

Abstract
Background
46,XX male disorders of sex development are rare. Approximately 80% of cases of testicular tissue differentiation may be due to translocation of SRY to the X chromosome or an autosome. SRY-negative 46,XX males show overexpression of pro-testis genes, such as SOX9 and SOX3, or failure of pro-ovarian genes, such as WNT4 and RSPO1, which induces testis differentiation, however, almost all testicles exhibit dysgenesis. Following inadequate exposure to androgens during the embryo stage, remnants of the Mullerian duct and incomplete closure of the urogenital sinus lead to enlargement of prostatic utricles. This condition is associated with proximal hypospadias and disorders of sex development. Many cases are asymptomatic, but show increased rates of postoperative complications and surgical failure.
Case presentation
A 5-year-old Chinese boy with scrotal hypospadias and bilateral cryptorchidism with prostatic utricles was presented. Gonadal histology showed ovo-testicular tissue on the right side and testicular tissue on the left side; all testicular tissue exhibited dysgenesis. Furthermore, chromosome karyotype analysis revealed 46,XX and, the presence of SRY was ruled out by polymerase chain reaction analysis. Whole-genome analysis showed the boy has a 1.4-Mb duplication in the Xq27.1q27.2 region (arr[hg19]Xq27.1q27.2:139585794–140996652) involving SOX3. No SOX3 duplication was observed in the parents, who had a normal phenotype.
Conclusions
We report the first case of an SRY-negative 46 XX male with prostatic utricle caused by SOX3 duplication. SOX3 duplication may cause sex reversal, and all 46,XX SRY-negative males should be screened for SOX3 mutations. Gonadal biopsy is recommended to evaluate ovarian and testicular tissue development. Testicular dysgenesis and low exposure to male hormones during fetal development can lead to enlarged prostatic utricles. Thus endoscopic examination should be performed preoperatively to detect prostatic utricles in SRY-negative 46,XX males to determine the surgical plan and reduce postoperative complications.
Keywords: Disorder of sex development, SOX3, Prostatic utricle, Case report
Background
Ovotesticular male disorders of sex development (OT-DSD) are rare worldwide. These disorders show differing prevalence and karyotypes; however, the 46,XX karyotype is the most common(65–90%) [1]. This presentation results from the translocation of SRY to the X chromosome or, more rarely, to the autosomes. The SRY-negative phenotype is rare and can be explained by two different mechanisms: overexpression of pro-testis genes, such as SOX9 and SOX3, and failure of pro-ovarian genes, such as WNT4 and RSPO1 [2]. SOX3 mutation-induced sex reversal may be driven by the shared analogous function of SOX3 and SRY, cooperation with SF1 to upregulate SOX9 expression, and induction of Sertoli cell differentiation in the bipotential gonad ridges [2]. A previous study reported cases of hypospadias, bilateral cryptorchidism, and kidney abnormality [3]. In our case, we additionally observed an enlarged prostatic utricle, which to our knowledge is the first instance of this phenotype observed in this specific type of DSD. The embryological origin of the prostatic utricle remains controversial. It was previously considered as a remnant of the Müllerian ducts, however, a recent study showed it to be derived from the urogenital sinus [4]. Either way, the prostatic utricle dilates when exposure to male hormones is low during fetal development [5]. In an SRY-negative 46,XX male, lack of the Y chromosome results in inadequate differentiation of the testis, an insufficient hormone dosage during critical periods, induction of remnant Mullerian ducts and incomplete closure of the urogenital sinus [6].
Case presentation
The patient was 5 years old and was referred to our department for ambiguous sex; he was the second child of a non-consanguineous couple. His parents and sister were healthy. The patient’s birth weight was 3.5 kg and length was 50 cm, which were within normal ranges; his intelligence was normal, and he had no pertinent medical history. At 5 years old, the patient’s height was 117 cm and weight was 20.5 kg. Physical examination revealed scrotal hypospadias and bilateral cryptorchidism (Fig. 1a). The bilateral gonads (approximately 1.0 × 0.6 cm) were palpated in the upper inguinal region. Ultrasonography indicated bilateral cryptorchidism with a normal testicular morphology (Fig. 1c, d). Abdominal ultrasound did not show internal female genitalia, and echocardiogram results were normal. Hormonal laboratory tests showed low basal levels of testosterone (T; < 2.5 ng/dL); the post-3-day human chorionic gonadotropin stimulation test showed an increase in T to 62.90 ng/dL, and the post-human chorionic gonadotropin test showed a normal level of double hydrogen testosterone (80.37 pg/mL). Thus, there was a good T response to human chorionic gonadotropin stimulation. Luteinizing hormone was low (< 0.1 IU/L) and follicle stimulating hormone was normal (0.4 IU/L). Anti-Mullerian hormone (48.48 ng/mL), prolactin (4.87 ng/mL), and 17-α-hydroxyprogesterone (0.8 nmol/L)levels were also normal (Table 1). As patient growth and development were normal, we did not screen for growth hormone or adreno-cortico-tropic hormone.
Fig. 1.
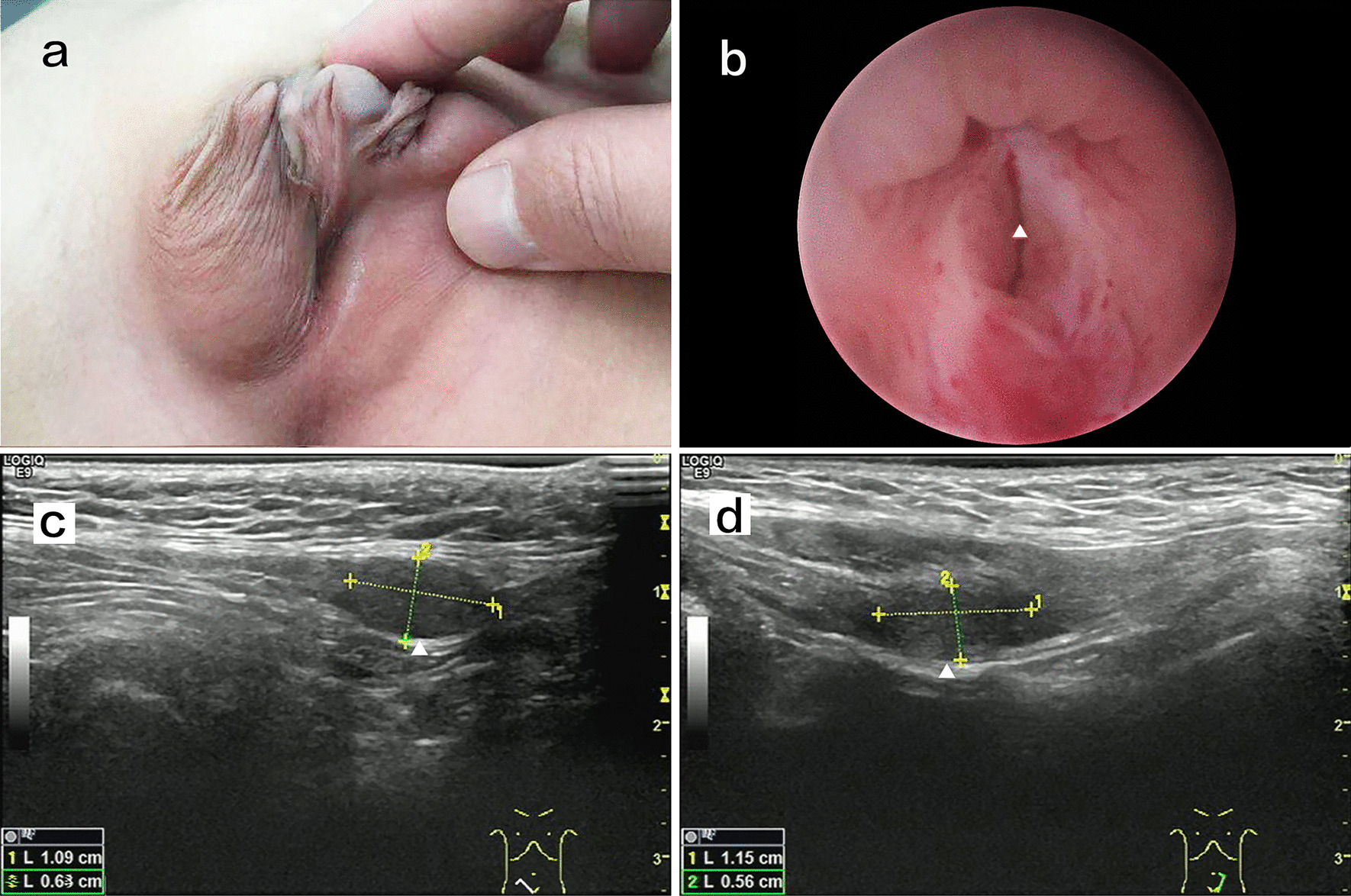
a Physical examination revealed scrotal hypospadias and bilateral cryptorchidism. b Pre─operation cystoscopy showing an orifice in the prostatic urethra; the white arrow indicates the prostatic urethra orifice. c Ultrasonography showing that the right testicle is in the inguinal canal; the morphology was normal. d Ultrasonography showing that the left testicle is in the inguinal canal; the morphology was normal
Table 1.
Hormonal laboratory tests
| Hormonal laboratory tests | Result | Reference range |
|---|---|---|
| testosterone | ||
| basal | < 2.5 ng/dl | |
| Post HCG | 62.90 ng/dl | |
| T difference value > 10 ng/dl | ||
| Post HCG DHT | 80.37 pg/ml | |
| T/DHT < 10 | ||
| AMH | 48.48 ng/ml |
Male(> 4 years):37.88–298.52 ng/ml Female(> 4yeays):0.05–7.02 ng/ml |
| LH | < 0.1 IU/L | 0- 4.10 IU/L |
| FSH | 0.4 IU/L | 0–1.90 IU/L |
| Prolactin | 4.87 ng/ml | 2.00–43.00 ng/ml |
| 17-α-hydroxyprogesterone | 0.8 nmol/l | 0–11.5 nmol/l |
HCG: Human Chorionic Gonadotropin. DHT: Double Hydrogen Testosterone. AMH: Anti-Mullerian hormone. LH: Luteinizing Hormone. FSH: Follicle Stimulating Hormone
The chromosome karyotype was 46,XX, and the presence of SRY was ruled out by polymerase chain reaction analysis. Therefore, we performed whole-genome analysis for genetic detection. Genomic DNA was extracted from blood samples using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). The DNA quantity of sequencing library was assessed by Qubit 2.0 fluorometer (Thermo Fisher Scientific, Massachusetts, United States). The quality and size of libraries were measured by 2100 Bioanalyzer High Sensitivity DNA Assay (Agilent Technologies, California, United States). The qualified libraries were sequenced using the 2 × 150-bp paired-end sequencing on the Illumina NovaSeq platform (Illumina, San Diego, USA). FASTQ files were aligned to the human reference genome (hg19/ GRCh37) using BWA v0.7.13 Variants (single nucleotide variants and indels) were genotyped from recalibrated BAM files using GATK 4.0 and annotated against multiple databases, including HGVS variant description, population frequency, disease or phenotype and variant functional prediction, using ANNOVAR. Variants were classified as pathogenic, likely pathogenic, variant of unknown significance, likely benign, or benign following the American College of Medical Genetics (ACMG) guidelines. Copy number variants were called by DNAcopy R package, filtered classified as per the ACMG guidelines, and manually checked by using the Integrative Genomics Viewer. Target capture area was 50 M, detection range capture rate 99.9%, detection data volume 10G, average sequencing depth ≥ 100X, and the proportion of Q30 was not less than 90%. The results revealed no pathogenic or likely pathogenic single-nucleotide variation or insertion-deletion variant related to sexual development, but revealed a 1.4-Mb duplication in the Xq27.1q27.2 region (arr[hg19]Xq27.1q27.2:139,585,794–140,996,652). The duplication contained CDR1, SOX3, and SPANXA1 (Fig. 2). According to the ACMG, this duplication is of uncertain significance. The parents did not show SOX3 duplication and had normal phenotypes.
Fig. 2.

Whole genome analysis showing 1.4-Mb duplication in the Xq27.1q27.2 region (arr[hg19]Xq27.1q27.2:139,585,794–140,996,652). The duplication contained CDR1, SOX3, and SPANXA1
After clinical consultation, the family decided to raise the child as a male. The patient underwent surgical procedures for the correction of hypospadias and gonad biopsy, followed by orchiopexy. Preoperative cystoscopy showed an orifice in the prostatic urethra. Upon probing with a camera, enlarged prostatic utricles were observed (Fig. 1b). The bilateral gonads were biopsied, followed by orchiopexy. Gonadal histology showed ovotesticular tissue on the right side and testicular tissue on the left side; both testicular tissues exhibited features of testicular dysgenesis. The ovarian tissue had primordial and primary follicles (Fig. 3a, b). Owing to this complicated condition, we performed penile straightening and planned to perform urethroplasty 6 months later. After degloving of the penis, the corpus cavernosum was well developed and the penis length and diameter were 4.5 and 1.5 cm, respectively.
Fig. 3.
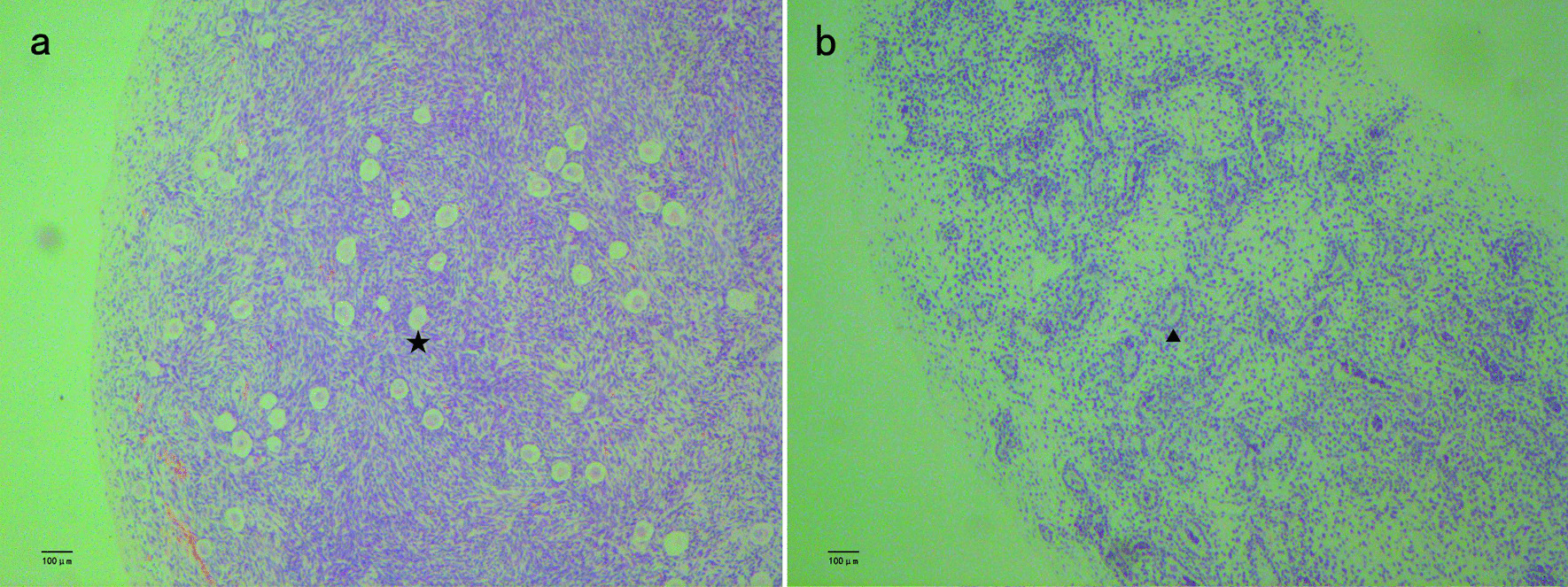
Hematoxylin and eosin staining of bilateral gonadal sample. a: The right side showing ovotesticular tissue; the ovarian tissue had primordial and primary follicle(star). b: The left side showing testicular dysgenesis(arrow). These images were obtained using the following equipment: microscope DM300 and camera DFC45O(leica, Germany). Scanner Hamamatsu, Nanozoomer S210-NDP. View 2 version 2.9.29, was used as acquisition software and the measurement resolution was 1200dpi
Discussion and conclusion
SOX3 is a single exon gene located in Xq27.1 and is required for normal brain, pituitary and craniofacial development in humans. Recent reports showed that mutation of SOX3 can cause developmental delay or intellectual disturbance [7, 8]. Although SOX3 is not required for normal sex development, it affects testis differentiation and oocyte development [9], Mutation of SOX3 induces SRY- negative 46,XX male sex reversal has been comfirmed by previous reports [10]. However, the mechanism underlying SOX3 mutation inducing SRY-negative 46 XX male sex reversal was not well understood. Through a literature review, we found 10 similar cases (Table 2) [2, 3, 10–15]. Seven of these cases contained SOX3 duplication mutations, ranging from 550 kb to 6 Mb. The mechanism may involve overexpression of SOX3 to upregulate SOX9 expression and induce testis differentiation [1]. Our patient had a 1.4-Mb duplication in the Xq27.1q27.2 region, which contained SOX3, supporting this prediction. In three of the cases, the condition was associated with rearrangements in the regulatory region of SOX3, which may weaken the inhibition of SOX3 [15].
Table 2.
Cases of SOX3 related XX male reversal
| Suttou et. al Patients A |
Suttou et. al Patients B |
Suttou et. al Patients C |
4: Moalen et. al | 5: Haines.et al | 6: Vetro.et | 7: Grisponetal.et | 8: Tasic.et | 9: Zhuang.J et | 10: Qin,S et | Our patient | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disorders of sex developmen | XX male reversal | XX male reversal | XX male reversal | XX male reversal | OT-DSD | XX male reversal | OT-DSD | XX male reversal | OT-DSD | XX male reversal | OT-DSD |
| Age | 30 years | Endocrine:19 years histology:26 years | 1.5 years | 1 years | 1.5 months | 8 years | 2.5 years | 11 years | 7 years | 31 years | 5 years |
| Growth and developmental issues | Normal | Developmental delay;microcephaly | Developmental and growth delay,microcephaly | Normal | normal | Mild intellectual disability | Normal | Normal | Normal | Normal | Normal |
| Hormone analysis |
FSH:22.0mIU/ml LH:11.IIU/L Prolactin 17.9ug/l Free T:1.92 ng/dl |
FSH:69.0 mIU/ml LH:35.0 IU/L Prolactin 3.0ug/l Free T:0.86 ng/dl |
Unknow | FSH/LH/T:normal |
T:9.2 nmol/L Post-HCG:T:2.1 → 24.6 nmol/l DHT:2.0 → 7.5 nmol/l Androstenedione0.6 → 1.1 nmol/l |
Unknow |
T(nmol/l) Basal: < 0.35 Post-HCG:1.05 AMH:216 LH: < 0.1 FSH0.73 |
Post HCG: T:146 ng/ml A:T: < 1 T:DHT was 5.6 |
LH < 0.2mIU/ml FSH:1.21 mIU/ml T: < 0.1 ng/ml Serum progesterone/prolactin:normal |
T:1.75 ng/ml Progesterone:0.11 ng/ml Prolactin:208.44uIU/ml LH:29.32mIU/ml FSH:37.88mIU/ml |
LH0.4 IU/L FSH0.1 IU/L Prolactin:4.87 ng/ml T: < 2.5 ng/dl AMH:48.48 ng/ml Post-HCG:T:62.9 ng/dl |
| Genitals | Unkown | penile development with small testis.Shaft length, 10.2 cm; shaft diameter, 2.6 cm | Right testicles appear smaller than left;Hypoplastic scrotum;testes are retractile and can be brought down | Left cryptorchidism | Bifid scrotum; small phallus; distal hypopadias | Normal | Hypospadias and bilateral cryptorchidism | Moderate coronal hypospadias | Hypospadias and bilateral cryptorchidism | Normal | Scrotal hypospadias and bilateral cryptorchidism |
| Gonadal Histology | No details | Testicular dysgenesis | No details | No details | Right:ovotesticular | No details | The testicular tissue and ovarian tissue all exist.Andtesticular dysgenesis | No details | The ovotesticular tissue on the left side and the testicular tissue on the right side | No details |
Right:ovotesticular tissue; Left: testicular tissue Testicular dysgenesis |
| Associated anomalies | – | – | – | Normal | Fallopian tube, hemiuterus and hemivagina on the right gonads | – | – | Kidney hypodysplasia | – | – | Prostatic utricle |
| Genotypes |
SRY negative Two microduplications were observed,the first of which spanned the entire sox3 gene |
Single 343-kb microdeletion on the X-chromosome immediately upstream of SOX3 |
SRY negative 6 Mb duplication that encompasses sox3 and at least 18 additional distally located genes |
SRY negative 0.494 Mb copy number gain in region Xq27.1 which contains the SOX3, RP1-177G6.2, CDR1 |
SRY negative carries 774 kb insertion translocation from chromosome 1 into a 82 kb distal to SOX3 |
A 5.6 Mb duplication of the long arm of a chromosome X, involving the SOX3 gene |
SRY negative, at Xq27.1. The duplicated region was around 0.5 Mb, and encompassed the SOX3 |
A unique 550 kb duplication involving SOX3 |
SRY negative 2.2 Mb duplication that encompasses SOX3 gene |
SRY negative, 867 kb heterozygous deletion in Xq27.1,located at 104 kb downstream of SOX3 |
SRY negative 1.4 Mb duplication that encompasses SOX3,CDR1,SPANXA1 |
Our patient’s bilateral gonads were biopsied, and histological analysis showed that the testicular component was dysgenetic and that ovarian tissue was present, Therefore, the diagnosis was revised to OT-DSD. Haines et al. [12], Grisponetal et al. [10], and Zhuang et al. [14] also performed gonadal biopsy and obtained similar results. The development of testicular tissue and ovarian tissue may influence the sex assignment of patients with DSD. If the patients are raised as males, then the ovarian portion must be removed before pubertal age to avoid gynecomastia and cyclic hemorrhage. If the patients are raised as females, testicular tissue must be removed to avoid virilization during puberty [1]. Therefore, at any age, when the patient is diagnosed with an SRY-negative 46 XX karyotype, the gonads must be biopsied.
Our case involved duplication of SOX3 in an SRY-negative 46,XX male associated with an enlarged prostatic utricle, which has not been reported previously. Tasicet et al. [3] reported patients with congenital anomalies of the kidneys. Haineset al [12] reported patients with fallopian tubes, hemiuterus, and hemivagina on the right gonads, which were removed through surgery. Such patients must also be screened for abnormalities in other organs. Our patient also underwent an ultrasound, but the results were normal. We routinely perform cystoscopic pre-operation in patients with proximal hypospadias, which revealed an enlarged prostatic utricle in the current patient. Ultrasonography and retrograde urethrography can detect the prostatic utricle, but it is difficult to detect a small prostatic utricle using ultrasonography, and retrograde urethrography is not useful because of the slit-like utricular opening [16]. Thus, endoscopy is suitable for detecting the prostatic utricle. An enlarged prostatic utricle can cause recurrent urinary tract infection, epididymo-orchitis, fistulas after urethroplasty, and calculi or malignancy [17]. Early diagnosis is important for providing support for individualized plans and reducing postoperative complications [18]. Therefore, endoscopic examination should be performed in SRY-negative 46,XX males with hypospadias, as recommended previously [6].
Our patient has a 1.4-Mb duplication in the Xq27.1q27.2 region. The duplication contained CDR1, SOX3, and SPANXA1. Cerebellar degeneration-related antigen 1(CDR1) is expressed in the nervous system, human epidermis, and cancer tissues, but its function is unclear [19]. SPANXA1 is expressed in the tumors or testis [20], but has not been reported to be associated with sexual reversal. Therefore, the relationship between these two genes and sex determination requires further analysis.
SOX3 duplication may causes sex reversal, and all 46,XX SRY-negative males should be screened for SOX3 mutations. Gonadal biopsy is recommended to determine the presence of ovarian and testicular tissue development. Testicular dysgenesis and low exposure to male hormones during fetal development can lead to enlarged prostatic utricles. Thus, endoscopic examination should be performed preoperatively to detect prostatic utricles in SRY-negative 46,XX males to guide the surgical plan and reduce postoperative complications.
Acknowledgements
The authors are grateful to the staff of Fuzhou Children’s Hospital of Fujian Province, who were responsible for the management of patients included in the study. We are also grateful to the patients and their families who participated in this study.
Abbreviations
- DSD
Disorders of sex development
- OT-DSD
Ovotesticular male disorders of sex development
- T
Testosterone
Author contributions
JW:Project design, data acquisition and analysis, and manuscript writing. CL: Data acquisition and analysis. MZ: Data acquisition and analysis, SL: Data acquisition and analysis, JF: Data Acquisition and Analysis, and PL: Data acquisition and analysis. All authors contributed to the editorial changes in the manuscript. All authors read and approved the final manuscript.
Funding
The study was supported by the Key Clinical Specialty Discipline Construction Program of Fuzhou, Fujian, P.R.C(2018-56). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets (whole-genome analysis, polymerase chain reaction results, hormonal laboratory tests) used and/ or analyzed during the current study are available from the corresponding author on reasonable request. The sequencing data has been deposited in NCBI Bioproject database, The data is accessible via the accession number: PRJNA864577 (https://www.ncbi.nlm.nih.gov/sra/PRJNA864577). The following databases are used in this study: Human Reference Genome (hg19/ GRCh37) (https://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml) GATK (https://www.broadinstitute.org/gatk/) ANNOVAR(https://annovar.openbioinformatics.org/en/latest/).
Declarations
Ethics approval and consent to participate
The study protocol was reviewed and approved by the Ethics Committee of the Fuzhou Children’s Hospital of Fujian Province (Approval Number: FCHFJ-2022–11). All analyses were conducted in accordance with the tenets of the Declaration of Helsinki. The patient and his parents provided written informed consent to participate in this study.
Consent for publication
All subjects who participated in this study signed written informed consent for publishing their own and their children’s genetic data and relevant information.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Grinspon RP, Rey RA. Disorders of sex development with testicular differentiation in SRY-negative 46, XX individuals: clinical and genetic aspects. Sex Dev. 2016;10(1):1–11. doi: 10.1159/000445088. [DOI] [PubMed] [Google Scholar]
- 2.Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. 2011;121(1):328–341. doi: 10.1172/JCI42580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasic V, Mitrotti A, Riepe FG, Kulle AE, Laban N, Polenakovic M, et al. Duplication of the SOX3 gene in an Sry-negative 46, XX male with associated congenital anomalies of kidneys and the urinary tract: case report and review of the literature. Balkan J Med Genet. 2019;22(1):81–88. doi: 10.2478/bjmg-2019-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapiro E, Huang H, McFADDEN DE, Masch RJ, Ng E, Lepor H, Wu XR. The prostatic utricle is not a Müllerian duct remnant: immunohistochemical evidence for a distinct urogenital sinus origin. J Urol. 2004;172:1753–56. doi: 10.1097/01.ju.0000140267.46772.7d. [DOI] [PubMed] [Google Scholar]
- 5.Furuya S, Hisasue S, Kato H, Shimamura S. Novel insight for midline cyst formation in prostate: the involvement of decreased prenatal testosterone suggested by second-to-fourth digit ratio study. Int J Urol. 2015;22(11):1063–1067. doi: 10.1111/iju.12892. [DOI] [PubMed] [Google Scholar]
- 6.Hester AG, Kogan SJ. The prostatic utricle: an under-recognized condition resulting in significant morbidity in boys with both hypospadias and normal external genitalia. J Pediatr Urol. 2017;13(5):492.e1–5. doi: 10.1016/j.jpurol.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 7.Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. 2005;76(5):833–849. doi: 10.1086/430134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du C, Wang F, Li Z, Zhang M, Yu X, Liang Y, et al. Xq26.3-q27.1 duplication including SOX3 gene in a Chinese boy with hypopituitarism: case report and two years treatment follow up. BMC Med Genom. 2022;15(1):19. doi: 10.1186/s12920-022-01167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss J, Meeks JJ, Hurley L, Raverot G, Frassetto A, Jameson JL, et al. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol. 2003;23(22):8084–8091. doi: 10.1128/MCB.23.22.8084-8091.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grinspon RP, Nevado J, Alvarez MDLÁM, Del Rey G, Castera R, Venara M, et al. 46, XX ovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin Endocrinol. 2016;85(4):673–675. doi: 10.1111/cen.13126. [DOI] [PubMed] [Google Scholar]
- 11.Moalem S, Babul-Hirji R, Stavropolous DJ, et al. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A. 2012;158(7):1759–64. doi: 10.1002/ajmg.a.35390. [DOI] [PubMed] [Google Scholar]
- 12.Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab. 2015;100(5):E815–20. doi: 10.1210/jc.2014-4383. [DOI] [PubMed] [Google Scholar]
- 13.Vetro A, Dehghani MR, Kraoua L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. 2015;23(8):1025–1032. doi: 10.1038/ejhg.2014.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhuang J, Chen C, Li J, Jiang Y, Wang J, Wang Y, et al. The 46, XX ovotesticular disorder of sex development With Xq27.1q27.2 duplication involving the SOX3 gene: a rare case report and literature review. Front Pediatr. 2021;9:682846. doi: 10.3389/fped.2021.682846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin S, Wang X, Wang J. Identification of an SRY-negative 46, XX infertility male with a heterozygous deletion downstream of SOX3 gene. Mol Cytogenet. 2022;15(1):2. doi: 10.1186/s13039-022-00580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kojima Y, Hayashi Y, Maruyama T, Sasaki S, Kohri K. Comparison between ultrasonography and retrograde urethrography for detection of prostatic utricle associated with hypospadias. Urology. 2001;57(6):1151–1155. doi: 10.1016/S0090-4295(01)00954-2. [DOI] [PubMed] [Google Scholar]
- 17.Şalvarcı A, İstanbulluoğlu O. Monosymptomatic persistent hematospermia due to rarely encountered prostatic utricle stones. Urol Int. 2015;95(3):370–372. doi: 10.1159/000354766. [DOI] [PubMed] [Google Scholar]
- 18.Gupta A, Khosa J, Barker A, Samnakay N. Clinical spectrum and management options for prostatic utricle in children. J Pediatr Surg. 2022 doi: 10.1016/j.jpedsurg.2022.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Wang LL, Gschwandtner M, Eckhart L, Tschachler E. Cerebellar degeneration-related antigen 1 is ubiquitously expressed in human epidermis and dermis. Curr Med Sci. 2020;40(3):570–573. doi: 10.1007/s11596-020-2192-2. [DOI] [PubMed] [Google Scholar]
- 20.Zendman AJ, Zschocke J, van Kraats AA, de Wit NJW, Kurpisz M, et al. The human SPANX multigene family: genomic organization, alignment and expression in male germ cells and tumor cell lines. Gene. 2003;309(2):125–133. doi: 10.1016/S0378-1119(03)00497-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets (whole-genome analysis, polymerase chain reaction results, hormonal laboratory tests) used and/ or analyzed during the current study are available from the corresponding author on reasonable request. The sequencing data has been deposited in NCBI Bioproject database, The data is accessible via the accession number: PRJNA864577 (https://www.ncbi.nlm.nih.gov/sra/PRJNA864577). The following databases are used in this study: Human Reference Genome (hg19/ GRCh37) (https://www.ncbi.nlm.nih.gov/projects/genome/guide/human/index.shtml) GATK (https://www.broadinstitute.org/gatk/) ANNOVAR(https://annovar.openbioinformatics.org/en/latest/).