

Abstract
Background
This is an updated version of the Cochrane Review first published in 2014 and last updated in 2020.
For nearly 30% of people with epilepsy, current treatments do not control seizures. Stiripentol is an antiepileptic drug (AED) that was developed in France and was approved by the European Medicines Agency (EMA) in 2007 as an adjunctive therapy with valproate and clobazam for the treatment of Dravet syndrome.
Objectives
To evaluate the efficacy and tolerability of stiripentol as add‐on treatment for people with drug‐resistant focal epilepsy who are taking AEDs.
Search methods
For the latest update, we searched the Cochrane Register of Studies (CRS Web) and MEDLINE on 28 March 2022. We contacted the manufacturer of stiripentol and epilepsy experts to identify published, unpublished and ongoing trials.
Selection criteria
Randomised controlled trials of add‐on stiripentol in people with drug‐resistant focal epilepsy.
Data collection and analysis
Review authors independently selected trials for inclusion and extracted data. We investigated outcomes including 50% or greater reduction in seizure frequency, seizure freedom, adverse effects, treatment withdrawal and changes in quality of life.
Main results
On the basis of our selection criteria, we included no new studies in the present review update. We included only one study from the original review (32 children with focal epilepsy). This study adopted a responder‐enriched design and found no clear evidence of a reduction of 50% or more in seizure frequency (risk ratio (RR) 1.51, 95% confidence interval (CI) 0.81 to 2.82; low‐certainty evidence) and no clear evidence of seizure freedom (RR 1.18, 95% CI 0.31 to 4.43; low‐certainty evidence) when comparing add‐on stiripentol with placebo.
Stiripentol led to a greater risk of adverse effects considered as a whole (RR 2.65, 95% CI 1.08 to 6.47; low‐certainty evidence). When we considered specific adverse effects, CIs were very wide and showed the possibility of substantial increases and small reductions in risks of neurological adverse effects (RR 2.65, 95% CI 0.88 to 8.01; low‐certainty evidence). Researchers noted no clear reduction in the risk of study withdrawal (RR 0.66, 95% CI 0.30 to 1.47; low‐certainty evidence), which was high in both groups (53.3% in placebo group and 35.3% in stiripentol group; low‐certainty evidence).
The external validity of this study was limited because only responders to stiripentol (i.e. participants experiencing a decrease in seizure frequency of 50% or greater during an open prerandomisation phase compared with baseline) were included in the randomised, add‐on, placebo‐controlled, double‐blind phase. Furthermore, carry‐over and withdrawal effects probably influenced outcomes related to seizure frequency. Very limited information derived from the only included study shows that adverse effects considered as a whole may occur more often with add‐on stiripentol than with add‐on placebo.
Authors' conclusions
We have found no new studies since the last version of this review was published. Hence, we have made no changes to the conclusions as presented in previous versions. We can draw no conclusions to support the use of stiripentol as add‐on treatment for drug‐resistant focal epilepsy. Additional large, randomised, well‐conducted trials are needed.
Plain language summary
Stiripentol as an add‐on treatment for drug‐resistant focal epilepsy (epilepsy affecting one side of the brain)
What is drug‐resistant epilepsy?
Epilepsy is one of the more common long‐lasting neurological disorders; it affects 1% of the population worldwide. Up to 30% of people with epilepsy continue to have seizures (sudden bursts of electrical activity in the brain that change how it works for a short time) despite adequate therapy with antiepileptic medicines. These people are regarded as having drug‐resistant epilepsy.
What did we want to find out?
Antiepileptic medicines can be used singularly (as monotherapy) or in combination (polytherapy). Stiripentol is an antiepileptic medicine that was developed in France and was approved in 2007 by the European Medicines Agency as add‐on therapy with valproate and clobazam for the treatment of Dravet syndrome (a rare, drug‐resistant epilepsy that begins in the first year of life in an otherwise healthy infant). This review assesses the evidence for the use of stiripentol as add‐on treatment for drug‐resistant focal epilepsy in people taking antiepileptic medicines.
What did we find?
On the basis of our review criteria, we included only one study in the review. In the included study, 67 children were given a dummy medicine (placebo) in addition to their usual treatment for one month, and were then given add‐on stiripentol (without the placebo) for four months. The 32 children who responded to stiripentol (who had half the number of seizures or fewer when taking stiripentol compared with during the first month) were included in the next part of the study. Seventeen of these children continued add‐on stiripentol, while the other 15 received add‐on placebo. This stage of the study was randomised and double‐blind (the treatment was allocated at random, and neither the children nor the doctors knew who was receiving what treatment).
After two months, the study authors found no clear differences between the two treatment groups in terms of seizure reduction (the number of children with half the number of seizures or fewer) or seizure freedom (the number of children who had no seizures). However, the children taking stiripentol were more likely to have harmful side effects than those taking the placebo. The results of the study may not apply to a more general population because it only included children who responded to stiripentol.
What are the limitations of the evidence?
We have little confidence in the evidence because the study included a small number of children, several children left the study before the end, and the treatment given to all children in the first part of the study might have affected the number of seizures in the randomised, double‐blind part of the study.
Currently, no available evidence supports the use of stiripentol as add‐on treatment for drug‐resistant focal epilepsy. Large, randomised, well‐conducted trials on this topic are needed.
The evidence is current to March 2022.
Summary of findings
Summary of findings 1. Add‐on stiripentol compared with add‐on placebo for drug‐resistant focal epilepsy.
| Add‐on stiripentol compared with add‐on placebo for drug‐resistant focal epilepsy | ||||||
|
Patient or population: people with drug‐resistant focal epilepsy Settings: community Intervention: add‐on stiripentol Comparison: add‐on placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Stiripentol | |||||
| ≥ 50% reduction in seizure frequency | 467 per 1000 | 705 per 1000 (378 to 1000) | RR 1.51 (0.81 to 2.82) | 32 (1) | ⊕⊕⊖⊖ Lowa |
— |
| Seizure freedom | 200 per 1000 | 236 per 1000 (62 to 886) | RR 1.18 (0.31 to 4.43) | 32 (1) | ⊕⊕⊖⊖ Lowa |
— |
| ≥ 1 adverse effects | 267 per 1000 | 707 per 1000 (288 to 1000) | RR 2.65 (1.08 to 6.47) | 32 (1) | ⊕⊕⊖⊖ Lowa | — |
| Neurological adverse effects | 200 per 1000 | 530 per 1000 (176 to 1000) | RR 2.65 (0.88 to 8.01) | 32 (1) | ⊕⊕⊖⊖ Lowa | — |
| Gastrointestinal adverse effects | 0 events occurred in the placebo group | 6 events occurred in the stiripentol group | Not estimable | 32 (1) | See comment | 0 participants receiving placebo developed gastrointestinal adverse effects. |
| Dropouts | 533 per 1000 | 352 per 1000 (160 to 784) | RR 0.66 (0.30 to 1.47) | 32 (1) | ⊕⊕⊖⊖ Lowa | — |
| Quality of life | See comment | See comment | See comment | See comment | See comment | The included study did not assess quality of life. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) and is calculated according to the following formula: corresponding intervention risk, per 1000 = 1000 × ACR × RR. CI: confidence interval; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level for risk of bias due to high number of dropouts (high risk of attrition bias) and probable carry‐over effect, and one level for imprecision due to small sample size.
Background
This is an updated version of the Cochrane Review first published in 2014 (Brigo 2014), and last updated in 2020 (Brigo 2020).
Description of the condition
Epilepsy is one of the more common chronic neurological disorders; it affects 1% of the population worldwide.
Up to 30% of people with epilepsy continue to have seizures despite adequate therapy with antiepileptic drugs (AEDs), used singularly or in combination (Cockerell 1995; Granata 2009). These individuals are regarded as having drug‐resistant epilepsy. Although there is no universal definition of drug‐resistant epilepsy, most definitions refer to continued seizures despite AED treatment. In 2010, a task force of the International League Against Epilepsy (ILAE) defined this condition as "failure of adequate trials of two tolerated, appropriately chosen and used AED schedules (whether given as monotherapy or in combination) to achieve sustained seizure freedom" (Kwan 2010). The most frequently used definition encompasses continued seizures despite frequent medication changes (French 2006).
Seizures may occur within (and may rapidly engage) bilaterally distributed networks (generalised seizures), or networks that are discretely localised or more widely distributed within a single hemisphere (focal seizures; Berg 2010).
Description of the intervention
Standard drugs (e.g. carbamazepine, phenytoin, valproate) do not control all people’s seizures. Since the late 1990s and early 2000s, however, numerous newly available AEDs have offered promise for the treatment of drug‐resistant epilepsy. Stiripentol is an AED that was developed in France and approved by the European Medicines Agency (EMA) in 2007 for the treatment of Dravet syndrome as adjunctive therapy with valproate and clobazam (Chiron 2007).
The safety profile of stiripentol is good, with most adverse effects related to a significant increase in plasma concentrations of valproate and clobazam (Perez 1999). Adverse effects include drowsiness, ataxia, nausea, abdominal pain and loss of appetite with weight loss. Asymptomatic neutropenia is occasionally observed (Chiron 2007).
How the intervention might work
Stiripentol is structurally unrelated to any other marketed AED. In vitro research has shown that stiripentol increases gamma‐aminobutyric acid (GABA) transmission (Quilichini 2006), probably owing to allosteric modulation of the GABAA receptor (Fisher 2009). The efficacy of stiripentol could therefore be related to potentiation of GABAergic inhibitory neurotransmission (Quilichini 2006), and enhancement of the action of benzodiazepines (Fisher 2009). In humans, stiripentol also inhibits cytochrome P450 enzymes (CYP) in the liver, resulting in increased plasma concentrations of concomitant AEDs metabolised by CYP (Chiron 2005). In people affected by severe myoclonic epilepsy in infancy, now usually known as Dravet syndrome, there is a pharmacokinetic interaction between stiripentol and clobazam (Giraud 2006).
Why it is important to do this review
To date, no systematic reviews have synthesised the available evidence on the effect of stiripentol on any condition other than Dravet syndrome.
In this systematic review, we aimed to assess and summarise the existing evidence on the efficacy and tolerability of stiripentol as add‐on treatment for people with drug‐resistant focal epilepsy.
Objectives
To evaluate the efficacy and tolerability of stiripentol as add‐on treatment for people with drug‐resistant focal epilepsy who are taking AEDs.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) – whether double‐blind, single‐blind or unblinded – as they are considered to provide the most effective evidence for evaluating benefits and risks of treatment (Strauss 2005).
We excluded all other study designs, including cohort studies, cross‐over studies, case‐control studies, outcomes research, case studies, case series and expert opinions.
We analysed different treatment groups and controls separately.
We applied no language restrictions.
Types of participants
Eligible studies included people with focal epilepsy defined according to ILAE criteria (International League Against Epilepsy 1989). We applied no restrictions based on age, sex, ethnicity or disability. As there is no universally accepted definition of drug‐resistant epilepsy, for the purpose of this review we included all trials that assessed stiripentol in drug‐resistant epilepsy regardless of defining criteria, but we noted the definition used by the trial authors. We excluded people affected by Dravet syndrome, as another of our systematic reviews specifically assesses the role of stiripentol in this epileptic condition (Brigo 2013).
Types of interventions
The treatment group received stiripentol in addition to conventional AED treatment.
The control group received a matching add‐on placebo or other AED in addition to conventional AED treatment.
Types of outcome measures
For each outcome, we performed an intention‐to‐treat primary analysis, including all participants in the treatment group to which they were allocated, irrespective of the treatment they actually received.
Primary outcomes
Reduction in seizure frequency: proportion of participants with a reduction of at least 50% in seizure frequency at the end of the study compared with baseline
Seizure freedom: proportion of participants achieving total cessation of seizures. If the length of follow‐up was long enough, we used the most current ILAE‐proposed definition of seizure freedom: no seizures of any type for 12 months, or three times the longest (pre‐intervention) seizure‐free interval, whichever was longer (Kwan 2010).
Secondary outcomes
-
Adverse effects
Proportion of participants who experienced at least one adverse effect
Proportion of participants who experienced individual adverse effects (to be listed separately)
Proportion of dropouts or withdrawals due to adverse effects, lack of efficacy or other reasons
Improvement in quality of life as assessed by validated and reliable rating scales (e.g. the 31‐item Quality of Life in Epilepsy Inventory (QOLIE‐31))
Search methods for identification of studies
Electronic searches
We ran the original searches for this review in May 2012, and ran subsequent searches in August 2013, August 2015, August 2017, and February 2020. For the latest update, we searched the following databases on 28 March 2022.
Cochrane Register of Studies (CRS Web), using the search strategy set out in Appendix 1.
MEDLINE (Ovid, 1946 to March 25, 2022), using the search strategy set out in Appendix 2.
CRS Web includes RCTs and quasi‐RCTs from PubMed, Embase, ClinicalTrials.gov, the World Health Organization International Clinical Trials Registry Platform (ICTRP), the Cochrane Central Register of Controlled Trials (CENTRAL), and the Specialized Registers of Cochrane Review Groups, including Epilepsy. In MEDLINE (Ovid), the coverage end date always lags a few days behind the search date.
We applied no language restrictions.
Searching other resources
We contacted the manufacturers of stiripentol (Biocodex) and experts in the field by email on 31 May 2012, 13 August 2015, 22 August 2017, and 29 March 2022, for information about unpublished or ongoing studies. We reviewed the reference lists of retrieved studies to identify any additional reports of relevant studies. We also considered ILAE conference proceedings.
Data collection and analysis
As we only identified one eligible study, we were unable to implement the methods set out in the protocol of this review for assessing heterogeneity, reporting biases, synthesising data and performing subgroup and sensitivity analyses (Brigo 2012). If we identify more studies in future updates, we may be able to analyse the data according to the published protocol.
Selection of studies
Two review authors (FB and SCI) independently screened the titles and abstracts of all publications identified by the searches, and excluded those that were clearly ineligible. The same two review authors then assessed the full text articles of the remaining records against the inclusion criteria, resolving any disagreements by discussion.
Data extraction and management
Two review authors (FB and SCI) independently extracted data − in the rawest available form − from the published report of the included trial. We used data extraction forms and resolved disagreements by discussion. We contacted the trial authors to request missing information. The data we planned to record are listed below.
Participant factors
Age
Sex
Epileptic seizure type and epilepsy syndrome
Causes of epilepsy
Duration of epilepsy
Number of seizures or seizure frequency before randomisation
Presence of status epilepticus
Numbers and types of AEDs previously taken
Concomitant AEDs
Presence of neurological deficit/signs
Neuropsychological status
Electroencephalographic (EEG) findings
Neuroradiological findings (computed tomography (CT), magnetic resonance imaging (MRI))
Trial design
Criteria used to diagnose epilepsy
Definition of drug‐resistant or refractory epilepsy
Trial design (parallel group or cross‐over; unblinded, single‐blind or double‐blind)
Inclusion and exclusion criteria
Method of randomisation
Method of allocation concealment
Method of blinding
Stratification factors
Number of participants allocated to each group
Duration of different phases of the trial (baseline, titration, maintenance and optional open‐label extension (if any))
Intervention and control
Control treatment
Dosage of stiripentol
Duration of treatment period
Follow‐up data
Duration of follow‐up
Reasons for incomplete outcome data
Dropout or loss to follow‐up rates
Methods of analysis (e.g. intention‐to‐treat, per‐protocol, worst‐case or best‐case scenario)
Primary outcomes
Reduction in seizure frequency: number of participants with a reduction of at least 50% in seizure frequency at the end of the study (numerator)/number of participants at prerandomisation baseline period (denominator)
Seizure freedom: number of participants achieving total cessation of seizures (numerator)/number of participants at prerandomisation baseline period (denominator)
Secondary outcomes
Incidence of adverse effects of any type: numbers of adverse effects (numerator)/total number of participants at prerandomisation baseline period (denominator)
Quality of life as assessed by validated and reliable rating scales (e.g. QOLIE‐31)
Assessment of risk of bias in included studies
Two review authors (FB and NLB) assessed the risk of bias of the included trial according to approaches described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
The risk of bias domains included in this assessment were:
random sequence generation (selection bias);
allocation concealment (selection bias);
blinding of participants and personnel (performance bias);
blinding of outcome assessment (detection bias);
incomplete outcome data;
selective reporting (reporting bias); and
other bias (including outcome reporting bias).
The possible risk of bias judgements for each domain were:
low risk of bias;
uncertain risk of bias; and
high risk of bias.
Measures of treatment effect
Data for our chosen outcomes were dichotomous, and our preferred outcome statistic was the risk ratio (RR), calculated with 95% confidence intervals (CIs).
Unit of analysis issues
We planned to deal with any unit of analysis issues using the guidance in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021). Had we identified any studies that reported separate results for different doses of the same drug, we would have combined these data into a single treatment group to avoid a duplicative error with multiple‐armed trials.
Dealing with missing data
For each outcome, we performed an intention‐to‐treat primary analysis, including all participants in the treatment group to which they were allocated, irrespective of the treatment they actually received.
Assessment of heterogeneity
As only one study satisfied our inclusion criteria, we did not assess between‐study heterogeneity.
If we had included more than one study, we would have assessed heterogeneity of the intervention effects among trials using the standard Chi² statistic, rejecting the hypothesis of homogeneity if the P value was less than 0.10; and the I² statistic, interpreting the results as follows (in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021)).
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: considerable heterogeneity.
We would have combined trial outcomes to obtain a summary estimate of effect (and the corresponding CIs) using a fixed‐effect model, unless we had found considerable heterogeneity, in which case we would have explored the contributing factors for heterogeneity. Had we found considerable heterogeneity that could not readily be explained, we would have used a random‐effects model.
We would have assessed possible sources of heterogeneity (for example clinical heterogeneity, methodological heterogeneity or statistical heterogeneity) by performing a sensitivity analysis, as described in Sensitivity analysis.
Assessment of reporting biases
As only one study satisfied our inclusion criteria, we did not assess reporting biases.
If we had included 10 or more studies, we would have assessed reporting bias using a funnel plot (Brigo 2012). We would have examined the possible sources of funnel plot asymmetry (e.g. publication bias, language bias, citation bias, poor methodological quality, true heterogeneity) when interpreting the results.
Data synthesis
If we had included more than one study in our review, we would have synthesised the results in a meta‐analysis using Review Manager 5 (Review Manager 2020), provided we considered it clinically appropriate and had found no important clinical or methodological heterogeneity.
We would have synthesised data on all seizures and for each seizure type. We would have analysed different treatments and controls separately, including no treatment and placebo together.
Subgroup analysis and investigation of heterogeneity
As eligible data were limited, we did not perform any subgroup analyses. We had not planned to perform subgroup analyses to further investigate heterogeneity (Brigo 2012).
Sensitivity analysis
If we had included more than one study, we would have performed a sensitivity analysis as follows.
In the case of residual unexplained heterogeneity, we would have evaluated the robustness of the results of the meta‐analysis by comparing fixed‐effect and random‐effects model estimates after removing trials with low methodological quality or with large effect sizes.
We would have also used the worst‐case and best‐case scenarios wherever possible. If removing these trials did not affect the results, we would have considered the evidence to be robust.
Summary of findings and assessment of the certainty of the evidence
We included all outcomes assessed in this review in Table 1, using GRADE criteria to evaluate the certainty of the evidence (Guyatt 2008).
Results
Description of studies
See the Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The update of searches for this review yielded eight results (seven from Cochrane Register of Studies (CRS Web) and one from MEDLINE). After removing two duplicates and one obviously irrelevant item, we screened the titles and abstracts of the remaining five records, finding that none met our inclusion criteria (Figure 1). Hence, we found no additional studies for inclusion in this review update. In the original version of this review (Brigo 2014), we had identified one study that met our inclusion criteria (Chiron 2006). None of the subsequent updates included any further studies (Brigo 2015; Brigo 2018; Brigo 2020).
1.
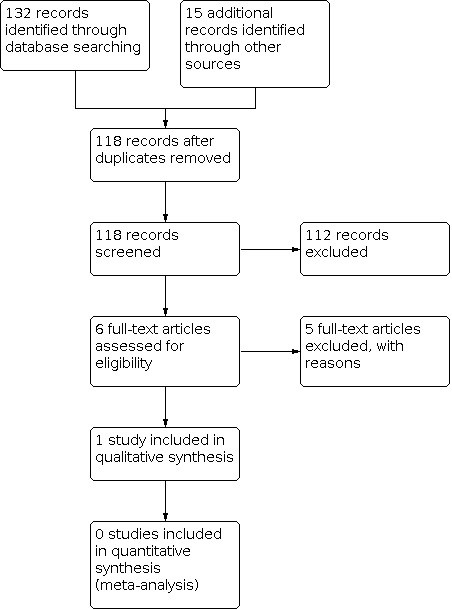
Study flow diagram. The results shown in this figure include the original searches conducted for the review and all subsequent updates.
Included studies
Chiron 2006
Chiron 2006 aimed to study stiripentol as add‐on therapy to carbamazepine for childhood focal epilepsy. It used a responder‐enriched design, whereby participants responding to stiripentol during a prerandomisation phase were randomly assigned to continue stiripentol or to receive a placebo instead. This trial therefore compared the effects of continuing versus withdrawing stiripentol.
The inclusion criteria were:
focal seizures;
carbamazepine as co‐medication, with a benzodiazepine (clobazam or clonazepam) or vigabatrin, or both, administered in association; and
carbamazepine dose of at least 400 mg/day.
The study authors excluded children receiving other drugs and children whose parents were unable to comply regularly with drug delivery and daily seizure diaries. Participants were defined as "refractory to the usual antiepileptic drugs (including valproate, carbamazepine, benzodiazepines and phenytoin), as well as to vigabatrin", although the study authors did not list drug‐resistant epilepsy as an inclusion criterion.
In the first study period (single‐blind baseline), 67 children received an add‐on placebo for one month. In the second (open) study period, the same 67 children received add‐on stiripentol for four months. These first two study periods adopted a non‐randomised before‐after design. At the end of the open period, responders (defined as children with at least a 50% decrease in seizure frequency during the third month of the open period compared with baseline) were randomly assigned to add‐on stiripentol or to add‐on placebo for a two‐month, double‐blind period.
We only included data from the randomised, double‐blind, placebo‐controlled portion of the trial in the present review. This third period included 32 children: 18 boys (7 in the stiripentol group and 11 in the placebo group) and 14 girls (10 in the stiripentol group and 4 in the placebo group). Mean age was 8.0 years (standard deviation (SD) 3.0 years) in the stiripentol group and 10.4 years (SD 3.4 years) in the placebo group.
At the end of the study, all 32 responders were put on long‐term open stiripentol.
The study authors made no mention of any conflicts of interest or sources of funding.
Excluded studies
None of the records obtained through the updated search strategy met our eligibility criteria and were therefore considered irrelevant (see Results of the search).
In the previous versions of this review (Brigo 2014; Brigo 2015; Brigo 2018; Brigo 2020), we excluded a total of five studies. Three of them were non‐randomised trials with an uncontrolled before‐after design (Loiseau 1988; Perez 1999; Rascol 1989). Chiron 2000, published as a conference proceeding, provided preliminary results (interim analyses) of our included study (Chiron 2006), which was published a few years later as a full length paper with definitive results. The other excluded study was a randomised, double‐blind, parallel‐group trial that evaluated the efficacy of stiripentol as add‐on therapy to carbamazepine versus carbamazepine monotherapy in people with epilepsy uncontrolled by carbamazepine monotherapy (Loiseau 1990). We excluded this study for two reasons: firstly, the study authors did not clearly specify whether any participants had focal epilepsy; and secondly, the participants had epilepsy that was "uncontrolled by carbamazepine monotherapy", whereas most available definitions of drug‐resistant epilepsy require failure of at least two AEDs (Berg 2006). As a consequence, we did not consider the participants in this study to be affected by drug‐resistant epilepsy, even when we applied the internationally accepted definition: failure of adequate trials of two tolerated, appropriately chosen and used AED schedules (whether given as monotherapy or in combination) to achieve sustained seizure freedom (Kwan 2010).
Risk of bias in included studies
See Characteristics of included studies table.
Allocation
Chiron 2006 used a computer‐generated list to randomly assign participants, and a pharmacist dosed the tablets, to ensure blinding of the investigators. However, through its responder‐enriched design, this study conducted a primary efficacy evaluation of an enriched population of participants, as the result of random assignment only of participants who responded to open‐label treatment (high risk of selection bias).
Blinding
The study authors described the second part of the trial as double‐blinded (low risk of performance bias). To maintain blinding and to mask the increase of the carbamazepine dose as stiripentol was gradually withdrawn in the placebo group, each participant received tablets of both stiripentol and placebo of stiripentol and tablets of both carbamazepine and placebo of carbamazepine. Part of the carbamazepine schedule was administered as "open carbamazepine"; however, the dose could be decreased when necessary.
Incomplete outcome data
The study authors reported the number of dropouts and specified reasons for dropout. Although these reasons were similar among participants in the two groups, and although strict escape criteria are necessary in a responder‐enriched study, the number of dropouts in both arms far exceeded 20% (35.5% in the stiripentol group, and 53.3% in the placebo group). We therefore considered the study to be at high risk of attrition bias.
Selective reporting
The study authors reported on all prespecified outcomes (low risk of reporting bias).
Other potential sources of bias
Risk of a carry‐over effect in the add‐on placebo group seemed to be high, because add‐on stiripentol was withdrawn from the placebo participants over three weeks (a long period, especially given that the overall length of the randomised, double‐blind portion of the trial was only two months). As a consequence, a carry‐over effect may have reduced seizure frequency in the add‐on placebo group.
Effects of interventions
See: Table 1
Add‐on stiripentol versus add‐on placebo
See Table 1.
We found one study that compared add‐on stiripentol with add‐on placebo in 32 participants (Chiron 2006). As outlined under Description of studies, this trial used a responder‐enriched design, whereby participants responding to stiripentol during a prerandomisation phase were randomly assigned to continue stiripentol or to receive a placebo instead. This trial therefore compared the effects of continuing versus withdrawing stiripentol.
Primary outcomes
See Data and analyses.
Reduction of 50% or more in seizure frequency
The study provided no clear evidence of a reduction in seizure frequency when comparing add‐on stiripentol with add‐on placebo (RR 1.51, 95% CI 0.81 to 2.82; Analysis 1.1). Seven participants dropped out because of increased seizure frequency compared to the baseline in the add‐on placebo group and five in the add‐on stiripentol group.
1.1. Analysis.

Comparison 1: Add‐on stiripentol versus add‐on placebo, Outcome 1: ≥ 50% seizure reduction
Seizure freedom
Add‐on stiripentol may have little or no effect on seizure freedom compared with add‐on placebo (RR 1.18, 95% CI 0.31 to 4.43; Analysis 1.2).
1.2. Analysis.

Comparison 1: Add‐on stiripentol versus add‐on placebo, Outcome 2: Seizure freedom
Secondary outcomes
See Data and analyses.
Adverse effects
Add‐on stiripentol led to greater risk of adverse effects considered as a whole when compared with placebo (RR 2.65, 95% CI 1.08 to 6.47; Analysis 1.3). When we considered specific adverse events, CIs were very wide and included the possibility of substantial increases and small reductions in risk of neurological adverse effects (RR 2.65, 95% CI 0.88 to 8.01; Analysis 1.4). We could not calculate the RR for gastrointestinal adverse effects because there were no events in the comparator group.
1.3. Analysis.

Comparison 1: Add‐on stiripentol versus add‐on placebo, Outcome 3: ≥ 1 adverse effects
1.4. Analysis.

Comparison 1: Add‐on stiripentol versus add‐on placebo, Outcome 4: Neurological adverse effects
Proportion of dropouts or withdrawals due to side effects, lack of efficacy or other reasons
We noted no clear reduction in the risk of study withdrawal, which was high in both groups (RR 0.66, 95% CI 0.30 to 1.47; Analysis 1.5). In the placebo group, eight participants (53.3%) dropped out because of loss of response (seven for an increase in seizure frequency and one for an increase in seizure severity), and four experienced worsening compared with baseline. Six participants in the stiripentol group (35.3%) dropped out (five because of an increase in seizure frequency and one for an increase in seizure severity).
1.5. Analysis.

Comparison 1: Add‐on stiripentol versus add‐on placebo, Outcome 5: Dropouts
Improvement in quality of life as assessed by validated and reliable rating scales
The included study did not assess improvement in quality of life.
Discussion
This review aimed to assess the efficacy and tolerability of stiripentol as add‐on treatment for drug‐resistant epilepsy.
In this systematic review update, we identified no additional studies for inclusion. Hence, we have made no changes to the conclusions presented in previous versions of this review (Brigo 2014; Brigo 2015; Brigo 2018; Brigo 2020).
Summary of main results
We included only one study, which we had identified in the first version of this review (Chiron 2006). This study adopted a responder‐enriched design. Although all included participants were "refractory to the usual antiepileptic drugs (including valproate, carbamazepine, benzodiazepines and phenytoin), as well as to vigabatrin as a new drug", the presence of drug‐resistant epilepsy was not listed among the inclusion criteria. Furthermore, the study authors did not provide a definition of refractory epilepsy.
Chiron 2006 provided no clear evidence of seizure reduction (of at least 50%) or of seizure freedom with add‐on stiripentol compared with placebo. Add‐on stiripentol led to greater risk of adverse effects considered as a whole compared with placebo; however we are uncertain of this effect, because the results are imprecise. There was no clear difference in neurological adverse effects or in gastrointestinal adverse effects between add‐on stiripentol and placebo. The study showed no clear differences in the proportion of dropouts between add‐on stiripentol and add‐on placebo, although with a trend towards increased dropouts among add‐on placebo participants.
Overall completeness and applicability of evidence
Despite an overall low risk of bias, the responder‐enriched design of the included trial raises several ethical and methodological concerns. This design shifts the focus to a participant subgroup that benefits most from the treatment, according to the accumulating data. Only the third phase of Chiron 2006 met the inclusion criteria of this systematic review (randomised, add‐on, placebo‐controlled, double‐blind trial), whereas the first two phases adopted a non‐randomised, before‐after design. By including only responders to add‐on stiripentol (i.e. those experiencing a decrease in seizure frequency of 50% or more during the third month of the open period versus baseline) in the randomised portion of the study, the study authors may have severely reduced the external validity of the results, limiting their generalisability to a wider population. This study design has therefore resulted in a primary efficacy evaluation of a highly selected enriched population of participants due to random assignment only of those who responded to open‐label treatment (high risk of selection bias).
Furthermore, implicit in the responder‐enriched design is the risk of a carry‐over effect in the add‐on placebo group. A carry‐over effect occurs when the effects of an intervention given during one period persist into a subsequent period, thus interfering with the effects of the subsequent intervention. Risk of a carry‐over effect in the add‐on placebo group of Chiron 2006 seems high, because add‐on stiripentol was withdrawn from the placebo participants over three weeks (a long period, especially given that the overall length of the randomised, double‐blind portion of the trial was only two months). As a consequence, a carry‐over effect may have reduced seizure frequency in the add‐on placebo group. Ideally, the investigators should have included a washout period before the randomised, double‐blind phase to reduce the carry‐over effect.
Conversely, the responder‐enriched design in this study carries the risk of a withdrawal effect secondary to stiripentol withdrawal in the placebo group during the randomised phase. This withdrawal effect may be responsible for an increase in seizure frequency (which, unlike reduction in seizure frequency, becomes a relevant endpoint within such a study design). This should be carefully taken into account when strict escape criteria are defined, to prevent exposure of participants in the add‐on placebo group to seizures that may become more severe or more prolonged and may even evolve into status epilepticus. Regarding this last aspect, it is noteworthy to consider that the percentage of dropouts was extremely high in both treatment arms as a result of increased seizure frequency or severity.
Length of follow‐up for the randomised, double‐blind study (only two months) was probably inadequate to permit evaluation of changes in seizure frequency, and was inadequate to assess seizure freedom (according to the ILAE definition; Kwan 2010). As RCTs tend to have short follow‐up, it is unlikely that they can report data on seizure freedom.
Additional research is needed to assess the efficacy and tolerability of add‐on stiripentol for the treatment of drug‐resistant focal epilepsy. Future studies should be randomised and double‐blinded, should aim to recruit a sufficiently large number of participants and should assess clinically meaningful outcome measures, while adopting an internationally accepted definition of drug‐resistant epilepsy (Kwan 2010).
Quality of the evidence
We cannot generalise the results of Chiron 2006 to a wider population for two main reasons: first, because the study authors only included responders to add‐on stiripentol in the randomised, add‐on, placebo‐controlled, double‐blind portion of the study; and second, because the sample size was very small and the dropout rates very high.
Because of the adopted design, carry‐over and withdrawal effects probably influenced outcomes related to seizure frequency. Using the GRADE methodology, we judged the certainty of evidence as low for all outcomes. We downgraded one level for risk of bias due to high number of dropouts (high risk of attrition bias), and due to the probable carry‐over effect, and by one level for imprecision due to the small sample size (Table 1).
Potential biases in the review process
Although we made every effort to identify all RCTs on the use of stiripentol for the treatment of drug‐resistant focal epilepsy through a comprehensive search of the literature, it is possible that we missed small studies published in the less accessible literature.
Agreements and disagreements with other studies or reviews
No other studies or reviews on the same topic have been published so far.
Authors' conclusions
Implications for practice.
In this review update, we found no new studies and therefore made no changes to the conclusions as presented in the initial review. Currently, no available evidence supports the use of add‐on stiripentol for treating drug‐resistant focal epilepsy. Although we derived very limited information from only one included study, we noted that adverse effects considered as a whole seemed to occur more frequently with add‐on stiripentol than with add‐on placebo.
Implications for research.
Additional research is needed to assess the efficacy and tolerability of add‐on stiripentol for the treatment of drug‐resistant focal epilepsy. Future research should consist of randomised, double‐blind studies and should aim to recruit sufficiently large numbers of participants and assess clinically meaningful outcome measures. Investigators should avoid a responder‐enriched design because of the risk of carry‐over and withdrawal effects in the add‐on placebo group, and because of the reduced external validity of this study design. Furthermore, they should adopt the internationally accepted definition of drug‐resistant epilepsy.
What's new
| Date | Event | Description |
|---|---|---|
| 28 March 2022 | New search has been performed | Searches updated 28 March 2022; no new relevant studies were identified. |
| 28 March 2022 | New citation required but conclusions have not changed | Conclusions are unchanged. |
History
Protocol first published: Issue 5, 2012 Review first published: Issue 1, 2014
| Date | Event | Description |
|---|---|---|
| 27 February 2020 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 27 February 2020 | New search has been performed | Searches updated 27 February 2020; no new studies were identified. |
| 21 August 2017 | New search has been performed | Searches updated 21 August 2017; no new studies were identified. |
| 21 August 2017 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 10 August 2015 | New search has been performed | Searches updated 10 August 2015 |
| 10 August 2015 | New citation required but conclusions have not changed | No new relevant studies identified; no changes made to conclusions |
Acknowledgements
This review update was supported by the National Institute for Health and Care Research, via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service or the Department of Health and Social Care.
We thank Monica Storti for contributions to the original protocol.
We are indebted to the following epilepsy experts, whom we contacted for information about unpublished or ongoing studies: Blaise Bourgeois, Barry Gidal, William H Theodore, Stefano Sartori, Jacqueline French, Eugen Trinka, Ronit Pressler, Pasquale Striano, Hans Hartmann, and Susanne Knake. We thank Marie‐Emmanuelle le Guern (Biocodex) for providing us with recent publications and for searching unpublished or ongoing trials related to use of add‐on stiripentol in epilepsy.
Cochrane Epilepsy supported the authors in the development of this review. The following people conducted the editorial process for this review.
Sign‐off Editor (final editorial decision): Tony Marson
Managing Editor (provided editorial guidance to authors, edited the update, conducted editorial policy checks): Rachael Kelly
Copy Editor (copy editing and production): Julia Turner
Appendices
Appendix 1. Cochrane Register of Studies (CRS Web) search strategy
1. (stiripentol or diacomit):AB,KW,KY,MC,MH,TI AND CENTRAL:TARGET
2. MESH DESCRIPTOR Epilepsies, Partial EXPLODE ALL AND CENTRAL:TARGET
3. ((partial or focal) and (seizure* or epilep*)):AB,KW,KY,MC,MH,TI AND CENTRAL:TARGET
4. (secondar* and (generalized or generalised) and seizure*):AB,KW,KY,MC,MH,TI AND CENTRAL:TARGET
5. #2 OR #3 OR #4
6. #1 AND #5
7. (monotherap* NOT (adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*)):TI AND CENTRAL:TARGET
8. #6 NOT #7
9. >21/08/2017:CRSCREATED AND CENTRAL:TARGET
10. #8 AND #9
Appendix 2. MEDLINE search strategy
This strategy includes a modification of the Cochrane Highly Sensitive Search Strategy for identifying randomised trials (Lefebvre 2022).
1. (stiripentol or Diacomit).mp.
2. exp Epilepsies, Partial/
3. ((partial or focal) and (seizure$ or epilep$)).mp.
4. (secondar$ and generali?ed and seizure$).mp.
5. 2 or 3 or 4
6. exp controlled clinical trial/ or (randomi?ed or placebo or randomly).ab.
7. clinical trials as topic.sh.
8. trial.ti.
9. 6 or 7 or 8
10. exp animals/ not humans.sh.
11. 9 not 10
12. 1 and 5 and 11
13. (monotherap$ not (adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$)).ti.
14. 12 not 13
15. limit 14 to ed=20170821‐20220328
16. 14 not (1$ or 2$).ed.
17. 16 and (2017$ or 2018$ or 2019$ or 2020$ or 2021$ or 2022$).dt.
18. 15 or 17
19. remove duplicates from 18
Data and analyses
Comparison 1. Add‐on stiripentol versus add‐on placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 ≥ 50% seizure reduction | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.51 [0.81, 2.82] |
| 1.2 Seizure freedom | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.31, 4.43] |
| 1.3 ≥ 1 adverse effects | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.65 [1.08, 6.47] |
| 1.4 Neurological adverse effects | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.65 [0.88, 8.01] |
| 1.5 Dropouts | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.30, 1.47] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Chiron 2006.
| Study characteristics | ||
| Methods |
|
|
| Participants | The 32 children who responded to add‐on stiripentol during the prerandomisation phase (≥ 50% decrease in seizure frequency during third month of open period versus baseline) were randomly assigned to continue add‐on stiripentol or to receive add‐on placebo. Add‐on stiripentol group: 7 boys, 10 girls (total 17 participants); mean age 8.0 years (SD 3.0 years) Add‐on placebo group: 11 boys, 4 girls (total 15 participants); mean age 10.4 years (SD 3.4 years) Inclusion criteria for baseline period
Exclusion criteria for baseline period
Inclusion criteria for randomised, placebo‐controlled, double‐blind, trial
Exclusion criteria (escape criteria) during randomised phase
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Participants were randomly assigned by a computer‐generated list. However, through its responder‐enriched design, this study resulted in a primary efficacy evaluation of an enriched population of participants, due to random assignment only of those who responded to open‐label treatment (high risk of selection bias). |
| Allocation concealment (selection bias) | Low risk | Central allocation (pharmacy‐controlled randomisation). "Each patient received tablets of both stiripentol and 'placebo of stiripentol' and tablets of both carbamazepine and 'placebo of carbamazepine'". "Individual tablets were prepared by the pharmacist". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Second part of the trial was defined as "double‐blind". "Each patient received tablets of both stiripentol and 'placebo of stiripentol' and tablets of both carbamazepine and 'placebo of carbamazepine'". "Individual tablets were prepared by the pharmacist". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Each patient received tablets of both stiripentol and 'placebo of stiripentol' and tablets of both carbamazepine and 'placebo of carbamazepine'". "Individual tablets were prepared by the pharmacist". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Numbers of dropouts from each group were reported, along with reasons for dropout. However, number of dropouts in both arms was high (35.3% in stiripentol group and 53.3% in placebo group). |
| Selective reporting (reporting bias) | Low risk | Published reports include all expected outcomes |
| Other bias | High risk | High risk of carry‐over and withdrawal effects |
AED: anti‐epileptic drug; SD: standard deviation.
Characteristics of excluded studies [ordered by year]
| Study | Reason for exclusion |
|---|---|
| Loiseau 1988 | Not randomised. Uncontrolled before‐after design. |
| Rascol 1989 | Not randomised. Uncontrolled before‐after design. |
| Loiseau 1990 | Not specified whether participants had focal epilepsy. Participants did not have refractory epilepsy. |
| Perez 1999 | Not randomised. Uncontrolled before‐after design. |
| Chiron 2000 | This study was published as a conference proceeding and provided preliminary results (interim analyses) of Chiron 2006, which is included in the review. |
Differences between protocol and review
We added the GRADE quality assessment criteria in the summary of findings table (Guyatt 2008).
Contributions of authors
FB wrote the text of the updated review, which was critically revised by SI and NB.
Sources of support
Internal sources
No sources of support provided
External sources
-
National Institute for Health Research, UK
This review update was supported by the National Institute for Health and Care Research, via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health and Social Care.
Declarations of interest
FB: none SI: none NB: none
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Chiron 2006 {published data only}
- Chiron C, Tonnelier S, Rey E, Brunet ML, Tran A, d'Athis P, et al. Stiripentol in childhood partial epilepsy: randomized placebo-controlled trial with enrichment and withdrawal design. Journal of Child Neurology 2006;21(6):496-502. [DOI: 10.1177/08830738060210062101] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Chiron 2000 {published data only}
- Chiron C, Tran A, Rey E, d'Athis P, Vincent J, Tonnelier S, et al. Stiripentol in childhood partial epilepsy: a placebo-controlled trial. Epilepsia 2000;41(Suppl 7):191, Abstract no: 3.070. [DOI] [PubMed] [Google Scholar]
Loiseau 1988 {published data only}
- Loiseau P, Strube E, Tor J, Levy RH, Dodrill C. Neurophysiological and therapeutic evaluation of stiripentol in epilepsy. Preliminary results. Revue Neurologique 1988;144(3):165-72. [PMID: ] [PubMed] [Google Scholar]
Loiseau 1990 {published data only}
- Loiseau P, Levy RJ, Houin G, Rascol O, Dordain G. Randomized double-blind, parallel, multicenter trial of stiripentol added to carbamazepine in the treatment of carbamazepine-resistant epilepsies. An interim analysis. Epilepsia 1990;31(5):618-9. [Google Scholar]
Perez 1999 {published data only}
- Perez J, Chiron C, Musial C, Rey E, Blehaut H, d'Athis P, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia 1999;40(11):1618-26. [DOI: 10.1111/j.1528-1157.1999.tb02048.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Rascol 1989 {published data only}
- Rascol O, Squalli A, Montastruc JL, Garat A, Houin G, Lachau S, et al. A pilot study of stiripentol, a new anticonvulsant drug, in complex partial seizures uncontrolled by carbamazepine. Clinical Neuropharmacology 1989;12(2):119-23. [DOI: 10.1097/00002826-198904000-00006] [PMID: ] [DOI] [PubMed] [Google Scholar]
Additional references
Berg 2006
- Berg AT, Kelly MM. Defining intractability: comparisons among published definitions. Epilepsia 2006;47(2):431-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Berg 2010
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, Van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51(4):676-85. [PMID: ] [DOI] [PubMed] [Google Scholar]
Brigo 2013
- Brigo F, Igwe SC, Bragazzi NL. Antiepileptic drugs for the treatment of infants with severe myoclonic epilepsy. Cochrane Database of Systematic Reviews 2017, Issue 5. Art. No: CD010483. [DOI: 10.1002/14651858.CD010483.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chiron 2005
- Chiron C. Stiripentol. Expert Opinion on Investigational Drugs 2005;14(7):905-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Chiron 2007
- Chiron C. Stiripentol. Neurotherapeutics 2007;4(1):123. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet 1995;346(8968):140-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Fisher 2009
- Fisher JL. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology 2009;56(1):190-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
French 2006
- French JA. Refractory epilepsy: one size does not fit all. Epilepsy Currents 2006;6(6):177-80. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giraud 2006
- Giraud C, Treluyer JM, Rey E, Chiron C, Vincent J, Pons G, et al. In vitro and in vivo inhibitory effect of stiripentol on clobazam metabolism. Drug Metabolism and Disposition 2006;34(4):608-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Granata 2009
- Granata T, Marchi N, Carlton E, Ghosh C, Gonzalez-Martinez J, Alexopoulos AV, et al. Management of the patient with medically refractory epilepsy. Expert Review of Neurotherapeutics 2009;9(12):1791-802. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al, GRADE Working Group. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Higgins 2021
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook/archive/v6.2/.
International League Against Epilepsy 1989
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30(4):389-99. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kwan 2010
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51(6):1069-77. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lefebvre 2022
- Lefebvre C, Glanville J, Briscoe S, Featherstone R, Littlewood A, Marshall C, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Quilichini 2006
- Quilichini PP, Chiron C, Ben-Ari Y, Gozlan H. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABA-A receptor channels. Epilepsia 2006;47(4):704-16. [PMID: ] [DOI] [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- Review Manager 5 (RevMan 5). Version 5.4. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2020.
Strauss 2005
- Strauss S, Richardson W, Glasziou P, Haynes R. Evidence-Based Medicine: How to Practice and Teach EBM. 3rd edition. Edinburgh (UK): Churchill Livingstone, 2005. [Google Scholar]
References to other published versions of this review
Brigo 2012
- Brigo F, Storti M. Stiripentol for focal refractory epilepsy. Cochrane Database of Systematic Reviews 2012, Issue 5. Art. No: CD009887. [DOI: 10.1002/14651858.CD009887] [DOI] [PubMed] [Google Scholar]
Brigo 2014
- Brigo F, Storti M. Stiripentol for focal refractory epilepsy. Cochrane Database of Systematic Reviews 2014, Issue 1. Art. No: CD009887. [DOI: 10.1002/14651858.CD009887.pub2] [DOI] [PubMed] [Google Scholar]
Brigo 2015
- Brigo F, Igwe SC, Bragazzi NL. Stiripentol for focal refractory epilepsy. Cochrane Database of Systematic Reviews 2015, Issue 10. Art. No: CD009887. [DOI: 10.1002/14651858.CD009887.pub3] [DOI] [PubMed] [Google Scholar]
Brigo 2018
- Brigo F, Igwe SC, Bragazzi NL. Stiripentol add-on therapy for focal refractory epilepsy. Cochrane Database of Systematic Reviews 2018, Issue 5. Art. No: CD009887. [DOI: 10.1002/14651858.CD009887.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brigo 2020
- Brigo F, Igwe SC, Bragazzi NL. Stiripentol add-on therapy for drug-resistant focal epilepsy. Cochrane Database of Systematic Reviews 2020, Issue 5. Art. No: CD009887. [DOI: 10.1002/14651858.CD009887.pub5] [DOI] [PMC free article] [PubMed] [Google Scholar]