Size-Dependent Increase in RNA Polymerase II Initiation Rates Mediates Gene Expression Scaling with Cell Size. Curr Biol 30, 1217–1230 e1217. [DOI] [PubMed] [Google Scholar]
- Swaffer MP, Kim J, Chandler-Brown D, Langhinrichs M, Marinov GK, Greenleaf WJ, Kundaje A, Schmoller KM, and Skotheim JM (2021a). Transcriptional and chromatin-based partitioning mechanisms uncouple protein scaling from cell size. Mol Cell 81, 4861–4875 e4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaffer MP, Marinov GK, Zheng H, Jones AW, Greenwood J, Kundaje A, Snijders AP, Greenleaf WJ, Reyes-Lamothe R, and Skotheim JM (2021b). RNA polymerase II dynamics and mRNA stability feedback determine mRNA scaling with cell size. bioRxiv, 2021.2009.2020.461005. [Google Scholar]
- Tan C, Ginzberg MB, Webster R, Iyengar S, Liu S, Papadopoli D, Concannon J, Wang Y, Auld DS, Jenkins JL, et al. (2021). Cell size homeostasis is maintained by CDK4-dependent activation of p38 MAPK. Dev Cell 56, 1756–1769 e1757. [DOI] [PubMed] [Google Scholar]
- Tzur A, Moore JK, Jorgensen P, Shapiro HM, and Kirschner MW (2011). Optimizing optical flow cytometry for cell volume-based sorting and analysis. PLoS One 6, e16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, and Scopes AW (1961). The distribution of nucleic acids and protein between different sized yeast cells. Exp Cell Res 24, 151–153. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Hein MY, Cox J, and Mann M (2014). A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 13, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al. (2020). Ensembl 2020. Nucleic Acids Res 48, D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatulovskiy E, and Skotheim JM (2020). On the Molecular Mechanisms Regulating Animal Cell Size Homeostasis. Trends Genet 36, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatulovskiy E, Zhang S, Berenson DF, Topacio BR, and Skotheim JM (2020). Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. Science 369, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecha J, Meng C, Zolg DP, Samaras P, Wilhelm M, and Kuster B (2018). Peptide Level Turnover Measurements Enable the Study of Proteoform Dynamics. Mol Cell Proteomics 17, 974–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecha J, Satpathy S, Kanashova T, Avanessian SC, Kane MH, Clauser KR, Mertins P, Carr SA, and Kuster B (2019). TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol Cell Proteomics 18, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky J, Leonhard K, Watt S, Marguerat S, Bahler J, and Nurse P (2010). A coordinated global control over cellular transcription. Curr Biol 20, 2010–2015. [DOI] [PubMed] [Google Scholar]
- Zlotek-Zlotkiewicz E, Monnier S, Cappello G, Le Berre M, and Piel M (2015). Optical volume and mass measurements show that mammalian cells swell during mitosis. J Cell Biol 211, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Slope values for size-sorted G1 cells (HLF and hTERT RPE-1), related to Figure 1
Table S2. Slope values for size-sorted G1 cells using SSC and Total protein dye, related to Figure 1 and S5
Table S4. Slope variance for proteins in protein complexes, related to Figure 2B
Table S5. 2D gene annotation enrichments for HLF and hTERT RPE-1, related to Figure 2D
Table S7. RNA and Protein Slope values for size-sorted G1 cells, related to Figure 3
Table S8. 2D gene annotation enrichments for linear model parameters, related to Figure 3D
Table S9. Slope values for size-sorted and ploidy-sorted G1 cells, related to Figure 7
Table S10. Proteomic Experiments list and quantitation channel information, related to STAR Methods
Table S11. MaxQuant spectral search parameter files, related to STAR Methods
Data Availability Statement
Proteomic and RNA-seq data have been deposited to PRIDE and GEO, respectively, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy and western blotting data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.