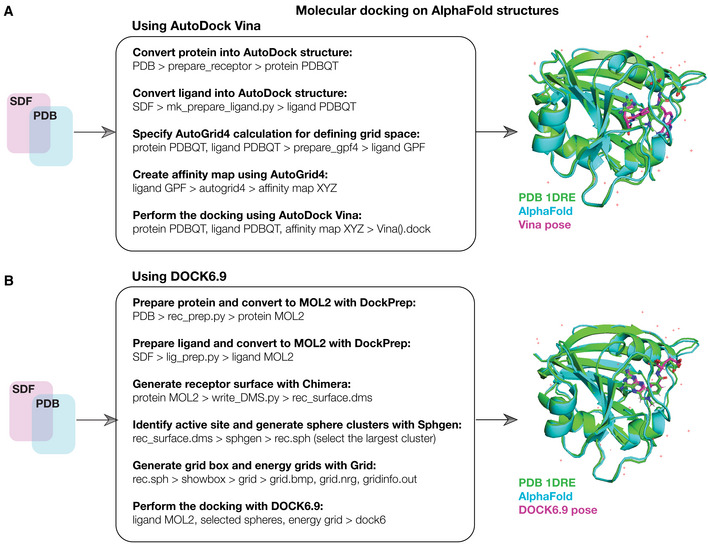
(Left) Schematic of the computational docking approach using AutoDock Vina. 296 essential proteins in
E. coli were identified, and their AlphaFold2‐predicted structures were curated. The 218 active compounds and 100 inactive compounds were represented in three dimensions in SDF files. All compounds and proteins were prepared for docking as shown and then docked using AutoDock Vina run on a high‐performance computing server. The resulting binding pose and thermodynamic binding affinity predictions for all 64,528 (active compounds) and 29,600 (inactive compounds) pairwise protein‐ligand interactions were analyzed and ranked. (Right) Superimposed predicted and experimental structures for methotrexate binding to
E. coli FolA (PDB 1DRE), which was used as a positive docking control from the Protein Data Bank (Dataset
EV2).